DELSTRIGO (doravirine 100 mg lamivudine 300 mg tenofovir disoproxil fumarate 300 mg) Dailymed
Generic: doravirine, lamivudine, and tenofovir disoproxil fumarate is used for the treatment of Hepatitis B, Chronic Acquired Immunodeficiency Syndrome

IMPRINT: LOGO 776
SHAPE: oval
COLOR: yellow
All Imprints
doravirine 100 mg / lamivudine 300 mg / tenofovir disoproxil fumarate 300 mg oral tablet [delstrigo] - logo 776 oval yellow
Boxed Warning
Warning: Posttreatment Acute Exacerbation Of Hepatitis B

Go PRO for all pill images
Recent Major Changes Section
Warnings and Precautions, Severe Skin Reactions ( 5.1 )11/2024 Warnings and Precautions, Bone Loss and Mineralization Defects ( 5.5 )01/2025
Warning: Posttreatment Acute Exacerbation Of Hepatitis B
Severe acute exacerbations of hepatitis B (HBV) have been reported in people with concomitant HIV-1 and HBV who have discontinued lamivudine or tenofovir disoproxil fumarate (TDF), which are components of DELSTRIGO. Hepatic function should be monitored closely with both clinical and laboratory follow-up for at least several months in patients who are coinfected with HIV-1 and HBV and discontinue DELSTRIGO. If appropriate, initiation of anti-hepatitis B therapy may be warranted [see Warnings and Precautions (5.2)].
WARNING: POSTTREATMENT ACUTE EXACERBATION OF HEPATITIS B
See full prescribing information for complete boxed warning.
Severe acute exacerbations of hepatitis B (HBV) have been reported in people with concomitant HIV-1 and HBV who have discontinued lamivudine or tenofovir disoproxil fumarate (TDF), two of the components of DELSTRIGO. Closely monitor hepatic function in these patients. If appropriate, initiation of anti-hepatitis B therapy may be warranted. (5.2 )
1 Indications And Usage
DELSTRIGO¬ģ is indicated as a complete regimen for the treatment of HIV-1 infection in adults and pediatric patients weighing at least 35 kg:
- with no prior antiretroviral treatment history, OR
- to replace the current antiretroviral regimen in those who are virologically-suppressed (HIV-1 RNA less than 50 copies per mL) on a stable antiretroviral regimen with no history of treatment failure and no known substitutions associated with resistance to the individual components of DELSTRIGO [see Clinical Studies (14)].
DELSTRIGO is a three-drug combination of doravirine (a non-nucleoside reverse transcriptase inhibitor [NNRTI]), lamivudine, and tenofovir disoproxil fumarate (both nucleoside analogue reverse transcriptase inhibitors) and is indicated as a complete regimen for the treatment of HIV-1 infection in adults and pediatric patients weighing at least 35 kg:
- with no antiretroviral treatment history, OR
- to replace the current antiretroviral regimen in those who are virologically-suppressed (HIV-1 RNA less than 50 copies per mL) on a stable antiretroviral regimen with no history of treatment failure and no known substitutions associated with resistance to the individual components of DELSTRIGO. (
1 )
2 Dosage And Administration
- Testing: Prior to or when initiating DELSTRIGO, test for HBV infection. Prior to or when initiating DELSTRIGO, and during treatment with DELSTRIGO, on a clinically appropriate schedule, assess serum creatinine, estimated creatinine clearance, urine glucose and urine protein in all patients. In patients with chronic kidney disease, also assess serum phosphorus. (
2.1 )- Recommended dosage: One tablet taken orally once daily with or without food in adults and pediatric patients weighing at least 35 kg. (
2.2 )- Renal impairment: Not recommended in patients with estimated creatinine clearance below 50 mL per minute. (
2.3 )- Dosage adjustment with rifabutin: Take one tablet of DELSTRIGO once daily, followed by one tablet of doravirine 100 mg (PIFELTRO) approximately 12 hours after the dose of DELSTRIGO. (
2.4 )2.1Testing When Initiating and During Treatment with DELSTRIGO
Prior to or when initiating DELSTRIGO, test patients for HBV infection [see Warnings and Precautions (5.2)].
Prior to or when initiating DELSTRIGO, and during treatment with DELSTRIGO, on a clinically appropriate schedule, assess serum creatinine, estimated creatinine clearance, urine glucose, and urine protein in all patients. In patients with chronic kidney disease, also assess serum phosphorus [see Warnings and Precautions (5.3)].
2.2Recommended Dosage
DELSTRIGO is a fixed-dose combination product containing 100 mg of doravirine (DOR), 300 mg of lamivudine (3TC), and 300 mg of TDF. The recommended dosage of DELSTRIGO in adults and pediatric patients weighing at least 35 kg is one tablet taken orally once daily with or without food [see Clinical Pharmacology (12.3)].
2.3Renal Impairment
Because DELSTRIGO is a fixed-dose combination tablet and the dosage of lamivudine and TDF cannot be adjusted, DELSTRIGO is not recommended in patients with estimated creatinine clearance less than 50 mL/min [see Warnings and Precautions (5.3) and Use in Specific Populations (8.6)].
2.4Dosage Adjustment with Rifabutin
If DELSTRIGO is co-administered with rifabutin, take one tablet of DELSTRIGO once daily, followed by one tablet of doravirine 100 mg (PIFELTRO) approximately 12 hours after the dose of DELSTRIGO for the duration of rifabutin co-administration [see Drug Interactions (7.2) and Clinical Pharmacology (12.3)].
3 Dosage Forms And Strengths
DELSTRIGO film-coated tablets are yellow, oval-shaped tablets, debossed with the corporate logo and 776 on one side and plain on the other side. Each tablet contains 100 mg doravirine, 300 mg lamivudine, and 300 mg tenofovir disoproxil fumarate (equivalent to 245 mg of tenofovir disoproxil).
- Tablets: 100 mg of doravirine, 300 mg of lamivudine, and 300 mg of tenofovir disoproxil fumarate. (
3 )
4 Contraindications
- DELSTRIGO is contraindicated when co-administered with drugs that are strong cytochrome P450 (CYP)3A enzyme inducers as significant decreases in doravirine plasma concentrations may occur, which may decrease the effectiveness of DELSTRIGO [see Warnings and Precautions (5.4), Drug Interactions (7.2), and Clinical Pharmacology (12.3)]. These drugs include, but are not limited to, the following:
- the anticonvulsants carbamazepine, oxcarbazepine, phenobarbital, phenytoin- the androgen receptor inhibitor enzalutamide- the antimycobacterials rifampin, rifapentine- the cytotoxic agent mitotane- St. John's wort (Hypericum perforatum)- DELSTRIGO is contraindicated in patients with a previous hypersensitivity reaction to lamivudine.
- DELSTRIGO is contraindicated when co-administered with drugs that are strong cytochrome P450 (CYP)3A enzyme inducers as significant decreases in doravirine plasma concentrations may occur, which may decrease the effectiveness of DELSTRIGO. (
4 )- DELSTRIGO is contraindicated in patients with a previous hypersensitivity reaction to lamivudine.
5 Warnings And Precautions
- Severe skin reactions, including Stevens-Johnson syndrome (SJS)/toxic epidermal necrolysis (TEN), have been reported during the postmarketing experience with doravirine-containing regimens. Discontinue DELSTRIGO, and other medications known to be associated with severe skin reactions, immediately if a painful rash with mucosal involvement or a progressive severe rash develops, and closely monitor clinical status. (
5.1 )- New onset or worsening renal impairment: Prior to or when initiating DELSTRIGO, and during treatment with DELSTRIGO, on a clinically appropriate schedule, assess serum creatinine, estimated creatinine clearance, urine glucose, and urine protein in all patients. Avoid administering DELSTRIGO with concurrent or recent use of nephrotoxic drugs. (
5.3 )- Bone loss and mineralization defects: Consider monitoring BMD in patients with a history of pathologic fracture or other risk factors of osteoporosis or bone loss. (
5.5 )- Monitor for Immune Reconstitution Syndrome. (
5.6 )5.1 Severe Skin Reactions
Severe skin reactions, including Stevens-Johnson syndrome (SJS)/toxic epidermal necrolysis (TEN), have been reported during the postmarketing experience with doravirine-containing regimens [see Adverse Reactions (6.2)]. Discontinue DELSTRIGO, and other medications known to be associated with severe skin reactions, immediately if a painful rash with mucosal involvement or a progressive severe rash develops. Clinical status should be closely monitored, and appropriate therapy should be initiated.
5.2 Severe Acute Exacerbation of Hepatitis B in People with Concomitant HIV-1 and HBV
All patients with HIV-1 should be tested for the presence of HBV before initiating antiretroviral therapy.
Severe acute exacerbations of hepatitis B (e.g., liver decompensated and liver failure) have been reported in people with concomitant HIV-1 and HBV who have discontinued products containing lamivudine and/or TDF, and may occur with discontinuation of DELSTRIGO. Patients who are coinfected with HIV-1 and HBV who discontinue DELSTRIGO should be closely monitored with both clinical and laboratory follow-up for at least several months after stopping treatment with DELSTRIGO. If appropriate, initiation of anti-hepatitis B therapy may be warranted, especially in patients with advanced liver disease or cirrhosis, since post-treatment exacerbation of hepatitis may lead to hepatic decompensation and liver failure.
5.3New Onset or Worsening Renal Impairment
Renal impairment, including cases of acute renal failure and Fanconi syndrome (renal tubular injury with severe hypophosphatemia), has been reported with the use of TDF, a component of DELSTRIGO.
DELSTRIGO should be avoided with concurrent or recent use of a nephrotoxic agent (e.g., high-dose or multiple nonsteroidal anti-inflammatory drugs [NSAIDs]) [see Drug Interactions (7.1)]. Cases of acute renal failure after initiation of high-dose or multiple NSAIDs have been reported in patients living with HIV with risk factors for renal dysfunction who appeared stable on TDF. Some patients required hospitalization and renal replacement therapy. Alternatives to NSAIDs should be considered, if needed, in patients at risk for renal dysfunction.
Persistent or worsening bone pain, pain in extremities, fractures, and/or muscular pain or weakness may be manifestations of proximal renal tubulopathy and should prompt an evaluation of renal function in at-risk patients.
Prior to or when initiating DELSTRIGO, and during treatment with DELSTRIGO, on a clinically appropriate schedule, assess serum creatinine, estimated creatinine clearance, urine glucose and urine protein in all patients. In patients with chronic kidney disease, also assess serum phosphorus. Discontinue DELSTRIGO in patients who develop clinically significant decreases in renal function or evidence of Fanconi syndrome.
