LEXIVA (fosamprenavir calcium 700 mg) Dailymed
Generic: fosamprenavir calcium is used for the treatment of Acquired Immunodeficiency Syndrome

All Imprints
lexiva (fosamprenavir calcium) 700 mg - gxll7 oval pink
fosamprenavir calcium 700 mg - gx ll7 capsule pink

Go PRO for all pill images
1 Indications And Usage
LEXIVA is indicated in combination with other antiretroviral agents for the treatment of human immunodeficiency virus (HIV-1) infection.
The following points should be considered when initiating therapy with LEXIVA plus ritonavir in protease inhibitor-experienced patients:
‚ÄĘ The protease inhibitor-experienced patient trial was not large enough to reach a definitive conclusion that LEXIVA plus ritonavir and lopinavir plus ritonavir are clinically equivalent [see Clinical Studies (14.2)].‚ÄĘ Once-daily administration of LEXIVA plus ritonavir is not recommended for adult protease inhibitor-experienced patients or any pediatric patients [see Dosage and Administration (2.2, 2.3), Clinical Studies (14.2, 14.3)].‚ÄĘ Dosing of LEXIVA plus ritonavir is not recommended for protease inhibitor-experienced pediatric patients younger than 6¬†months [see Clinical Pharmacology (12.3)].
LEXIVA is an HIV protease inhibitor indicated in combination with other antiretroviral agents for the treatment of HIV-1 infection. (1 )
2 Dosage And Administration
‚ÄĘ Therapy-Naive Adults: LEXIVA 1,400¬†mg twice daily; LEXIVA 1,400¬†mg once daily plus ritonavir 200¬†mg once daily; LEXIVA 1,400¬†mg once daily plus ritonavir 100¬†mg once daily; LEXIVA 700¬†mg twice daily plus ritonavir 100¬†mg twice daily. (2.2 )‚ÄĘ Protease Inhibitor-Experienced Adults: LEXIVA 700¬†mg twice daily plus ritonavir 100¬†mg twice daily. (2.2 )‚ÄĘ Pregnant Patients: LEXIVA 700 mg twice daily plus ritonavir 100 mg twice daily should only be considered in women who are already on a stable twice-daily regimen of LEXIVA/ritonavir 700 mg/100 mg prior to pregnancy and who are virologically suppressed (HIV-1 RNA less than 50 copies per mL). (2.2 )‚ÄĘ Pediatric Patients (aged at least 4 weeks to 18¬†years): Dosage should be calculated based on body weight (kg) and should not exceed adult dose. (2.3 )‚ÄĘ Hepatic Impairment: Recommended adjustments for patients with mild, moderate, or severe hepatic impairment. (2.4 )
Dosing Considerations
‚ÄĘ LEXIVA tablets may be taken with or without food. (2.1 )‚ÄĘ LEXIVA suspension: Adults should take without food; pediatric patients should take with food. (2.1 )2.1 General Dosing Information
LEXIVA tablets may be taken with or without food.
Adults should take LEXIVA oral suspension without food. Pediatric patients should take LEXIVA oral suspension with food [see Clinical Pharmacology (12.3)]. If emesis occurs within 30 minutes after dosing, re-dosing of LEXIVA oral suspension should occur.
Higher-than-approved dose combinations of LEXIVA plus ritonavir are not recommended due to an increased risk of transaminase elevations [see Overdosage (10)].
When LEXIVA is used in combination with ritonavir, prescribers should consult the full prescribing information for ritonavir.
2.2 Adults
Therapy-Naive Adults
‚ÄĘ LEXIVA 1,400¬†mg twice daily (without ritonavir).‚ÄĘ LEXIVA 1,400¬†mg once daily plus ritonavir 200¬†mg once daily.‚ÄĘ LEXIVA 1,400¬†mg once daily plus ritonavir 100¬†mg once daily.
‚ÄĘ Dosing of LEXIVA 1,400¬†mg once daily plus ritonavir 100¬†mg once daily is supported by pharmacokinetic data [see Clinical Pharmacology (12.3)].‚ÄĘ LEXIVA 700¬†mg twice daily plus ritonavir 100¬†mg twice daily.
‚ÄĘ Dosing of LEXIVA 700¬†mg twice daily plus ritonavir 100¬†mg twice daily is supported by pharmacokinetic and safety data [see Clinical Pharmacology (12.3)].
Protease Inhibitor-Experienced Adults
‚ÄĘ LEXIVA 700¬†mg twice daily plus ritonavir 100¬†mg twice daily.
Pregnancy
‚ÄĘ LEXIVA 700 mg twice daily plus ritonavir 100 mg twice daily.