The lamivudine and TDF components of DELSTRIGO are primarily excreted by the kidney. Discontinue DELSTRIGO if estimated creatinine clearance declines below 50 mL/min as dose interval adjustment required for lamivudine and TDF cannot be achieved with the fixed-dose combination tablet [see Use in Specific Populations (8.6)].
5.4Risk of Adverse Reactions or Loss of Virologic Response Due to Drug Interactions
The concomitant use of DELSTRIGO and certain other drugs may result in known or potentially significant drug interactions, some of which may lead to [see Dosage and Administration (2.4), Contraindications (4), and Drug Interactions (7.2)]:
- Loss of therapeutic effect of DELSTRIGO and possible development of resistance.
- Possible clinically significant adverse reactions from greater exposures of a component of DELSTRIGO.
See Table 6 for steps to prevent or manage these possible and known significant drug interactions, including dosing recommendations. Consider the potential for drug interactions prior to and during DELSTRIGO therapy, review concomitant medications during DELSTRIGO therapy, and monitor for adverse reactions.
5.5Bone Loss and Mineralization Defects
Bone Mineral Density
In clinical trials in adults living with HIV, TDF (a component of DELSTRIGO) was associated with slightly greater decreases in bone mineral density (BMD) and increases in biochemical markers of bone metabolism, suggesting increased bone turnover relative to comparators. Serum parathyroid hormone levels and 1,25 Vitamin D levels were also higher in participants receiving TDF.
Clinical trials evaluating TDF in pediatric participants were conducted. Under normal circumstances, BMD increases rapidly in pediatric patients. In participants 2 years to less than 18 years of age living with HIV, bone effects were similar to those observed in adult participants and suggest increased bone turnover. Total body BMD gain was less in the TDF-treated pediatric participants living with HIV as compared to the control groups. Similar trends were observed in chronic HBV-infected pediatric participants 2 years to less than 18 years of age. In all pediatric trials, normal skeletal growth (height) was not affected for the duration of the clinical trials.
The effects of TDF-associated changes in BMD and biochemical markers on long-term bone health and future fracture risk in adults and pediatric participants 2 years and older are unknown. The long-term effect of lower spine and total body BMD on skeletal growth in pediatric patients, and in particular, the effects of long-duration exposure in younger children are unknown.
Assessment of BMD should be considered for adult and pediatric patients living with HIV who have a history of pathologic bone fracture or other risk factors for osteoporosis or bone loss. Although the effect of supplementation with calcium and vitamin D was not studied, such supplementation may be beneficial in all patients. If bone abnormalities are suspected, then appropriate consultation should be obtained.
Mineralization Defects
Cases of osteomalacia associated with proximal renal tubulopathy, manifested as bone pain or pain in extremities and which may contribute to fractures, have been reported in association with the use of TDF [see Adverse Reactions (6.2)]. Arthralgias and muscle pain or weakness have also been reported in cases of proximal renal tubulopathy. Hypophosphatemia and osteomalacia secondary to proximal renal tubulopathy should be considered in patients at risk of renal dysfunction who present with persistent or worsening bone or muscle symptoms while receiving products containing TDF [see Warnings and Precautions (5.3)].
5.6Immune Reconstitution Syndrome
Immune reconstitution syndrome has been reported in patients treated with combination antiretroviral therapy. During the initial phase of combination antiretroviral treatment, patients whose immune system responds may develop an inflammatory response to indolent or residual opportunistic infections (such as Mycobacterium avium infection, cytomegalovirus, Pneumocystis jirovecii pneumonia (PCP), or tuberculosis), which may necessitate further evaluation and treatment.
Autoimmune disorders (such as Graves' disease, polymyositis, Guillain-Barré syndrome, and autoimmune hepatitis) have also been reported to occur in the setting of immune reconstitution; however, the time to onset is more variable and can occur many months after initiation of treatment.
6 Adverse Reactions
The following adverse reactions are discussed in other sections of the labeling:
- Severe Acute Exacerbation of Hepatitis B in people with concomitant HIV-1 and HBV [see Warnings and Precautions (5.2)]
- New Onset or Worsening Renal Impairment [see Warnings and Precautions (5.3)]
- Bone Loss and Mineralization Defects [see Warnings and Precautions (5.5)]
- Immune Reconstitution Syndrome [see Warnings and Precautions (5.6)]
Most common adverse reactions (incidence greater than or equal to 5%, all grades) are dizziness, nausea, and abnormal dreams. (6.1 )
To report SUSPECTED ADVERSE REACTIONS, contact Merck Sharp & Dohme LLC at 1-877-888-4231 or FDA at 1-800-FDA-1088 orwww.fda.gov/medwatch .
6.1Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Adverse Reactions in Adults with No Antiretroviral Treatment History
The safety assessment of DELSTRIGO is based on Week 96 data from two Phase 3, randomized, international, multicenter, double-blind, active-controlled trials. A total of 747 participants received doravirine either as the single entity in combination with other antiretroviral drugs as background regimens (n=383) or as the fixed-dose DELSTRIGO (n=364), and a total of 747 participants were randomized to control arms.
In DRIVE-AHEAD (Protocol 021), 728 adult participants received either DELSTRIGO (n=364) or EFV/FTC/TDF once daily (n=364). By Week 96, 3% in the DELSTRIGO group and 7% in the EFV/FTC/TDF group had adverse events leading to discontinuation of study medication.
Adverse reactions reported in greater than or equal to 5% of participants in any treatment group in DRIVE-AHEAD are presented in Table 1.
Table 1: Adverse Reactions Frequencies of adverse reactions are based on all adverse events attributed to trial drugs by the investigator. (All Grades) Reported in ‚Č•5%No adverse reactions of Grade 2 or higher (moderate or severe) occurred in ‚Č•2% of participants treated with DELSTRIGO. of Participants in Any Treatment Group in Adults with No Antiretroviral Treatment History in DRIVE-AHEAD (Week 96)DELSTRIGOOnce DailyN=364 EFV/FTC/TDFOnce DailyN=364 Dizziness 7% 32% Nausea 5% 7% Abnormal Dreams 5% 10% Headache 4% 5% Insomnia 4% 5% Diarrhea 4% 6% Somnolence 3% 7% Rash Rash: includes rash, rash erythematous, rash generalized, rash macular, rash maculo-papular, rash papular, rash pruritic. 2% 12%
The majority (66%) of adverse reactions associated with DELSTRIGO occurred at severity Grade 1 (mild).
Neuropsychiatric Adverse Events
For DRIVE-AHEAD, the analysis of participants with neuropsychiatric adverse events by Week 48 is presented in Table 2. The proportion of participants who reported one or more neuropsychiatric adverse events was 24% and 57% in the DELSTRIGO and EFV/FTC/TDF groups, respectively.
A statistically significantly lower proportion of DELSTRIGO-treated participants compared to EFV/FTC/TDF-treated participants reported neuropsychiatric adverse events by Week 48 in the three pre-specified categories of dizziness, sleep disorders and disturbances, and altered sensorium.
Table 2: DRIVE-AHEAD - Analysis of Participants with Neuropsychiatric Adverse Events All causality and all grade events were included in the analysis. (Week 48)DELSTRIGOOnce DailyN=364 EFV/FTC/TDFOnce DailyN=364 Treatment Difference(DELSTRIGO - EFV/FTC/TDF)Estimate (95% CI) The 95% CIs were calculated using Miettinen and Nurminen's method. Categories pre-specified for statistical testing were dizziness (p <0.001), sleep disorders and disturbances (p <0.001), and altered sensorium (p=0.033). Sleep disorders and disturbances Predefined using MedDRA preferred terms including: abnormal dreams, hyposomnia, initial insomnia, insomnia, nightmare, sleep disorder, somnambulism. 12% 26% -13.5 (-19.1, -7.9) Dizziness 9% 37% -28.3 (-34.0, -22.5) Altered sensorium Predefined using MedDRA preferred terms including: altered state of consciousness, lethargy, somnolence, syncope. 4% 8% -3.8 (-7.6, -0.3)
Neuropsychiatric adverse events in the pre-defined category of depression and suicide/self-injury were reported in 4% and 7% of participants, in the DELSTRIGO and EFV/FTC/TDF groups, respectively.
In DRIVE-AHEAD through 48 weeks of treatment, the majority of participants who reported neuropsychiatric adverse events reported events that were mild to moderate in severity (97% [83/86] and 96% [198/207], in the DELSTRIGO and EFV/FTC/TDF groups, respectively) and the majority of participants reported these events in the first 4 weeks of treatment (72% [62/86] in the DELSTRIGO group and 86% [177/207] in the EFV/FTC/TDF group).
Neuropsychiatric adverse events led to treatment discontinuation in 1% (2/364) and 1% (5/364) of participants in the DELSTRIGO and EFV/FTC/TDF groups, respectively. The proportion of participants who reported neuropsychiatric adverse events through Week 4 was 17% (62/364) in the DELSTRIGO group and 49% (177/364) in the EFV/FTC/TDF group. At Week 48, the prevalence of neuropsychiatric adverse events was 12% (44/364) in the DELSTRIGO group and 22% (81/364) in the EFV/FTC/TDF group. At Week 96, the prevalence of neuropsychiatric adverse events was 13% (47/364) in the DELSTRIGO group and 23% (82/364) in the EFV/FTC/TDF group.
Laboratory Abnormalities
The percentages of participants with selected laboratory abnormalities (that represent a worsening from baseline) who were treated with DELSTRIGO or EFV/FTC/TDF in DRIVE-AHEAD are presented in Table 3.
Table 3: Selected Laboratory Abnormalities Reported in Adult Participants with No Antiretroviral Treatment History in DRIVE-AHEAD (Week 96) Laboratory Parameter Preferred Term (Unit)/Limit DELSTRIGOOnce DailyN=364 EFV/FTC/TDFOnce DailyN=364 Blood Chemistry Each participant is only counted once per parameter at the highest toxicity grade. Only participants with a baseline value and at least one on-treatment value for a given laboratory parameter are included. ULN = Upper limit of normal range. Total bilirubin (mg/dL) 1.1 - <1.6 √ó ULN 5% 0% 1.6 - <2.6 √ó ULN 2% 0% ‚Č•2.6 √ó ULN 1% <1% Creatinine (mg/dL) >1.3 - 1.8 √ó ULN or Increase of >0.3 mg/dL above baseline 3% 2% >1.8 √ó ULN or Increase of ‚Č•1.5 √ó above baseline 3% 2% Aspartate aminotransferase (IU/L) 2.5 - <5.0 √ó ULN 3% 3% ‚Č•5.0 √ó ULN 1% 4% Alanine aminotransferase (IU/L) 2.5 - <5.0 √ó ULN 4% 4% ‚Č•5.0 √ó ULN 1% 3% Alkaline phosphatase (IU/L) 2.5 - <5.0 √ó ULN <1% 1% ‚Č•5.0 √ó ULN 0% <1% Lipase 1.5 - <3.0 √ó ULN 6% 4% ‚Č•3.0 √ó ULN 2% 3% Creatine kinase (IU/L) 6.0 - <10.0 √ó ULN 3% 3% ‚Č•10.0 √ó ULN 4% 6% Cholesterol, fasted (mg/dL) ‚Č•300 mg/dL 1% <1% LDL cholesterol, fasted (mg/dL) ‚Č•190 mg/dL <1% 2% Triglycerides, fasted (mg/dL) >500 mg/dL 1% 3%
Change in Lipids from Baseline
For DRIVE-AHEAD, changes from baseline at Week 48 in LDL-cholesterol, non-HDL-cholesterol, total cholesterol, triglycerides, and HDL-cholesterol are shown in Table 4. Changes from baseline at Week 96 were similar to findings at Week 48.
The LDL and non-HDL comparisons were pre-specified and are summarized in Table 4. The differences were statistically significant, showing superiority of DELSTRIGO for both parameters. The clinical benefit of these findings has not been demonstrated.
Table 4: Mean Change from Baseline in Fasting Lipids in Adult Participants with No Antiretroviral Treatment History in DRIVE-AHEAD (Week 48) Laboratory Parameter Preferred Term DELSTRIGOOnce DailyN=320 EFV/FTC/TDFOnce DailyN=307 Difference Estimates(DELSTRIGO - EFV/FTC/TDF) Baseline Change Baseline Change Difference (95% CI) Participants on lipid-lowering agents at baseline were excluded from these analyses (DELSTRIGO n=15 and EFV/FTC/TDF n=10).Participants initiating a lipid-lowering agent post-baseline had their last fasted on-treatment value (prior to starting the agent) carried forward (DELSTRIGO n=3 and EFV/FTC/TDF n=8). LDL-Cholesterol (mg/dL) p-value for the pre-specified hypothesis testing for treatment difference was <0.0001. 91.7 -2.1 91.3 8.3 -10.2 (-13.8, -6.7) Non-HDL Cholesterol (mg/dL) 114.7 -4.1 115.3 12.7 -16.9 (-20.8, -13.0) Total Cholesterol (mg/dL) Not pre-specified for hypothesis testing. 156.8 -2.2 156.8 21.1 - Triglycerides (mg/dL) 118.7 -12.0 122.6 21.6 - HDL-Cholesterol (mg/dL) 42.1 1.8 41.6 8.4 -
Adverse Reactions in Virologically-Suppressed Adults
The safety of DELSTRIGO in virologically-suppressed adults was based on Week 48 data from 670 participants in the DRIVE-SHIFT trial (Protocol 024), a randomized, international, multicenter, open-label trial in which virologically-suppressed participants were switched from a baseline regimen consisting of two nucleoside reverse transcriptase inhibitors (NRTIs) in combination with a protease inhibitor (PI) plus either ritonavir or cobicistat, or elvitegravir plus cobicistat, or an NNRTI to DELSTRIGO. Overall, the safety profile in virologically-suppressed adult participants was similar to that in participants with no antiretroviral treatment history.
Laboratory Abnormalities
Change in Lipids from Baseline
Changes from baseline at Week 24 in LDL-cholesterol, non-HDL-cholesterol, total cholesterol, triglycerides, and HDL-cholesterol in participants on a PI plus ritonavir-based regimen at baseline are shown in Table 5. The LDL and non-HDL comparisons were pre-specified, and the differences were statistically significant, showing superiority for an immediate switch to DELSTRIGO for both parameters. The clinical benefit of these findings has not been demonstrated.
Table 5: Mean Change from Baseline in Fasting Lipids in Adult Virologically-Suppressed Participants on a PI plus Ritonavir-based Regimen at Baseline in DRIVE-SHIFT (Week 24) Laboratory Parameter Preferred Term DELSTRIGO(Week 0-24)Once DailyN=244 PI+ritonavir(Week 0-24)Once DailyN=124 Difference Estimates Baseline Change Baseline Change Difference (95% CI) Participants on lipid-lowering agents at baseline were excluded from these analyses (DELSTRIGO n=26 and PI+ritonavir n=13). Participants initiating a lipid-lowering agent post-baseline had their last fasted on-treatment value (prior to starting the agent) carried forward (DELSTRIGO n=4 and PI+ritonavir n=2). LDL-Cholesterol (mg/dL) p-value for the pre-specified hypothesis testing for treatment difference was <0.0001. 108.7 -16.3 110.5 -2.6 -14.5 (-18.9, -10.1) Non-HDL Cholesterol (mg/dL) 138.6 -24.8 138.8 -2.1 -22.8 (-27.9, -17.7) Total Cholesterol (mg/dL) Not pre-specified for hypothesis testing. 188.5 -26.1 187.4 -0.2 - Triglycerides (mg/dL) 153.1 -44.4 151.4 -0.4 - HDL-Cholesterol (mg/dL) 50.0 -1.3 48.5 1.9 -
Adverse Reactions in Pediatric Participants
The safety of DELSTRIGO was evaluated in 45 virologically-suppressed or treatment-na√Įve pediatric participants 12 to less than 18 years of age living with HIV, through Week 24 in an open-label trial (IMPAACT 2014 (Protocol 027)) [see Clinical Studies (14.3)]. The safety profile in pediatric participants was similar to that in adults. There were no serious or Grade 3 or 4 adverse reactions. No participants discontinued due to an adverse event.
6.2Postmarketing Experience
The following adverse reactions have been identified during postmarketing experience in patients receiving doravirine-, lamivudine- or TDF-containing regimens. Because postmarketing reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Doravirine:
Skin and Subcutaneous Tissue Disorders: Stevens-Johnson syndrome (SJS)/toxic epidermal necrolysis (TEN)
Lamivudine:
Body as a Whole: redistribution/accumulation of body fat
Endocrine and Metabolic: hyperglycemia
General: Weakness
Hemic and Lymphatic: anemia (including pure red cell aplasia and severe anemias progressing on therapy)
Hepatic and Pancreatic: lactic acidosis and hepatic steatosis, posttreatment exacerbations of hepatitis B
Hypersensitivity: anaphylaxis, urticaria
Musculoskeletal: muscle weakness, CPK elevation, rhabdomyolysis
Skin: alopecia, pruritus
TDF:
Immune System Disorders: allergic reaction, including angioedema
Metabolism and Nutrition Disorders: lactic acidosis, hypokalemia, hypophosphatemia
Respiratory, Thoracic, and Mediastinal Disorders: dyspnea
Gastrointestinal Disorders: pancreatitis, increased amylase, abdominal pain
Hepatobiliary Disorders: hepatic steatosis, hepatitis, increased liver enzymes (most commonly AST, ALT gamma GT)
Skin and Subcutaneous Tissue Disorders: rash
Musculoskeletal and Connective Tissue Disorders: rhabdomyolysis, osteomalacia (manifested as bone pain and which may contribute to fractures), muscular weakness, myopathy
Renal and Urinary Disorders: acute renal failure, renal failure, acute tubular necrosis, Fanconi syndrome, proximal renal tubulopathy, interstitial nephritis (including acute cases), nephrogenic diabetes insipidus, renal insufficiency, increased creatinine, proteinuria, polyuria
General Disorders and Administration Site Conditions: asthenia
The following adverse reactions, uled under the body system headings above, may occur as a consequence of proximal renal tubulopathy: rhabdomyolysis, osteomalacia, hypokalemia, muscular weakness, myopathy, hypophosphatemia.
7 Drug Interactions
- Because DELSTRIGO is a complete regimen, co-administration with other antiretroviral medications for treatment of HIV-1 infection is not recommended. (
7.1 )- Consult the full prescribing information prior to and during treatment for important potential drug-drug interactions. (
4 ,5.4 ,7 )7.1Concomitant Use with Other Antiretroviral Medications
Because DELSTRIGO is a complete regimen for the treatment of HIV-1 infection, co-administration with other antiretroviral medications for treatment of HIV-1 infection is not recommended. Information regarding potential drug-drug interactions with other antiretroviral medications is not provided.
7.2Effect of Other Drugs on DELSTRIGO
Co-administration of DELSTRIGO with a CYP3A inducer decreases doravirine plasma concentrations, which may reduce DELSTRIGO efficacy [see Contraindications (4), Warnings and Precautions (5.4), and Clinical Pharmacology (12.3)].
Co-administration of DELSTRIGO and drugs that are inhibitors of CYP3A may result in increased plasma concentrations of doravirine.
Table 6 shows the significant drug interactions with the components of DELSTRIGO. The drug interactions described are based on studies conducted with either DELSTRIGO or the components of DELSTRIGO as individual agents.
Table 6: Drug Interactions with DELSTRIGO This table is not all-inclusive Concomitant Drug Class: Drug Name Effect on Concentration Clinical Comment ‚ÜĎ = increase, ‚Üď = decrease All other drug-drug interactions shown are anticipated based on the known metabolic and elimination pathways. Androgen Receptors enzalutamide ‚Üď doravirine Co-administration is contraindicated with enzalutamide.At least a 4-week cessation period is recommended prior to initiation of DELSTRIGO. Anticonvulsants carbamazepineoxcarbazepinephenobarbitalphenytoin ‚Üď doravirine Co-administration is contraindicated with these anticonvulsants. At least a 4-week cessation period is recommended prior to initiation of DELSTRIGO. Antimycobacterials rifampin The interaction between doravirine and the concomitant drug was evaluated in a clinical study. rifapentine‚Üď doravirine Co-administration is contraindicated with rifampin or rifapentine. At least a 4-week cessation period is recommended prior to initiation of DELSTRIGO. rifabutin ‚Üď doravirine If DELSTRIGO is co-administered with rifabutin, one tablet of doravirine (PIFELTRO) should be taken approximately 12 hours after the dose of DELSTRIGO [seeDosage and Administration (2.4)]. Cytotoxic Agents mitotane ‚Üď doravirine Co-administration is contraindicated with mitotane. At least a 4-week cessation period is recommended prior to initiation of DELSTRIGO. Hepatitis C Antiviral Agents ledipasvir/sofosbuvirsofosbuvir/velpatasvir ‚ÜĎ tenofovir Monitor for adverse reactions associated with TDF. Herbal Products St. John's wort ‚Üď doravirine Co-administration is contraindicated with St. John's wort. At least a 4-week cessation period is recommended prior to initiation of DELSTRIGO. Other Agents sorbitol ‚Üď lamivudine Co-administration of single doses of lamivudine and sorbitol resulted in a sorbitol dose-dependent reduction in lamivudine exposures. When possible, avoid use of sorbitol-containing medicines with lamivudine-containing medicines.