‚ÄĘ Dosing of LEXIVA 700 mg twice daily plus ritonavir 100 mg twice daily should only be considered in pregnant patients who are already on a stable twice-daily regimen of LEXIVA/ritonavir 700 mg/100 mg prior to pregnancy and who are virologically suppressed (HIV-1 RNA less than 50 copies per mL). Lower exposures of amprenavir were observed during pregnancy; therefore, viral load should be monitored closely to ensure viral suppression is maintained [see Use in Specific Populations (8.1), Clinical Pharmacology ( 12.3 )]. Data regarding use of other regimens of LEXIVA (with or without ritonavir) in pregnancy are not available.2.3 Pediatric Patients (Aged at Least 4 Weeks to 18 Years)
The recommended dosage of LEXIVA in patients aged at least 4 weeks to 18 years should be calculated based on body weight (kg) and should not exceed the recommended adult dose (Table 1).
Table 1. Twice-Daily Dosage Regimens by Weight for Protease Inhibitor-Naive Pediatric Patients (Aged 4 Weeks and Older) and for Protease Inhibitor-Experienced Pediatric Patients (Aged 6 Months and Older) Using LEXIVA Oral Suspension with Concurrent Ritonavir a When dosing with ritonavir, do not exceed the adult dose of LEXIVA 700 mg/ritonavir 100 mg twice-daily dose.
Weight
Twice-Daily Dosage Regimen
<11 kg
LEXIVA 45 mg/kg plus ritonavir 7 mg/kga
11 kg - <15 kg
LEXIVA 30 mg/kg plus ritonavir 3 mg/kga
15 kg - <20 kg
LEXIVA 23 mg/kg plus ritonavir 3 mg/kga
‚Č•20¬†kg
LEXIVA 18 mg/kg plus ritonavir 3 mg/kga
Alternatively, protease inhibitor-naive children aged 2 years and older can be administered LEXIVA (without ritonavir) 30 mg per kg twice daily.
LEXIVA should only be administered to infants born at 38 weeks’ gestation or greater and who have attained a postnatal age of 28 days.
For pediatric patients, pharmacokinetic and clinical data:
‚ÄĘ do not support once-daily dosing of LEXIVA alone or in combination with ritonavir [see Clinical Studies (14.3)].‚ÄĘ do not support administration of LEXIVA alone or in combination with ritonavir for protease inhibitor‚ÄĎexperienced children younger than 6¬†months [see Clinical Pharmacology (12.3)].‚ÄĘ do not support twice-daily dosing of LEXIVA without ritonavir in pediatric patients younger than 2¬†years [see Clinical Pharmacology (12.3)].
Other Dosing Considerations
‚ÄĘ When administered without ritonavir, the adult regimen of LEXIVA tablets 1,400¬†mg twice daily may be used for pediatric patients weighing at least 47¬†kg.‚ÄĘ When administered in combination with ritonavir, LEXIVA tablets may be used for pediatric patients weighing at least 39¬†kg; ritonavir capsules may be used for pediatric patients weighing at least 33¬†kg.2.4 Patients with Hepatic Impairment
See Clinical Pharmacology (12.3).
Mild Hepatic Impairment (Child-Pugh Score Ranging from 5 to 6)
LEXIVA should be used with caution at a reduced dosage of 700 mg twice daily without ritonavir (therapy-naive) or 700 mg twice daily plus ritonavir 100 mg once daily (therapy-naive or protease inhibitor-experienced).
Moderate Hepatic Impairment (Child-Pugh Score Ranging from 7 to 9)
LEXIVA should be used with caution at a reduced dosage of 700 mg twice daily without ritonavir (therapy-naive) or 450 mg twice daily plus ritonavir 100 mg once daily (therapy-naive or protease inhibitor-experienced).
Severe Hepatic Impairment (Child-Pugh Score Ranging from 10 to 15)
LEXIVA should be used with caution at a reduced dosage of 350 mg twice daily without ritonavir (therapy-naive) or 300 mg twice daily plus ritonavir 100 mg once daily (therapy-naive or protease inhibitor-experienced).
There are no data to support dosing recommendations for pediatric patients with hepatic impairment.
3 Dosage Forms And Strengths
LEXIVA tablets, 700¬†mg, are pink, film-coated, capsule-shaped, biconvex tablets with ‚ÄúGX¬†LL7‚ÄĚ debossed on one face.
LEXIVA oral suspension, 50 mg per mL, is a white to off-white suspension that has a characteristic grape-bubblegum-peppermint flavor.
‚ÄĘ 700-mg tablets (3 )‚ÄĘ 50-mg-per-mL oral suspension (3 )
4 Contraindications
‚ÄĘ LEXIVA is contraindicated in patients with previously demonstrated clinically significant hypersensitivity (e.g., Stevens-Johnson syndrome) to any of the components of this product or to amprenavir.¬† LEXIVA is contraindicated when coadministered with drugs that are highly dependent on cytochrome P450 (CYP)3A4 for clearance and for which elevated plasma concentrations are associated with serious and/or life‚ÄĎthreatening events. These drugs and other contraindicated drugs (which may lead to reduced efficacy of LEXIVA and possible resistance) are uled below [see Drug Interactions (7), Clinical Pharmacology (12.3)]. The ul of contraindicated drugs applies to the use of LEXIVA with or without ritonavir, unless otherwise indicated. If LEXIVA is coadministered with ritonavir, reference should be made to the full prescribing information for ritonavir for additional contraindications.