Co-administration of DELSTRIGO with drugs that reduce renal function or compete for active tubular secretion may increase serum concentrations of lamivudine, tenofovir, and/or other renally eliminated drugs. Some examples of drugs that are eliminated by active tubular secretion include, but are not limited to, acyclovir, cidofovir, ganciclovir, valacyclovir, valganciclovir, aminoglycosides (e.g., gentamicin), and high-dose or multiple NSAIDs [see Warnings and Precautions (5.3) and Clinical Pharmacology (12.3)].
No clinically significant changes in concentration were observed for doravirine when co-administered with the following agents: TDF, lamivudine, elbasvir and grazoprevir, ledipasvir and sofosbuvir, ritonavir, ketoconazole, aluminum hydroxide/magnesium hydroxide/simethicone containing antacid, pantoprazole, or methadone [see Clinical Pharmacology (12.3)].
No clinically significant changes in concentration were observed for tenofovir when co-administered with tacrolimus or entecavir [see Clinical Pharmacology (12.3)].
7.3Effect of DELSTRIGO on Other Drugs
No clinically significant changes in concentration were observed for the following agents when co-administered with doravirine: lamivudine, TDF, elbasvir and grazoprevir, ledipasvir and sofosbuvir, atorvastatin, an oral contraceptive containing ethinyl estradiol and levonorgestrel, metformin, methadone, or midazolam.
No clinically significant drug interactions have been observed between TDF and the following medications: entecavir, methadone, oral contraceptives, sofosbuvir, or tacrolimus in studies conducted in healthy participants.
Lamivudine is not significantly metabolized by CYP enzymes nor does it inhibit or induce this enzyme system; therefore, it is unlikely that clinically significant drug interactions will occur through these pathways [see Clinical Pharmacology (12.3)].
8 Use In Specific Populations
- Pediatrics: Not recommended for patients weighing less than 35 kg. (
8.4 )8.1 Pregnancy
Pregnancy Exposure Registry
There is a pregnancy exposure registry that monitors pregnancy outcomes in individuals exposed to DELSTRIGO during pregnancy. Healthcare providers are encouraged to register patients by calling the Antiretroviral Pregnancy Registry (APR) at 1-800-258-4263.
Risk Summary
There is insufficient prospective pregnancy data from the APR to adequately assess the risk of birth defects and miscarriage. Doravirine use in individuals during pregnancy has not been evaluated; however, lamivudine and TDF use during pregnancy has been evaluated in a limited number of individuals reported to the APR. Available data from the APR show no difference in the overall risk of major birth defects for lamivudine and TDF compared with the background rate for major birth defects of 2.7% in the U.S. reference population of the Metropolitan Atlanta Congenital Defects Program (MACDP) (see Data ). The rate of miscarriage is not reported in the APR. The estimated background rate of miscarriage in the clinically recognized pregnancies in the U.S. general population is 15-20%. Methodological limitations of the APR include the use of MACDP as the external comparator group. The MACDP population is not disease-specific, evaluates individuals and infants from the limited geographic area, and does not include outcomes for births that occurred at less than 20 weeks gestation.
In animal reproduction studies, oral administration of lamivudine to pregnant rabbits during organogenesis resulted in embryolethality at systemic exposure (AUC) similar to the recommended clinical dose; however, no adverse development effects were observed with oral administration of lamivudine to pregnant rats during organogenesis at plasma concentrations (Cmax) 35 times the recommended clinical dose.
No adverse developmental effects were observed when doravirine and TDF were administered separately at doses/exposures ‚Č•8 (doravirine) and ‚Č•14 (TDF) times those of the recommended human dose (RHD) of DELSTRIGO (see Data ).
Data
Human Data
Animal Data
8.2 Lactation
Risk Summary
Based on limited published data, both lamivudine and tenofovir are present in human milk. It is unknown whether doravirine is present in human milk, but doravirine is present in the milk of lactating rats (see Data ). It is not known whether DELSTRIGO or the components of DELSTRIGO affects human milk production, or has effects on the breastfed infant. Potential risks of breastfeeding include: (1) HIV-1 transmission (in HIV-1-negative infants), (2) developing viral resistance (in HIV-1-positive infants), and (3) serious adverse reactions in a breastfed infant similar to those seen in adults.
Data
Doravirine : Doravirine was excreted into the milk of lactating rats following oral administration (450 mg/kg/day) from GD 6 to lactation day 14, with milk concentrations approximately 1.5 times that of maternal plasma concentrations observed 2 hours post dose on lactation day 14.
8.4 Pediatric Use
The safety and efficacy of DELSTRIGO for the treatment of HIV-1 infection have been established in pediatric patients weighing at least 35 kg [see Indications and Usage (1) and Dosage and Administration (2.2)].
Use of DELSTRIGO in this group is supported by evidence from adequate and well-controlled trials in adults with additional pharmacokinetic, safety, and efficacy data from an open-label trial in virologically-suppressed or treatment-na√Įve pediatric participants 12 to less than 18 years of age. The safety and efficacy of DELSTRIGO in these pediatric participants were similar to that in adults, and there was no clinically significant difference in exposure for the components of DELSTRIGO. [see Adverse Reactions (6.1), Clinical Pharmacology (12.3), and Clinical Studies (14.3)].
Safety and efficacy of DELSTRIGO in pediatric patients weighing less than 35 kg have not been established.
8.5 Geriatric Use
Clinical trials of doravirine, lamivudine, or TDF did not include sufficient numbers of participants aged 65 years and over to determine whether they respond differently from younger participants. In general, caution should be exercised in the administration of DELSTRIGO in elderly patients reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy [see Clinical Pharmacology (12.3)].
8.6Renal Impairment
Because DELSTRIGO is a fixed-dose combination tablet and the dosage of lamivudine and TDF, both components of DELSTRIGO, cannot be altered, DELSTRIGO is not recommended in patients with estimated creatinine clearance less than 50 mL/min [see Warnings and Precautions (5.3) and Clinical Pharmacology (12.3)].
8.7Hepatic Impairment
No dosage adjustment of DELSTRIGO is required in patients with mild (Child-Pugh Class A) or moderate (Child-Pugh Class B) hepatic impairment. DELSTRIGO has not been studied in patients with severe hepatic impairment (Child-Pugh Class C) [see Clinical Pharmacology (12.3)].
10 Overdosage
No data are available on overdose of DELSTRIGO in patients and there is no known specific treatment for overdose with DELSTRIGO. If overdose occurs, the patient should be monitored and standard supportive treatment applied as required.
Doravirine: There is no known specific treatment for overdose with doravirine.
Lamivudine: Because a negligible amount of lamivudine was removed via (4-hour) hemodialysis, continuous ambulatory peritoneal dialysis, and automated peritoneal dialysis, it is not known if continuous hemodialysis would provide clinical benefit in a lamivudine overdose event.
TDF: TDF is efficiently removed by hemodialysis with an extraction coefficient of approximately 54%. Following a single 300 mg dose of TDF, a 4-hour hemodialysis session removed approximately 10% of the administered tenofovir dose.
11 Description
DELSTRIGO is a fixed-dose combination, film-coated tablet, containing doravirine, lamivudine, and TDF for oral administration.
Doravirine is an HIV-1 non-nucleoside reverse transcriptase inhibitor (NNRTI).
Lamivudine is the (-)enantiomer of a dideoxy analogue of cytidine and is an HIV-1 nucleoside analogue reverse transcriptase inhibitor.
TDF (a prodrug of tenofovir) is a fumaric acid salt of the bis-isopropoxycarbonyloxymethyl ester derivative of tenofovir. In vivo TDF is converted to tenofovir, an acyclic nucleoside phosphonate (nucleotide) analog of adenosine 5'-monophosphate. Tenofovir is an HIV-1 reverse transcriptase inhibitor.
Each tablet contains 100 mg of doravirine, 300 mg of lamivudine, and 300 mg of TDF (equivalent to 245 mg of tenofovir disoproxil) as active ingredients. The tablets include the following inactive ingredients: colloidal silicon dioxide, croscarmellose sodium, hypromellose acetate succinate, magnesium stearate, microcrystalline cellulose, and sodium stearyl fumarate. The tablets are film coated with a coating material containing the following inactive ingredients: hypromellose, iron oxide yellow, lactose monohydrate, titanium dioxide, and triacetin. The coated tablets are polished with carnauba wax.
Doravirine:
The chemical name for doravirine is 3-chloro-5-[[1-[(4,5-dihydro-4-methyl-5-oxo-1H-1,2,4-triazol-3-yl)methyl]-1,2-dihydro-2-oxo-4-(trifluoromethyl)-3-pyridinyl]oxy]benzonitrile.
It has a molecular formula of C17H11ClF3N5O3 and a molecular weight of 425.75.
It has the following structural formula:
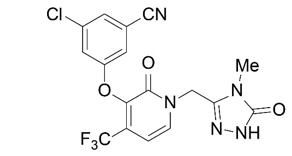
Doravirine is practically insoluble in water.
Lamivudine:
The chemical name for lamivudine is (-)-1-[(2R,5S)-2-(hydroxymethyl)-1,3-oxathiolan-5-yl]-cytosine.
It has a molecular formula of C8H11N3O3S and a molecular weight of 229.26.
It has the following structural formula:
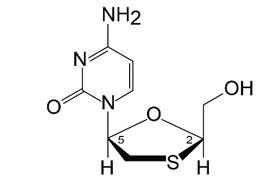
Lamivudine is soluble in water.
TDF:
The chemical name for TDF is 9-[(R)-2-[[bis[[(isopropoxycarbonyl)oxy]methoxy] phosphinyl]- methoxy]propyl]adenine fumarate (1:1).
It has a molecular formula of C19H30N5O10 P‚ąôC4H4O4 and a molecular weight of 635.52.
It has the following structural formula:
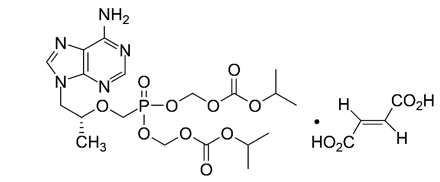
TDF is slightly soluble in water.
12 Clinical Pharmacology
12.1 Mechanism of Action
DELSTRIGO is a fixed-dose combination of the antiretroviral drugs doravirine, lamivudine, and TDF [see Microbiology (12.4)].
12.2 Pharmacodynamics
In a Phase 2 trial evaluating doravirine over a dose range of 0.25 to 2 times the recommended dose of doravirine in DELSTRIGO (in combination with FTC/TDF) in participants living with HIV with no antiretroviral treatment history, no exposure-response relationship for efficacy was identified for doravirine.
Cardiac Electrophysiology
At a doravirine dose of 1200 mg, which provides approximately 4 times the peak concentration observed following the recommended dose of doravirine in DELSTRIGO does not prolong the QT interval to any clinically relevant extent.
12.3 Pharmacokinetics
Single-dose administration of one DELSTRIGO tablet to healthy participants provided comparable exposures of doravirine, lamivudine, and tenofovir to administration of doravirine tablets (100 mg) plus lamivudine tablets (300 mg) plus TDF tablets (300 mg). Doravirine pharmacokinetics are similar in healthy participants and participants living with HIV. Pharmacokinetic properties of the components of DELSTRIGO are provided in Table 7.
Table 7: Pharmacokinetic Properties of the Components of DELSTRIGO Parameter Doravirine Lamivudine Tenofovir Abbreviations: NA=not available; AUC=area under the time concentration curve; Cmax=maximum concentration; C24=concentration at 24 hours; Tmax=time to Cmax; Vdss=apparent volume of distribution at steady state; t1/2=elimination half-life; CL/F=apparent clearance; CLrenal = renal clearance General ¬†¬†Steady State Exposure Presented as geometric mean (%CV: geometric coefficient of variation) or mean ¬Ī SD. AUC0-24 (mcg‚ąôh/mL) 16.1 (29) Doravirine 100 mg once daily to participants living with HIV. 8.87 ¬Ī 1.83 Lamivudine 300 mg once daily for 7 days to 60 healthy participants. 2.29 ¬Ī 0.69 Single 300 mg dose of TDF to participants living with HIV in the fasted state. Cmax (mcg/mL) 0.962 (19) 2.04 ¬Ī 0.54 0.30 ¬Ī 0.09 C24 (mcg/mL) 0.396 (63) NA NA Absorption Absolute Bioavailability 64% 86% 25% Tmax (h) 2 NA 1 ¬†¬†Effect of Food Geometric mean ratio [high-fat meal/fasting] and (90% confidence interval) for PK parameters. High fat meal is approximately 1000 kcal, 50% fat. The effect of food is not clinically relevant. AUC Ratio 1.10 (1.01, 1.20) 0.93 (0.84, 1.03) 1.27 (1.17, 1.37) Cmax Ratio 0.95 (0.80, 1.12) 0.81 (0.65, 1.01) 0.88 (0.74, 1.04) C24 Ratio 1.26 (1.13, 1.41) NA NA Distribution Vdss Based on IV dose . 60.5 L 1.3 L/kg 1.3 L/kg Plasma Protein Binding 76% < 36% <0.7% Elimination t1/2 (h) 15 5-7 17 CL/F (mL/ min) 106 (35.2) 398.5 ¬Ī 69.1 1,043.7 ¬Ī 115.4 CLrenal (mL/ min) 9.3 (18.6) 199.7 ¬Ī 56.9 243.5 ¬Ī 33.3 ¬†¬†Metabolism Primary Pathway(s) CYP3A Minor No CYP Metabolism ¬†¬†Excretion Major route of elimination Metabolism Glomerular filtration and active tubular secretion Glomerular filtration and active tubular secretion Urine (unchanged) 6% 71% 70-80% Biliary/Fecal (unchanged) Minor NA NA
Specific Populations
In adults, no clinically significant differences in the pharmacokinetics of certain DELSTRIGO components were observed based on age ‚Č•65 years (for doravirine), sex (for doravirine, lamivudine, tenofovir), and race/ethnicity (for doravirine, lamivudine). The effects of age (‚Č•65 years) on the pharmacokinetics of lamivudine and tenofovir, and the effect of race on the pharmacokinetics of tenofovir are unknown.
Patients with Renal Impairment
Patients with Hepatic Impairment
Pediatric Patients
Mean doravirine exposures were similar in 54 pediatric participants aged 12 to less than 18 years and weighing at least 35 kg who received doravirine or DELSTRIGO in IMPAACT 2014 (Protocol 027) relative to adults following administration of doravirine or DELSTRIGO. Exposures of lamivudine and tenofovir in pediatric participants following the administration of DELSTRIGO were similar to those in adults following administration of lamivudine and tenofovir (Table 8). For pediatric participants weighing ‚Č• 35 kg and < 45 kg who receive doravirine 100 mg or DELSTRIGO, the population pharmacokinetic model-predicted mean C24 of doravirine was comparable to that achieved in adults, whereas mean AUC0-24 and Cmax of doravirine were 25% and 36% higher than adult values, respectively. However, the predicted AUC0-24 and Cmax increases are not considered clinically significant.
Table 8: Steady State Pharmacokinetics for Doravirine, Lamivudine, and Tenofovir Following Administration of Doravirine or DELSTRIGO in Pediatric Participants Living with HIV Aged 12 to Less than 18 Years and Weighing at Least 35 kg Parameter Presented as geometric mean (%CV: geometric coefficient of variation) Doravirine From population PK analysis (n=53 weighing ‚Č•45 kg, n=1 weighing ‚Č•35 kg to <45 kg) Lamivudine From intensive PK analysis (n=10) Tenofovir Abbreviations: NA=not applicable; AUC=area under the time concentration curve; Cmax=maximum concentration; C24=concentration at 24 hours AUC0-24 (mcg‚ÄĘh/mL) 16.4 (24) 11.3 (28) 2.55 (14) Cmax (mcg/mL) 1.03 (16) 2.1 (24) 0.293 (37) C24 (mcg/mL) 0.379 (42) NA NA
Drug Interaction Studies
DELSTRIGO is a complete regimen for the treatment of HIV-1 infection; therefore, DELSTRIGO is not recommended to be administered with other HIV-1 antiretroviral medications. Information regarding potential drug-drug interactions with other antiretroviral medications is not provided.
The drug interaction trials described were conducted with doravirine, lamivudine and/or TDF, as single entities; no drug interaction trials have been conducted using the combination of doravirine, lamivudine, and TDF. No clinically relevant drug interactions were observed between doravirine, lamivudine, and TDF.
Doravirine: Doravirine is primarily metabolized by CYP3A, and drugs that induce or inhibit CYP3A may affect the clearance of doravirine. Co-administration of doravirine and drugs that induce CYP3A may result in decreased plasma concentrations of doravirine. Co-administration of doravirine and drugs that inhibit CYP3A may result in increased plasma concentrations of doravirine.
Doravirine is not likely to have a clinically relevant effect on the exposure of medicinal products metabolized by CYP enzymes. Doravirine did not inhibit major drug metabolizing enzymes in vitro, including CYPs 1A2, 2B6, 2C8, 2C9, 2C19, 2D6, 3A4, and UGT1A1 and is not likely to be an inducer of CYP1A2, 2B6, or 3A4. Based on in vitro assays, doravirine is not likely to be an inhibitor of OATP1B1, OATP1B3, P-glycoprotein, BSEP, OAT1, OAT3, OCT2, MATE1, and MATE2K. Drug interaction studies were performed with doravirine and other drugs likely to be co-administered or commonly used as probes for pharmacokinetic interactions. The effects of co-administration with other drugs on the exposure (Cmax, AUC, and C24) of doravirine are summarized in Table 9. A single doravirine 100 mg dose was administered in these studies unless otherwise noted.
Table 9: Drug Interactions: Changes in Pharmacokinetic Parameter Values of Doravirine in the Presence of Co-administered Drug Co-administered Drug Regimen of Co-administered Drug N Geometric Mean Ratio (90% CI) of Doravirine Pharmacokinetics with/without Co-administered Drug (No Effect=1.00) AUC AUC0-‚ąě for single-dose, AUC0-24 for once daily. Cmax C24 CI = confidence interval; QD = once daily; BID = twice daily Azole Antifungal Agents ketoconazole Changes in doravirine pharmacokinetic values are not clinically relevant. 400 mg QD 10 3.06 (2.85, 3.29) 1.25 (1.05, 1.49) 2.75 (2.54, 2.98) Antimycobacterials rifampin 600 mg QD 10 0.12 (0.10, 0.15) 0.43 (0.35, 0.52) 0.03 (0.02, 0.04) rifabutin 300 mg QD 12 0.50 (0.45, 0.55) 0.99 (0.85, 1.15) 0.32 (0.28, 0.35) 300 mg QD Doravirine 100 mg BID resulted in similar pharmacokinetic values when compared to 100 mg QD without rifabutin. 15 1.03 (0.94, 1.14) 0.97 (0.87, 1.08) 0.98 (0.88, 1.10) HIV Antiviral Agents ritonavir , A single doravirine 50 mg dose (0.5 times the recommended approved dose) was administered. 100 mg BID 8 3.54 (3.04, 4.11) 1.31 (1.17, 1.46) 2.91 (2.33, 3.62) efavirenz 600 mg QD The first day following the cessation of efavirenz therapy and initiation of doravirine 100 mg QD. 17 0.38 (0.33, 0.45) 0.65 (0.58, 0.73) 0.15 (0.10, 0.23) 600 mg QD 14 days following the cessation of efavirenz therapy and initiation of doravirine 100 mg QD. 17 0.68 (0.58, 0.80) 0.86 (0.77, 0.97) 0.50 (0.39, 0.64)
Based on drug interaction studies conducted with doravirine, no clinically significant drug interactions have been observed following the co-administration of doravirine and the following drugs: dolutegravir, TDF, lamivudine, elbasvir and grazoprevir, ledipasvir and sofosbuvir, ketoconazole, ritonavir, aluminum hydroxide/magnesium hydroxide/simethicone containing antacid, pantoprazole, atorvastatin, an oral contraceptive containing ethinyl estradiol and levonorgestrel, metformin, methadone, and midazolam.
Lamivudine:
TDF:
No clinically significant changes in exposure were observed for tenofovir when co-administered with tacrolimus or entecavir.
No clinically significant changes in exposure were observed for the following drugs when co-administered with tenofovir: tacrolimus, entecavir, methadone, or ethinyl estradiol/norgestimate.
12.4Microbiology
Mechanism of Action
Doravirine: Doravirine is a pyridinone non-nucleoside reverse transcriptase inhibitor of HIV-1 and inhibits HIV-1 replication by non-competitive inhibition of HIV-1 reverse transcriptase (RT). The inhibitory concentration at 50% (IC50) of doravirine for RNA-dependent DNA polymerization of recombinant wild-type HIV-1 RT in a biochemical assay was 12.2¬Ī2.0 nM (n=3). Doravirine does not inhibit the human cellular DNA polymerases őĪ, ő≤, and mitochondrial DNA polymerase ő≥.