¬†¬†¬†¬†‚Äʬ†¬†¬†¬†LEXIVA is contraindicated when coadministered with the following drugs:
              o     Alpha 1-adrenoreceptor antagonist: Alfuzosin
              o     Antiarrhythmics: Flecainide (with ritonavir), propafenone (with ritonavir)
              o     Antimycobacterial: Rifampin
              o     Antipsychotic: Lurasidone (with ritonavir), pimozide
              o     Ergot derivatives: Dihydroergotamine, ergonovine, ergotamine, methylergonovine
              o     GI motility agent: Cisapride
              o     Herbal product: St. John’s wort (Hypericum perforatum)
              o     Lipid modifying agents: Lomitapide, lovastatin, simvastatin
              o     Non-nucleoside reverse transcriptase inhibitor: Delavirdine
              o     PDE5 inhibitor: Sildenafil (REVATIO) (for treatment of pulmonary arterial hypertension)
              o     Sedative/hypnotics: Midazolam, triazolam
‚ÄĘ Hypersensitivity to LEXIVA or amprenavir (e.g., Stevens-Johnson syndrome). (4 )‚ÄĘ Drugs highly dependent on cytochrome P450 (CYP)3A4 for clearance and for which elevated plasma levels may result in serious and/or life-threatening events. (4 )‚ÄĘ Review ritonavir contraindications when used in combination. (4 )
5 Warnings And Precautions
‚ÄĘ The concomitant use of LEXIVA with ritonavir and certain other drugs may result in known or potentially significant drug interactions. Consult the full prescribing information prior to and during treatment for potential drug interactions. (5.1 , 7.3)‚ÄĘ LEXIVA should be discontinued for severe skin reactions including Stevens-Johnson syndrome. (5.2 )‚ÄĘ LEXIVA should be used with caution in patients with a known sulfonamide allergy. (5.3 )‚ÄĘ Use of higher-than-approved doses may lead to transaminase elevations. Patients with hepatitis B or C are at increased risk of transaminase elevations. (5.4 )‚ÄĘ Patients receiving LEXIVA may develop new onset or exacerbations of diabetes mellitus, hyperglycemia (5.5 ), immune reconstitution syndrome (5.6 ), increase of body fat (5.7 ), and elevated triglyceride and cholesterol concentrations (5.8 ). Monitor cholesterol and triglycerides prior to therapy and periodically thereafter.‚ÄĘ Acute hemolytic anemia has been reported with amprenavir. (5.9 )‚ÄĘ Hemophilia: Spontaneous bleeding may occur, and additional factor VIII may be required. (5.10 )‚ÄĘ Nephrolithiasis: Cases of nephrolithiasis have been reported with fosamprenavir. (5.11 )5.1 Risk of Serious Adverse Reactions Due to Drug Interactions
Initiation of LEXIVA/ritonavir, a CYP3A inhibitor, in patients receiving medications metabolized by CYP3A, or initiation of medications metabolized by CYP3A in patients already receiving LEXIVA/ritonavir may increase plasma concentrations of medications metabolized by CYP3A. Initiation of medications that inhibit or induce CYP3A may increase or decrease concentrations of LEXIVA/ritonavir, respectively. These interactions may lead to:
‚ÄĘ clinically significant adverse reactions, potentially leading to severe, life-threatening, or fatal events from greater exposures of concomitant medications.‚ÄĘ clinically significant adverse reactions from greater exposures of LEXIVA/ritonavir.‚ÄĘ loss of therapeutic effect of LEXIVA/ritonavir and possible development of resistance.
See Table 6 for steps to prevent or manage these possible and known significant drug interactions, including dosing recommendations [see Drug Interactions (7)]. Consider the potential for drug interactions prior to and during therapy with LEXIVA/ritonavir; review concomitant medications during therapy with LEXIVA/ritonavir, and monitor for the adverse reactions associated with the concomitant medications [see Contraindications (4), Drug Interactions (7)].
5.2 Skin Reactions
Severe and life-threatening skin reactions, including 1 case of Stevens-Johnson syndrome among 700 subjects treated with LEXIVA in clinical trials. Treatment with LEXIVA should be discontinued for severe or life-threatening rashes and for moderate rashes accompanied by systemic symptoms [see Adverse Reactions (6)].
5.3 Sulfa Allergy
LEXIVA should be used with caution in patients with a known sulfonamide allergy. Fosamprenavir contains a sulfonamide moiety. The potential for cross-sensitivity between drugs in the sulfonamide class and fosamprenavir is unknown. In a clinical trial of LEXIVA used as the sole protease inhibitor, rash occurred in 2 of 10 subjects (20%) with a history of sulfonamide allergy compared with 42 of 126 subjects (33%) with no history of sulfonamide allergy. In 2 clinical trials of LEXIVA plus low-dose ritonavir, rash occurred in 8 of 50 subjects (16%) with a history of sulfonamide allergy compared with 50 of 412 subjects (12%) with no history of sulfonamide allergy.
5.4 Hepatic Toxicity
Use of LEXIVA with ritonavir at higher-than-recommended dosages may result in transaminase elevations and should not be used [see Dosage and Administration (2), Overdosage (10)]. Patients with underlying hepatitis B or C or marked elevations in transaminases prior to treatment may be at increased risk for developing or worsening of transaminase elevations. Appropriate laboratory testing should be conducted prior to initiating therapy with LEXIVA and patients should be monitored closely during treatment.
5.5 Diabetes/Hyperglycemia
New onset diabetes mellitus, exacerbation of pre-existing diabetes mellitus, and hyperglycemia have been reported during postmarketing surveillance in HIV-1‚Äďinfected patients receiving protease inhibitor therapy. Some patients required either initiation or dose adjustments of insulin or oral hypoglycemic agents for treatment of these events. In some cases, diabetic ketoacidosis has occurred. In those patients who discontinued protease inhibitor therapy, hyperglycemia persisted in some cases. Because these events have been reported voluntarily during clinical practice, estimates of frequency cannot be made and causal relationships between protease inhibitor therapy and these events have not been established.
5.6 Immune Reconstitution Syndrome
Immune reconstitution syndrome has been reported in patients treated with combination antiretroviral therapy, including LEXIVA. During the initial phase of combination antiretroviral treatment, patients whose immune systems respond may develop an inflammatory response to indolent or residual opportunistic infections (such as Mycobacterium avium infection, cytomegalovirus, Pneumocystis jirovecii pneumonia [PCP], or tuberculosis), which may necessitate further evaluation and treatment.
Autoimmune disorders (such as Graves’ disease, polymyositis, and Guillain-Barré syndrome) have also been reported to occur in the setting of immune reconstitution; however, the time to onset is more variable, and can occur many months after initiation of treatment.
5.7 Increase in Body Fat
Increase of body fat has been observed in patients receiving protease inhibitors, including LEXIVA. The mechanism and long-term consequences of these events are currently unknown. A causal relationship has not been established.
5.8 Lipid Elevations
Treatment with LEXIVA plus ritonavir has resulted in increases in the concentration of triglycerides and cholesterol [see Adverse Reactions (6)]. Triglyceride and cholesterol testing should be performed prior to initiating therapy with LEXIVA and at periodic intervals during therapy. Lipid disorders should be managed as clinically appropriate [see Drug Interactions (7)].
5.9 Hemolytic Anemia
Acute hemolytic anemia has been reported in a patient treated with amprenavir.
5.10 Patients with Hemophilia
There have been reports of spontaneous bleeding in patients with hemophilia A and B treated with protease inhibitors. In some patients, additional factor VIII was required. In many of the reported cases, treatment with protease inhibitors was continued or restarted. A causal relationship between protease inhibitor therapy and these episodes has not been established.
5.11 Nephrolithiasis
Cases of nephrolithiasis were reported during postmarketing surveillance in HIV‚Äď1‚Äďinfected patients receiving LEXIVA. Because these events were reported voluntarily during clinical practice, estimates of frequency cannot be made. If signs or symptoms of nephrolithiasis occur, temporary interruption or discontinuation of therapy may be considered.
5.12 Resistance/Cross-Resistance
Because the potential for HIV cross-resistance among protease inhibitors has not been fully explored, it is unknown what effect therapy with LEXIVA will have on the activity of subsequently administered protease inhibitors. LEXIVA has been studied in patients who have experienced treatment failure with protease inhibitors [see Clinical Studies (14.2)].
6 Adverse Reactions
‚ÄĘ Severe or life-threatening skin reactions have been reported with the use of LEXIVA [see Warnings and Precautions (5.2)].‚ÄĘ The most common moderate to severe adverse reactions in clinical trials of LEXIVA were diarrhea, rash, nausea, vomiting, and headache.‚ÄĘ Treatment discontinuation due to adverse events occurred in 6.4% of subjects receiving LEXIVA and in 5.9% of subjects receiving comparator treatments. The most common adverse reactions leading to discontinuation of LEXIVA (incidence less than or equal to 1% of subjects) included diarrhea, nausea, vomiting, AST increased, ALT increased, and rash.
‚ÄĘ In adults the most common adverse reactions (incidence greater than or equal to 4%) are diarrhea, rash, nausea, vomiting, and headache. (6.1 )‚ÄĘ Vomiting and neutropenia were more frequent in pediatrics than in adults. (6.1 )
To report SUSPECTED ADVERSE REACTIONS, contact ViiV Healthcare at 1-877-844-8872 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared with rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
Adult Trials
The data for the 3¬†active-controlled clinical trials described below reflect exposure of 700¬†HIV-1‚Äďinfected subjects to LEXIVA tablets, including 599¬†subjects exposed to LEXIVA for greater than 24¬†weeks, and 409¬†subjects exposed for greater than 48¬†weeks. The population age ranged from 17 to 72¬†years. Of these subjects, 26% were female, 51% white, 31% black, 16% American Hispanic, and 70% were antiretroviral-naive. Sixty-one percent received LEXIVA 1,400¬†mg once daily plus ritonavir 200¬†mg once daily; 24% received LEXIVA 1,400¬†mg twice daily; and 15% received LEXIVA 700¬†mg twice daily plus ritonavir 100¬†mg twice daily.