Lamivudine: Lamivudine is a synthetic nucleoside analogue. Intracellularly, lamivudine is phosphorylated to its active 5'-triphosphate metabolite, lamivudine triphosphate (3TC-TP). The principal mode of action of 3TC-TP is inhibition of RT via DNA chain termination after incorporation of the nucleotide analogue. Lamivudine triphosphate (3TC-TP) is a weak inhibitor of mammalian DNA polymerases őĪ, ő≤, and mitochondrial DNA polymerase ő≥.
TDF: TDF is an acyclic nucleoside phosphonate diester analog of adenosine monophosphate. TDF requires initial diester hydrolysis for conversion to tenofovir and subsequent phosphorylations by cellular enzymes to form tenofovir diphosphate. Tenofovir diphosphate inhibits the activity of HIV-1 RT by competing with the natural substrate deoxyadenosine 5'-triphosphate and, after incorporation into DNA, by DNA chain termination. Tenofovir diphosphate is a weak inhibitor of mammalian DNA polymerases őĪ, ő≤, and mitochondrial DNA polymerase ő≥.
Antiviral Activity in Cell Culture
Doravirine: Doravirine exhibited an EC50 value of 12.0¬Ī4.4 nM against wild-type laboratory strains of HIV-1 when tested in the presence of 100% normal human serum (NHS) using MT4-GFP reporter cells and a median EC50 value for HIV-1 subtype B primary isolates (n=118) of 4.1 nM (range: 1.0 nM-16.0 nM). Doravirine demonstrated antiviral activity against a broad panel of primary HIV-1 isolates (A, A1, AE, AG, B, BF, C, D, G, H) with EC50 values ranging from 1.2 nM to 10.0 nM. The antiviral activity of doravirine was not antagonistic when combined with lamivudine and TDF.
Lamivudine: The antiviral activity of lamivudine against HIV-1 was assessed in a number of cell lines including monocytes and peripheral blood mononuclear cells (PBMCs) using standard susceptibility assays. EC50 values were in the range of 3 to 15,000 nM (1,000 nM = 230 ng per mL). The median EC50 values of lamivudine were 60 nM (range: 20 to 70 nM), 35 nM (range: 30 to 40 nM), 30 nM (range: 20 to 90 nM), 20 nM (range: 3 to 40 nM), 30 nM (range: 1 to 60 nM), 30 nM (range: 20 to 70 nM), 30 nM (range: 3 to 70 nM), and 30 nM (range: 20 to 90 nM) against HIV-1 clades A-G and group O viruses (n = 3 except n = 2 for clade B) respectively. Ribavirin (50,000 nM) used in the treatment of chronic HCV infection decreased the anti-HIV-1 activity of lamivudine by 3.5-fold in MT-4 cells.
TDF: The antiviral activity of tenofovir against laboratory and clinical isolates of HIV-1 was assessed in T lymphoblastoid cell lines, primary monocyte/macrophage cells and peripheral blood lymphocytes. The EC50 values for tenofovir were in the range of 40-8,500 nM. Tenofovir displayed antiviral activity in cell culture against HIV-1 clades A, B, C, D, E, F, G, and O (EC50 values ranged from 500-2,200 nM).
Resistance
In Cell Culture
In Clinical Trials
Cross-Resistance
No significant cross-resistance has been demonstrated between doravirine-resistant HIV-1 variants and lamivudine/emtricitabine or tenofovir or between lamivudine or tenofovir-resistant variants and doravirine.
Doravirine: Cross-resistance has been observed among NNRTIs. Treatment-emergent doravirine resistance-associated substitutions can confer cross resistance to efavirenz, etravirine, nevirapine, and rilpivirine. Of the 6 virologic failure participants who developed doravirine phenotypic resistance, all had phenotypic resistance to efavirenz and nevirapine, 4 had phenotypic resistance to rilpivirine, and 4 had resistance to etravirine in the Monogram PhenoSense assay. Of the 11 virologic failure participants phenotypically resistant to efavirenz, 2 (18%) had decreased susceptibility to doravirine (18- and 36-fold).
The treatment-emergent doravirine resistance-associated substitution Y318F did not confer reduced susceptibility to efavirenz, etravirine, or rilpivirine.
A panel of 96 diverse clinical isolates containing NNRTI resistance-associated substitutions was evaluated for susceptibility to doravirine. Clinical isolates containing the Y188L substitution alone or in combination with K103N or V106I, V106A in combination with G190A and F227L, or E138K in combination with Y181C and M230L showed greater than 100-fold reduced susceptibility to doravirine.
Lamivudine: Cross-resistance has been observed among NRTIs. The M184I/V lamivudine resistance-associated substitution confers resistance to abacavir, didanosine and emtricitabine. Lamivudine also has reduced susceptibility against the K65R substitution.
TDF: Cross-resistance has been observed among NRTIs. The K65R substitution in HIV-1 RT selected by tenofovir is also selected in some patients living with HIV treated with abacavir or didanosine. HIV-1 isolates with the K65R substitution also showed reduced susceptibility to emtricitabine and lamivudine. Therefore, cross-resistance among these NRTIs may occur in patients whose virus harbors the K65R substitution. The K70E substitution selected clinically by TDF results in reduced susceptibility to abacavir, didanosine, emtricitabine, lamivudine, and tenofovir. HIV-1 isolates from patients (n=20) whose HIV-1 expressed a mean of 3 zidovudine resistance-associated substitutions (M41L, D67N, K70R, L210W, T215Y/F, or K219Q/E/N) showed a 3.1-fold decrease in the susceptibility to tenofovir. Participants whose virus expressed an RT L74V substitution without zidovudine resistance-associated substitutions (n=8) had reduced response to TDF. Limited data are available for patients whose virus expressed a Y115F substitution (n=3), Q151M substitution (n=2), or T69 insertion (n=4) in HIV-1 RT, all of whom had a reduced response in clinical trials.
13 Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Doravirine: Doravirine was not carcinogenic in long-term oral carcinogenicity studies in mice and rats at exposures up to 6 and 7 times, respectively, the human exposures at the RHD. A statistically significant incidence of thyroid parafollicular cell adenoma and carcinoma seen only in female rats at the high dose was within the range observed in historical controls.
Lamivudine: Long-term carcinogenicity studies with lamivudine in mice and rats showed no evidence of carcinogenic potential at exposures up to 10 times (mice) and 58 times (rats) the human exposures at the RHD.
TDF: Long-term oral carcinogenicity studies of TDF in mice and rats were carried out at exposures up to approximately 16 times (mice) and 5 times (rats) those observed in humans at the RHD. At the high dose in female mice, liver adenomas were increased at exposures 16 times of that in humans. In rats, the study was negative for carcinogenic findings at exposures up to 5 times that observed in humans at the RHD.
Mutagenesis
Doravirine: Doravirine was not genotoxic in a battery of in vitro or in vivo assays, including microbial mutagenesis, chromosomal aberration in Chinese hamster ovary cells, and in in vivo rat micronucleus assays.
Lamivudine: Lamivudine was mutagenic in an L5178Y mouse lymphoma assay and clastogenic in a cytogenetic assay using cultured human lymphocytes. Lamivudine was not mutagenic in a microbial mutagenicity assay, in an in vitro cell transformation assay, in a rat micronucleus test, in a rat bone marrow cytogenetic assay, and in an assay for unscheduled DNA synthesis in rat liver. Lamivudine showed no evidence of in vivo genotoxic activity in the rat at oral doses of up to 2,000 mg per kg, producing plasma levels of 35 to 45 times those in humans at the recommended dose for HIV-1 infection.
TDF: TDF was mutagenic in the in vitro mouse lymphoma assay and negative in an in vitro bacterial mutagenicity test (Ames test). In an in vivo mouse micronucleus assay, TDF was negative when administered to male mice.
Impairment of Fertility
Doravirine: There were no effects on fertility, mating performance or early embryonic development when doravirine was administered to rats up to the highest dose tested. Systemic exposures (AUC) to doravirine were approximately 7 times the exposure in humans at the RHD.
Lamivudine: In a study of reproductive performance, lamivudine administered to rats at doses up to 4,000 mg per kg per day, producing plasma levels 47 to 70 times those in humans, revealed no evidence of impaired fertility and no effect on the survival, growth, and development to weaning of the offspring.
TDF: There were no effects on fertility, mating performance or early embryonic development when TDF was administered to male rats at a dose equivalent to 10 times the RHD based on body surface area comparisons for 28 days prior to mating and to female rats for 15 days prior to mating through day 7 of gestation. There was, however, an alteration of the estrous cycle in female rats.
14 Clinical Studies
14.1Clinical Trial Results in Adults with No Antiretroviral Treatment History
The efficacy of DELSTRIGO is based on the analyses of 96-week data from a randomized, multicenter, double-blind, active controlled Phase 3 trial (DRIVE-AHEAD, NCT02403674) in participants living with HIV with no antiretroviral treatment history (n=728).
Participants were randomized and received at least 1 dose of either DELSTRIGO or EFV 600 mg/FTC 200 mg/TDF 300 mg once daily. At baseline, the median age of participants was 31 years, 15% were female, 52% were Non-White, 3% had hepatitis B or C coinfection, 14% had a history of AIDS, 21% had HIV-1 RNA greater than 100,000 copies/mL, and 88% had CD4+ T-cell count greater than 200 cells/mm3; these characteristics were similar between treatment groups. Week 96 outcomes for DRIVE-AHEAD are provided in Table 10.
Mean CD4+ T-cell counts in the DELSTRIGO and EFV/FTC/TDF groups increased from baseline by 238 and 223 cells/mm3, respectively.