Selected adverse reactions reported during the clinical efficacy trials of LEXIVA are shown in Tables 2 and 3. Each table presents adverse reactions of moderate or severe intensity in subjects treated with combination therapy for up to 48 weeks.
Table 2. Selected Moderate/Severe Clinical Adverse Reactions Reported in Greater than or Equal to 2% of Antiretroviral-Naive Adult Subjects a All subjects also received abacavir and lamivudine twice daily.
Adverse Reaction
APV30001a
APV30002a
LEXIVA
1,400 mg Twice Daily
(n = 166)
Nelfinavir
1,250 mg Twice Daily
(n = 83)
LEXIVA 1,400 mg and
Ritonavir 200 mg
Once Daily
(n = 322)
Nelfinavir
1,250 mg Twice Daily
(n = 327)
Gastrointestinal
  Diarrhea
5%
18%
10%
18%
  Nausea
7%
4%
7%
5%
  Vomiting
2%
4%
6%
4%
  Abdominal pain
1%
0%
2%
2%
Skin
  Rash
8%
2%
3%
2%
General disorders
  Fatigue
2%
1%
4%
2%
Nervous system
  Headache
2%
4%
3%
3%
Table 3. Selected Moderate/Severe Clinical Adverse Reactions Reported in Greater than or Equal to 2% of Protease Inhibitor-Experienced Adult Subjects (Trial APV30003) a All subjects also received 2 reverse transcriptase inhibitors.
Adverse Reaction
LEXIVA 700 mg and
Ritonavir 100 mg
Twice Dailya
(n = 106)
Lopinavir 400 mg and
Ritonavir 100 mg
Twice Daily a
(n = 103)
Gastrointestinal
  Diarrhea
13%
11%
  Nausea
3%
9%
  Vomiting
3%
5%
  Abdominal pain
<1%
2%
Skin
  Rash
3%
0%
Nervous system
  Headache
4%
2%
Skin rash (without regard to causality) occurred in approximately 19% of subjects treated with LEXIVA in the pivotal efficacy trials. Rashes were usually maculopapular and of mild or moderate intensity, some with pruritus. Rash had a median onset of 11 days after initiation of LEXIVA and had a median duration of 13 days. Skin rash led to discontinuation of LEXIVA in less than 1% of subjects. In some subjects with mild or moderate rash, dosing with LEXIVA was often continued without interruption; if interrupted, reintroduction of LEXIVA generally did not result in rash recurrence.
The percentages of subjects with Grade 3 or 4 laboratory abnormalities in the clinical efficacy trials of LEXIVA are presented in Tables 4 and 5.
Table 4. Grade 3/4 Laboratory Abnormalities Reported in Greater than or Equal to 2% of Antiretroviral-Naive Adult Subjects in Trials APV30001 and APV30002 a All subjects also received abacavir and lamivudine twice daily. b Fasting specimens.ULN = Upper limit of normal.
Laboratory Abnormality
APV30001a
APV30002a
LEXIVA
1,400 mg Twice Daily
(n = 166)
Nelfinavir
1,250 mg Twice Daily
(n = 83)
LEXIVA 1,400 mg and
Ritonavir 200 mg Once Daily
(n = 322)
Nelfinavir
1,250 mg Twice Daily
(n = 327)
ALT (>5 x ULN)
6%
5%
8%
8%
AST (>5 x ULN)
6%
6%
6%
7%
Serum lipase (>2 x ULN)
8%
4%
6%
4%
Triglyceridesb
(>750 mg/dL)
0%
1%
6%
2%
Neutrophil count, absolute
(<750 cells/mm3)
3%
6%
3%
4%
The incidence of Grade 3 or 4 hyperglycemia in antiretroviral-naive subjects who received LEXIVA in the pivotal trials was less than 1%.
Table 5. Grade 3/4 Laboratory Abnormalities Reported in Greater than or Equal to 2% of Protease Inhibitor-Experienced Adult Subjects in Trial APV30003 a All subjects also received 2 reverse transcriptase inhibitors. b Fasting specimens. c n = 100 for LEXIVA plus ritonavir, n = 98 for lopinavir plus ritonavir.ULN = Upper limit of normal.