Table 10: Virologic Outcome in DRIVE-AHEAD at Week 96 in HIV-1 Adult Participants with No Antiretroviral Treatment History Outcome DELSTRIGOOnce DailyN=364 EFV/FTC/TDFOnce DailyN=364 HIV-1 RNA <50 copies/mL 77% 74% ¬†¬†Treatment Difference (95% CI) The 95% CI for the treatment difference was calculated using stratum-adjusted Mantel-Haenszel method. 3.8% (-2.4%, 10.0%) HIV-1 RNA ‚Č• 50 copies/mL Includes participants who discontinued study drug or study before Week 96 for lack or loss of efficacy and participants with HIV-1 RNA equal to or above 50 copies/mL in the Week 96 window. 15% 12% No Virologic Data at Week 96 Window 7% 14% ¬†¬†Discontinued study due to AE or Death Includes participants who discontinued because of adverse event (AE) or death if this resulted in no virologic data in the Week 96 window. 3% 8% ¬†¬†Discontinued study for Other Reasons Other reasons include: lost to follow-up, non-compliance with study drug, physician decision, pregnancy, protocol deviation, screen failure, withdrawal by participant. 4% 5% ¬†¬†On study but missing data in window 1% 1% Proportion (%) of Participants With HIV-1 RNA <50 copies/mL at Week 96 by Baseline and Demographic Category Gender ¬†¬†Male 78% (N = 305) 73% (N = 311) ¬†¬†Female 75% (N = 59) 75% (N = 53) Race ¬†¬†White 80% (N = 176) 74% (N = 170) ¬†¬†Non-White 76% (N = 188) 74% (N = 194) Ethnicity Does not include participants whose ethnicity or viral subtypes were unknown. ¬†¬†Hispanic or Latino 81% (N = 126) 77% (N = 119) ¬†¬†Not Hispanic or Latino 76% (N = 238) 72% (N = 239) Baseline HIV-1 RNA (copies/mL) ¬†¬†‚ȧ100,000 copies/mL 80% (N = 291) 77% (N = 282) ¬†¬†>100,000 copies/mL 67% (N = 73) 62% (N = 82) CD4+ T-cell Count (cells/mm3) ¬†¬†‚ȧ200 cells/mm3 59% (N = 44) 70% (N = 46) ¬†¬†>200 cells/mm3 80% (N = 320) 74% (N = 318) Viral Subtype ¬†¬†Subtype B 80% (N = 232) 72% (N = 253) ¬†¬†Subtype Non-B 73% (N = 130) 77% (N = 111) 14.2Clinical Trial Results in Virologically-Suppressed Adults
The efficacy of switching from a baseline regimen consisting of two NRTIs in combination with a PI plus either ritonavir or cobicistat, or elvitegravir plus cobicistat, or an NNRTI to DELSTRIGO was evaluated in a randomized, open-label trial (DRIVE-SHIFT, NCT02397096), in virologically-suppressed adults living with HIV. Participants must have been virologically-suppressed (HIV-1 RNA <50 copies/mL) on their baseline regimen for at least 6 months prior to trial entry, with no history of virologic failure. Participants were randomized to either switch to DELSTRIGO at baseline [n = 447, Immediate Switch Group (ISG)], or stay on their baseline regimen until Week 24, at which point they switched to DELSTRIGO [n = 223, Delayed Switch Group (DSG)].
At baseline, the median age of participants was 43 years, 16% were female, and 24% were Non-White, 21% were of Hispanic or Latino ethnicity, 3% had hepatitis B and/or C virus co-infection, 17% had a history of AIDS, 96% had CD4+ T-cell count greater than or equal to 200 cells/mm3, 70% were on a regimen containing a PI plus ritonavir, 24% were on a regimen containing an NNRTI, 6% were on a regimen containing elvitegravir plus cobicistat, and 1% were on a regimen containing a PI plus cobicistat; these characteristics were similar between treatment groups.
Virologic outcome results are shown in Table 11.
Table 11: Virologic Outcomes in DRIVE-SHIFT in HIV-1 Virologically-Suppressed Participants Who Switched to DELSTRIGO Outcome DELSTRIGOOnce Daily ISG Week 48N=447 Baseline Regimen DSG Week 24N=223 HIV-1 RNA ‚Č• 50 copies/mL Includes participants who discontinued study drug or study before Week 48 for ISG or before Week 24 for DSG for lack or loss of efficacy and participants with HIV-1 RNA ‚Č•50 copies/mL in the Week 48 window for ISG and in the Week 24 window for DSG 2% 1% ISG-DSG, Difference (95% CI) The 95% CI for the treatment difference was calculated using stratum-adjusted Mantel-Haenszel method. ,Assessed using a non-inferiority margin of 4%. 0.7% (-1.3%, 2.6%) HIV-1 RNA <50 copies/mL 91% 95% No Virologic Data Within the Time Window 8% 4% ¬†¬†Discontinued study due to AE or Death Includes participants who discontinued because of adverse event (AE) or death if this resulted in no virologic data on treatment during the specified window. 3% <1% ¬†¬†Discontinued study for Other Reasons Other reasons include: lost to follow-up, non-compliance with study drug, physician decision, protocol deviation, withdrawal by participant. 4% 4% ¬†¬†On study but missing data in window 0 0 Proportion (%) of Participants With HIV-1 RNA <50 copies/mL by Baseline and Demographic Category Age (years) ¬†¬†<50 90% (N = 320) 95% (N = 157) ¬†¬†‚Č•50 94% (N = 127) 94% (N = 66) Gender ¬†¬†Male 91% (N = 372) 94% (N = 194) ¬†¬†Female 91% (N = 75) 100% (N = 29) Race ¬†¬†White 90% (N = 344) 95% (N = 168) ¬†¬†Non-White 93% (N = 103) 93% (N = 55) Ethnicity ¬†¬†Hispanic or Latino 88% (N = 99) 91% (N = 45) ¬†¬†Not Hispanic or Latino 91% (N = 341) 95% (N = 175) CD4+ T-cell Count (cells/mm3) ¬†¬†<200 cells/mm3 85% (N = 13) 75% (N = 4) ¬†¬†‚Č•200 cells/mm3 91% (N = 426) 95% (N = 216) Baseline Regimen Baseline Regimen = PI plus either ritonavir or cobicistat (specifically atazanavir, darunavir, or lopinavir), or elvitegravir plus cobicistat, or NNRTI (specifically efavirenz, nevirapine, or rilpivirine), each administered with two NRTIs. ¬†¬†PI plus either ritonavir or cobicistat 90% (N = 316) 94% (N = 156) ¬†¬†elvitegravir plus cobicistat or NNRTI 93% (N = 131) 96% (N = 67) 14.3 Clinical Trial Results in Pediatric Participants
The efficacy of DELSTRIGO was evaluated in cohort 2 of an open-label, single-arm 2-cohort trial in pediatric participants 12 to less than 18 years of age living with HIV (IMPAACT 2014 (Protocol 027), NCT03332095). In cohort 1, virologically-suppressed participants (n=9) received a single 100 mg dose of doravirine followed by intensive PK sampling. In cohort 2, virologically-suppressed participants (n=43) were switched to DELSTRIGO and treatment-na√Įve participants (n=2) were started on DELSTRIGO.
In cohort 2, at baseline the median age of participants was 15 years (range: 12 to 17), the median weight was 52 kg (range: 45 to 80), 58% were female, 78% were Asian and 22% were Black, and the median CD4+ T-cell count was 713 cells per mm3 (range 84 to 1397). After switching to DELSTRIGO, 95% (41/43) of virologically-suppressed participants remained suppressed (HIV-1 RNA <50 copies/mL) at Week 24. One of the two treatment-na√Įve participants achieved HIV-1 RNA <50 copies/mL at Week 24. The other treatment-na√Įve participant met the protocol-defined virologic failure criteria (defined as 2 consecutive plasma HIV-1 RNA test results ‚Č•200 copies/mL at or after Week 24) and was evaluated for the development of resistance; no emergence of genotypic or phenotypic resistance to doravirine, lamivudine, or tenofovir was detected.
16 How Supplied/storage And Handling
Each DELSTRIGO tablet contains 100 mg of doravirine, 300 mg of lamivudine, and 300 mg of tenofovir disoproxil fumarate (equivalent to 245 mg of tenofovir disoproxil), is yellow, oval-shaped, film-coated, and is debossed with the corporate logo and 776 on one side and plain on the other side. Each bottle contains 30 tablets (NDC 0006-5007-01) and silica gel desiccants, and is closed with a child-resistant closure.
STORAGE AND HANDLING SECTION
Store DELSTRIGO in the original bottle. Keep the bottle tightly closed to protect from moisture. Do not remove the desiccants.
Store DELSTRIGO at 20¬įC to 25¬įC (68¬įF to 77¬įF); excursions permitted to 15¬įC to 30¬įC (59¬įF to 86¬įF) [see USP Controlled Room Temperature].
17 Patient Counseling Information
Advise the patient to read the FDA-approved patient labeling (Patient Information).
Severe Skin Reactions
Inform patients that severe skin reactions including Stevens-Johnson syndrome (SJS)/toxic epidermal necrolysis (TEN) have been reported with DELSTRIGO. Advise patients to immediately contact their healthcare provider if they develop a rash. Instruct patients to immediately stop taking DELSTRIGO and seek medical attention if a painful rash with mucosal involvement develops [see Warnings and Precautions (5.1)].
Severe Acute Exacerbation of Hepatitis B in people with concomitant HIV-1 and HBV
Inform patients that severe acute exacerbations of hepatitis B have been reported in people with concomitant HIV-1 and HBV who have discontinued lamivudine or TDF and may occur with discontinuation of DELSTRIGO [see Warnings and Precautions (5.2)]. Advise patients not to discontinue DELSTRIGO without first informing their healthcare provider.
Drug Interactions
Inform patients that DELSTRIGO may interact with certain other drugs; therefore, advise patients to report to their healthcare provider the use of any other prescription or nonprescription medication or herbal products, including St. John's wort [see Contraindications (4), Warnings and Precautions (5.4), and Drug Interactions (7)].
For patients concomitantly receiving rifabutin, take one tablet of doravirine (PIFELTRO) 100 mg approximately 12 hours after the dose of DELSTRIGO [see Dosage and Administration (2.4)].
New Onset or Worsening Renal Impairment
Inform patients that renal impairment, including cases of acute renal failure and Fanconi syndrome, has been reported in association with the use of TDF. Advise patients to avoid DELSTRIGO with concurrent or recent use of a nephrotoxic agent (e.g., high-dose or multiple NSAIDS) [see Warnings and Precautions (5.3)].
Bone Loss and Mineralization Defects
Inform patients that decreases in bone mineral density have been observed with the use of TDF, a component of DELSTRIGO. Assessment of bone mineral density (BMD) should be considered in patients who have a history of pathologic bone fracture or other risk factors for osteoporosis or bone loss [see Warnings and Precautions (5.5)].
Immune Reconstitution Syndrome
Inform patients that in some patients with advanced HIV infection (AIDS), signs and symptoms of inflammation from previous infections may occur soon after anti-HIV treatment is started. It is believed that these symptoms are due to an improvement in the body's immune response, enabling the body to fight infections that may have been present with no obvious symptoms. Advise patients to inform their healthcare provider immediately of any symptoms of infection [see Warnings and Precautions (5.6)].
Dosing Instructions
Advise patients to take DELSTRIGO every day at a regularly scheduled time with or without food. Inform patients that it is important not to miss or skip doses as it can result in development of resistance. If a patient forgets to take DELSTRIGO, tell the patient to take the missed dose right away, unless it is almost time for the next dose. Advise the patient not to take 2 doses at one time and to take the next dose at the regularly scheduled time.
Pregnancy Registry
Inform patients that there is an antiretroviral pregnancy registry to monitor fetal outcomes of pregnant individuals exposed to DELSTRIGO [see Use in Specific Populations (8.1)].
Lactation
Inform individuals with HIV-1 infection that the potential risks of breastfeeding include: (1) HIV-1 transmission (in HIV-1-negative infants), (2) developing viral resistance (in HIV-1-positive infants), and (3) serious adverse reactions in a breastfed infant similar to those seen in adults [see Use in Specific Populations (8.2)].
Manufactured for: Merck Sharp & Dohme LLC Rahway, NJ 07065, USA
For patent information: www.msd.com/research/patent
The trademarks depicted herein are owned by their respective companies.
Copyright © 2018-2025 Merck & Co., Inc., Rahway, NJ, USA, and its affiliates. All rights reserved.
uspi-mk1439a-t-2501r008
Spl Patient Package Insert Section
Patient InformationDELSTRIGO¬ģ (del-STREE-go)(doravirine, lamivudine, and tenofovir disoproxil fumarate)tablets This Patient Information has been approved by the U.S. Food and Drug Administration. Revised: 11/2024 What is the most important information I should know about DELSTRIGO?
DELSTRIGO can cause serious side effects, including:Worsening of hepatitis B virus infection (HBV). If you have Human Immunodeficiency Virus-1 (HIV-1) and HBV infection, your HBV infection may get worse (flare-up) if you stop taking DELSTRIGO. A "flare-up" is when your HBV infection suddenly returns in a worse way than before. Your healthcare provider will test you for HBV infection before you start treatment with DELSTRIGO.
- Do not run out of DELSTRIGO. Refill your prescription or talk to your healthcare provider before your DELSTRIGO is all gone.
- Do not stop taking DELSTRIGO without first talking to your healthcare provider. If you stop taking DELSTRIGO, your healthcare provider will need to check your health often and do blood tests regularly for several months to check your liver. Tell your healthcare provider about any new or unusual symptoms you may have after you stop taking DELSTRIGO.
For more information about side effects, see "What are the possible side effects of DELSTRIGO?" What is DELSTRIGO? DELSTRIGO is a prescription medicine that is used without other HIV-1 medicines to treat HIV-1 infection in adults and children who weigh at least 77 pounds (35 kg):
- who have not received HIV-1 medicines in the past, or
- to replace their current HIV-1 medicines for people whose healthcare provider determines that they meet certain requirements.
HIV-1 is the virus that causes Acquired Immune Deficiency Syndrome (AIDS). DELSTRIGO contains the prescription medicines doravirine, lamivudine and tenofovir disoproxil fumarate. It is not known if DELSTRIGO is safe and effective in children who weigh less than 77 pounds (35 kg). Who should not take DELSTRIGO? Do not take DELSTRIGO if you take any of the following medicines:
- carbamazepine
- oxcarbazepine
- phenobarbital
- phenytoin
- enzalutamide
- rifampin
- rifapentine
- mitotane
- St. John's wort
Ask your healthcare provider or pharmacist if you are not sure if your medicine is one that is uled above. If you have taken any of the medicines in the past 4 weeks, talk to your healthcare provider or pharmacist before starting treatment with DELSTRIGO. Do not take DELSTRIGO if you have ever had an allergic reaction to lamivudine. What should I tell my healthcare provider before treatment with DELSTRIGO? Before treatment with DELSTRIGO, tell your healthcare provider about all of your medical conditions, including if you:
- have hepatitis B virus infection
- have kidney problems
- have bone problems, including a history of bone fractures
- are pregnant or plan to become pregnant. It is not known if DELSTRIGO can harm your unborn baby. Tell your healthcare provider if you become pregnant during treatment with DELSTRIGO. Pregnancy Registry: There is a pregnancy registry for people who take DELSTRIGO during pregnancy. The purpose of this registry is to collect information about the health of you and your baby. Talk to your healthcare provider about how you can take part in this registry.
- are breastfeeding or plan to breastfeed. Two of the medicines in DELSTRIGO (lamivudine and tenofovir) can pass into your breast milk. Doravirine may pass to your baby in your breast milk. Talk with your healthcare provider about the following risks to your baby from breastfeeding during treatment with DELSTRIGO:
- the HIV-1 virus may pass to your baby if your baby does not have HIV-1 infection.
- the HIV-1 virus may become harder to treat if your baby has HIV-1 infection.
- your baby may get side effects from DELSTRIGO.
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements.
- Some medicines interact with DELSTRIGO. Keep a ul of your medicines to show your healthcare provider and pharmacist.
- Tell your healthcare provider if you have taken rifabutin in the past 4 weeks.
- You can ask your healthcare provider or pharmacist for a ul of medicines that interact with DELSTRIGO.
- Do not start taking a new medicine without telling your healthcare provider. Your healthcare provider can tell you if it is safe to take DELSTRIGO with other medicines.
How should I take DELSTRIGO?
- Take DELSTRIGO every day exactly as your healthcare provider tells you to take it.
- Take DELSTRIGO 1 time each day, at about the same time every day.
- DELSTRIGO is usually taken by itself (without other HIV-1 medicines).
- If you take the medicine rifabutin during treatment with DELSTRIGO, your healthcare provider will also prescribe an additional dose of doravirine for you. You may not have enough doravirine in your blood if you take rifabutin during treatment with DELSTRIGO. Carefully follow your healthcare provider's instructions about when to take doravirine and how much to take. This is usually 1 tablet of doravirine about 12 hours after your last dose of DELSTRIGO.
- Take DELSTRIGO with or without food.
- Do not change your dose or stop taking DELSTRIGO without talking to your healthcare provider. Stay under a healthcare provider's care when taking DELSTRIGO.
- It is important that you do not miss or skip doses of DELSTRIGO.
- If you miss a dose of DELSTRIGO, take it as soon as you remember. If it is almost time for your next dose, skip the missed dose and take the next dose at your regular time. Do not take 2 doses of DELSTRIGO at the same time.
- If you have any questions, call your healthcare provider or pharmacist.
- If you take too much DELSTRIGO, call your healthcare provider or go to the nearest hospital emergency room right away.
- When your DELSTRIGO supply starts to run low, get more from your healthcare provider or pharmacy. This is very important because the amount of virus in your blood may increase if the medicine is stopped for even a short time. The virus may develop resistance to DELSTRIGO and become harder to treat.
What are the possible side effects of DELSTRIGO?
DELSTRIGO may cause serious side effects, including:
- See "What is the most important information I should know about DELSTRIGO?"
- Severe skin reactions have happened in people treated with DELSTRIGO. Call your healthcare provider right away if you develop a rash during treatment with DELSTRIGO. Stop taking DELSTRIGO and get medical help right away if you develop a painful rash with any of the following symptoms: fever, bulers or sores in the mouth, bulers or peeling of the skin, or redness or swelling of the eyes (conjunctivitis).
- New or worse kidney problems, including kidney failure. Your healthcare provider should do blood and urine tests to check your kidneys before you start and during treatment with DELSTRIGO. Your healthcare provider may tell you to stop taking DELSTRIGO if you develop new or worse kidney problems.
- Bone problems can happen in some people who take DELSTRIGO. Bone problems include bone pain, softening or thinning (which may lead to fractures). Your healthcare provider may need to do tests to check your bones.
Tell your healthcare provider if you have any of the following symptoms during treatment with DELSTRIGO: bone pain that does not go away or worsening bone pain, pain in your arms, legs, hands or feet, broken (fractured) bones, or muscle pain or weakness. These may be symptoms of a bone or kidney problem.
- Changes in your immune system (Immune Reconstitution Syndrome) can happen when you start taking HIV-1 medicines. Your immune system may get stronger and begin to fight infections that have been hidden in your body for a long time. Tell your healthcare provider right away if you start having any new symptoms after starting your HIV-1 medicine.
The most common side effects of DELSTRIGO include dizziness, nausea, and abnormal dreams. These are not all of the possible side effects of DELSTRIGO.Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. How should I store DELSTRIGO?
- Store DELSTRIGO tablets at room temperature between 68¬įF to 77¬įF (20¬įC to 25¬įC).
- Keep DELSTRIGO in the original bottle.
- Do not take the tablets out of the bottle to store in another container, such as a pill box.
- Keep the bottle tightly closed to protect DELSTRIGO from moisture.
- The DELSTRIGO bottle contains desiccants to help keep your medicine dry (protect it from moisture). Keep the desiccants in the bottle. Do not eat the desiccants.
Keep DELSTRIGO and all medicines out of the reach of children. General information about the safe and effective use of DELSTRIGO. Medicines are sometimes prescribed for purposes other than those uled in the Patient Information leaflet. Do not use DELSTRIGO for a condition for which it was not prescribed. Do not give DELSTRIGO to other people, even if they have the same symptoms that you have. It may harm them. You can ask your pharmacist or healthcare provider for information about DELSTRIGO that is written for healthcare professionals. What are the ingredients in DELSTRIGO? Active ingredients: doravirine, lamivudine, and tenofovir disoproxil fumarate. Inactive ingredients: colloidal silicon dioxide, croscarmellose sodium, hypromellose acetate succinate, magnesium stearate, microcrystalline cellulose, and sodium stearyl fumarate. The tablet film coating contains hypromellose, iron oxide yellow, lactose monohydrate, titanium dioxide, and triacetin. The coated tablets are polished with carnauba wax. Manufactured for: Merck Sharp & Dohme LLC Rahway, NJ 07065, USA For patent information: www.msd.com/research/patent The trademarks depicted herein are owned by their respective companies.Copyright © 2018-2024 Merck & Co., Inc., Rahway, NJ, USA, and its affiliates. All rights reserved.usppi-mk1439a-t-2411r005 For more information, go to www.DELSTRIGO.com or call 1-877-888-4231.
Principal Display Panel - 30 Tablet Bottle Label
NDC 0006-5007-01
Delstrigo¬ģ (doravirine, lamivudine, andtenofovir disoproxil fumarate) tablets
100mg/300mg /300mg
Each tablet contains 100 mg doravirine, 300 mglamivudine, and 300 mg tenofovir disoproxil fumarate(equivalent to 245 mg tenofovir disoproxil).
ALERT: Find out about medicines thatshould NOT be taken with Delstrigo¬ģ.
Rx only
30 Tablets

DISCLAIMER:
"This tool does not provide medical advice, and is for informational and educational purposes only, and is not a substitute for professional medical advice, treatment or diagnosis. Call your doctor to receive medical advice. If you think you may have a medical emergency, please dial 911."
"Do not rely on openFDA to make decisions regarding medical care. While we make every effort to ensure that data is accurate, you should assume all results are unvalidated. We may limit or otherwise restrict your access to the API in line with our Terms of Service."
"This product uses publicly available data from the U.S. National Library of Medicine (NLM), National Institutes of Health, Department of Health and Human Services; NLM is not responsible for the product and does not endorse or recommend this or any other product."
PillSync may earn a commission via links on our site