Laboratory Abnormality
LEXIVA 700 mg and
Ritonavir 100 mg
Twice Daily a
(n = 104)
Lopinavir 400 mg and
Ritonavir 100 mg
Twice Daily a
(n = 103)
Triglyceridesb (>750 mg/dL)
11%c
6%c
Serum lipase (>2 x ULN)
5%
12%
ALT (>5 x ULN)
4%
4%
AST (>5 x ULN)
4%
2%
Glucose (>251 mg/dL)
2%c
2%c
Pediatric Trials
LEXIVA with and without ritonavir was studied in 237¬†HIV-1‚Äďinfected pediatric subjects aged at least 4 weeks to 18¬†years in 3¬†open‚ÄĎlabel trials; APV20002, APV20003, and APV29005 [see Clinical Studies (14.3)]. Vomiting and neutropenia occurred more frequently in pediatric subjects compared with adults. Other adverse events occurred with similar frequency in pediatric subjects compared with adults.
The frequency of vomiting among pediatric subjects receiving LEXIVA twice daily with ritonavir was 20% in subjects aged at least 4 weeks to younger than 2 years and 36% in subjects aged 2 to 18 years compared with 10% in adults. The frequency of vomiting among pediatric subjects receiving LEXIVA twice daily without ritonavir was 60% in subjects aged 2 to 5 years compared with 16% in adults.
The median duration of drug‚ÄĎrelated vomiting episodes in APV29005 was 1¬†day (range: 1 to 3¬†days), in APV20003 was 16 days (range: 1 to 38¬†days), and in APV20002 was 9¬†days (range: 4 to 13¬†days). Vomiting was treatment limiting in 4¬†pediatric subjects across all 3 trials.
The incidence of Grade 3 or 4 neutropenia (neutrophils less than 750 cells per mm3) seen in pediatric subjects treated with LEXIVA with and without ritonavir was higher (15%) than the incidence seen in adult subjects (3%). Grade 3/4 neutropenia occurred in 10% (5 of 51) of subjects aged at least 4 weeks to younger than 2 years and 16% (28 of 170) of subjects aged 2 to 18 years.
6.2 Postmarketing Experience
The following adverse reactions have been identified during postapproval use of LEXIVA. Because these reactions are reported voluntarily from a population of unknown size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure. These reactions have been chosen for inclusion due to a combination of their seriousness, frequency of reporting, or potential causal connection to LEXIVA.
Cardiac Disorders
Myocardial infarction.
Metabolism and Nutrition Disorders
Hypercholesterolemia.
Nervous System Disorders
Oral paresthesia.
Skin and Subcutaneous Tissue Disorders
Angioedema.
Urogenital
Nephrolithiasis.
7 Drug Interactions
‚ÄĘ Coadministration of LEXIVA with drugs that induce CYP3A4 may decrease amprenavir (active metabolite) concentrations leading to potential loss of virologic activity. (7 ,12.3 )‚ÄĘ Coadministration with drugs that inhibit CYP3A4 may increase amprenavir concentrations. (7 ,12.3 )‚ÄĘ Coadministration of LEXIVA or LEXIVA and ritonavir may result in clinically significant interactions with drugs metabolized by CYP3A4. (7 )‚ÄĘ Coadministration of LEXIVA and ritonavir may result in clinically significant interactions with drugs metabolized by CYP2D6. (7 )7.1 Cytochrome P450 Inhibitors and Inducers
Amprenavir, the active metabolite of fosamprenavir, is an inhibitor of CYP3A4 metabolism and therefore should not be administered concurrently with medications with narrow therapeutic windows that are substrates of CYP3A4. Data also suggest that amprenavir induces CYP3A4.
Amprenavir is metabolized by CYP3A4. Coadministration of LEXIVA and drugs that induce CYP3A4, such as rifampin, may decrease amprenavir concentrations and reduce its therapeutic effect. Coadministration of LEXIVA and drugs that inhibit CYP3A4 may increase amprenavir concentrations and increase the incidence of adverse effects.
The potential for drug interactions with LEXIVA changes when LEXIVA is coadministered with the potent CYP3A4 inhibitor ritonavir. The magnitude of CYP3A4-mediated drug interactions (effect on amprenavir or effect on coadministered drug) may change when LEXIVA is coadministered with ritonavir. Because ritonavir is a CYP2D6 inhibitor, clinically significant interactions with drugs metabolized by CYP2D6 are possible when coadministered with LEXIVA plus ritonavir. Ritonavir also appears to induce CYP3A, CYP1A2, CYP2C9, CYP2C19, and CYP2B6, as well as other enzymes, including glucuronosyl transferase.
There are other agents that may result in serious and/or life-threatening drug interactions [see Contraindications (4)].
7.2 Established and Other Potentially Significant Drug Interactions
If LEXIVA is used in combination with ritonavir, see full prescribing information for ritonavir for additional information on drug interactions [see Contraindications (4), Clinical Pharmacology (12.3)].
Table 6 provides a uling of established or potentially clinically significant drug interactions. Information in the table applies to LEXIVA with or without ritonavir, unless otherwise indicated.
Table 6. Established and Other Potentially Significant Drug Interactions a   See Clinical Pharmacology (12.3) Tables 10, 11, 12, or 13 for magnitude of interaction.
Concomitant Drug Class: Drug Name
Effect on Concentration of Amprenavir or Concomitant Drug
Clinical Comment
HCV/HIV-Antiviral Agents
HCV protease inhibitor:
Boceprevir
LEXIVA:
‚ÜďAmprenavir (predicted)
‚ÜĒ or ‚ÜďBoceprevir (predicted)
LEXIVA/ritonavir:
‚ÜďAmprenavir (predicted)
‚ÜďBoceprevir (predicted)
Coadministration of LEXIVA or LEXIVA/ritonavir and boceprevir is not recommended.
HCV protease inhibitor:
Simeprevir
LEXIVA:
‚ÜĒAmprenavir (predicted)
‚ÜĎ or ‚ÜďSimeprevir (predicted)
LEXIVA/ritonavir:
‚ÜĒAmprenavir (predicted)
‚ÜĎSimeprevir (predicted)
Coadministration of LEXIVA or LEXIVA/ritonavir and simeprevir is not recommended.
HCV protease inhibitor:
Paritaprevir (coformulated with ritonavir and ombitasvir and coadministered with dasabuvir)
LEXIVA:
‚ÜĎAmprenavir (predicted)
‚ÜĎ or ‚ÜĒParitaprevir (predicted)
LEXIVA/ritonavir:
‚ÜĎ or ‚ÜĒAmprenavir (predicted)
‚ÜĎParitaprevir (predicted)
Appropriate doses of the combinations with respect to safety and efficacy have not been established.
LEXIVA 1,400 mg once daily may be considered when coadministered with paritaprevir/ritonavir/ombitasvir/ dasabuvir.
Coadministration of LEXIVA/ritonavir and paritaprevir/ritonavir/ombitasvir/ dasabuvir is not recommended.
Non-nucleoside reverse transcriptase inhibitor:
Delavirdinea
LEXIVA:
‚ÜĎAmprenavir
‚ÜďDelavirdine
LEXIVA/ritonavir:
‚ÜĎAmprenavir
‚ÜďDelavirdine
Coadministration is contraindicated as it may lead to loss of virologic response and possible resistance to delavirdine [see Contraindications (4)].
Non‚ÄĎnucleoside reverse transcriptase inhibitor:
Efavirenza
LEXIVA:
‚ÜďAmprenavir
LEXIVA/ritonavir:
‚ÜďAmprenavir
Appropriate doses of the combinations with respect to safety and efficacy have not been established.
An additional 100 mg/day (300 mg total) of ritonavir is recommended when efavirenz is administered with LEXIVA/ritonavir once daily. No change in the ritonavir dose is required when efavirenz is administered with LEXIVA plus ritonavir twice daily.
Non‚ÄĎnucleoside reverse transcriptase inhibitor:
Nevirapinea
LEXIVA:
‚ÜďAmprenavir
‚ÜĎNevirapine
LEXIVA/ritonavir:
‚ÜďAmprenavir
‚ÜĎNevirapine
Coadministration of nevirapine and LEXIVA without ritonavir is not recommended.
No dosage adjustment required when nevirapine is administered with LEXIVA/ritonavir twice daily.
The combination of nevirapine administered with LEXIVA/ritonavir once‚ÄĎdaily regimen has not been studied.
HIV protease inhibitor:
Atazanavira
LEXIVA:
Interaction has not been evaluated.
LEXIVA/ritonavir:
‚ÜďAtazanavir
‚ÜĒAmprenavir
Appropriate doses of the combinations with respect to safety and efficacy have not been established.
HIV protease inhibitors:
Indinavira, nelfinavira
LEXIVA:
‚ÜĎAmprenavir
Effect on indinavir and nelfinavir is not well established.
LEXIVA/ritonavir:
Interaction has not been evaluated.
Appropriate doses of the combinations with respect to safety and efficacy have not been established.
HIV protease inhibitors:
Lopinavir/ritonavira
‚ÜďAmprenavir
‚ÜďLopinavir
An increased rate of adverse events has been observed. Appropriate doses of the combinations with respect to safety and efficacy have not been established.
HIV protease inhibitor:
Saquinavira
LEXIVA:
‚ÜďAmprenavir
Effect on saquinavir is not well established.
LEXIVA/ritonavir:
Interaction has not been evaluated.
Appropriate doses of the combination with respect to safety and efficacy have not been established.
HIV integrase inhibitor:
Raltegravira
LEXIVA:
‚ÜďAmprenavir
‚ÜďRaltegravir
LEXIVA/ritonavir:
‚ÜďAmprenavir
‚ÜďRaltegravir
Appropriate doses of the combination with respect to safety and efficacy have not been established.
HIV integrase inhibitor:
Dolutegravira
LEXIVA/ritonavir:
‚ÜďDolutegravir
The recommended dose of dolutegravir is 50 mg twice daily when coadministered with LEXIVA/ritonavir.
Use an alternative combination where possible in patients with known or suspected integrase inhibitor resistance.
HIV CCR5 co-receptor antagonist:
Maraviroca
LEXIVA/ritonavir:
‚ÜďAmprenavir
‚ÜĎMaraviroc
No dosage adjustment required for LEXIVA/ritonavir. The recommended dose of maraviroc is 150 mg twice daily when coadministered with LEXIVA/ritonavir. LEXIVA should be given with ritonavir when coadministered with maraviroc.
Other Agents
Alpha 1-adrenoreceptor antagonist:
Alfuzosin
‚ÜĎAlfuzosin
Coadministration is contraindicated due to potential hypotension [see Contraindications (4)].
Antacid:
MAALOX TC
‚ÜďAmprenavir
Use with caution when administered at the same time. LEXIVA may be less effective due to decreased amprenavir plasma concentrations.
Staggered coadministration of antacids and LEXIVA has not been evaluated.
Antiarrhythmics:
Amiodarone, disopyramide, lidocaine (systemic), and quinidine
‚ÜĎAntiarrhythmics
Therapeutic concentration monitoring, if available, is recommended for antiarrhythmics.
Antiarrhythmics:
Flecainide, propafenone
‚ÜĎAntiarrhythmics
Coadministration is contraindicated due to potential for serious and/or life‚ÄĎthreatening reactions such as cardiac arrhythmias if LEXIVA is co-prescribed with ritonavir [see Contraindications (4)].
Anticoagulant:
Warfarin
Concentrations of warfarin may be affected. It is recommended that INR (international normalized ratio) be monitored.
Anticonvulsants:
Carbamazepine, phenobarbital, phenytoin
LEXIVA:
‚ÜďAmprenavir
Use with caution. LEXIVA may be less effective due to decreased amprenavir plasma concentrations in patients taking these agents concomitantly.
Phenytoina
LEXIVA/ritonavir:
‚ÜĎAmprenavir
‚ÜďPhenytoin
Plasma phenytoin concentrations should be monitored and phenytoin dose should be increased as appropriate. No change in LEXIVA/ritonavir dose is recommended.
Antidepressant:
Paroxetine, trazodone
‚ÜďParoxetine
Any paroxetine dose adjustment should be guided by clinical effect (tolerability and efficacy).
‚ÜĎTrazodone
Adverse events of nausea, dizziness, hypotension, and syncope have been observed following coadministration of trazodone and ritonavir. If trazodone is used with a CYP3A4 inhibitor such as LEXIVA, the combination should be used with caution and a lower dose of trazodone should be considered.
Antifungals:
Ketoconazolea, itraconazole
‚ÜĎKetoconazole
‚ÜĎItraconazole
Increase monitoring for adverse events.
LEXIVA:
Dose reduction of ketoconazole or itraconazole may be needed for patients receiving more than 400 mg ketoconazole or itraconazole per day.
LEXIVA/ritonavir:
High doses of ketoconazole or itraconazole (greater than 200 mg/day) are not recommended.
Anti-gout:
Colchicine
‚ÜĎColchicine
Patients with renal or hepatic impairment should not be given colchicine with LEXIVA/ritonavir.
LEXIVA/ritonavir and coadministration of colchicine:
Treatment of gout flares:
0.6 mg (1 tablet) x 1 dose, followed by 0.3 mg (half tablet) 1 hour later. Dose to be repeated no earlier than 3 days.
Prophylaxis of gout flares:
If the original regimen was 0.6 mg twice a day, the regimen should be adjusted to 0.3 mg once a day. If the original regimen was 0.6 mg once a day, the regimen should be adjusted to 0.3 mg once every other day.
Treatment of familial Mediterranean fever (FMF):
Maximum daily dose of 0.6 mg (may be given as 0.3 mg twice a day).
LEXIVA and coadministration of colchicine:
Treatment of gout flares:
1.2 mg (2 tablets) x 1 dose. Dose to be repeated no earlier than 3 days.
Prophylaxis of gout flares:
If the original regimen was 0.6 mg twice a day, the regimen should be adjusted to 0.3 mg twice a day or 0.6 mg once a day. If the original regimen was 0.6 mg once a day, the regimen should be adjusted to 0.3 mg once a day.
Treatment of FMF:
Maximum daily dose of 1.2 mg (may be given as 0.6 mg twice a day).
Antimycobacterial:
Rifabutina
‚ÜĎRifabutin and rifabutin metabolite
A complete blood count should be performed weekly and as clinically indicated to monitor for neutropenia.
LEXIVA:
A dosage reduction of rifabutin by at least half the recommended dose is required.
LEXIVA/ritonavir:
Dosage reduction of rifabutin by at least 75% of the usual dose of 300 mg/day is recommended (a maximum dose of 150 mg every other day or 3 times per week).
Antimycobacterial:
Rifampina
‚ÜďAmprenavir
Coadministration is contraindicated as it may lead to loss of virologic response and possible resistance to LEXIVA or to the class of protease inhibitors [see Contraindications (4)].
Antineoplastics:
Dasatinib, nilotinib, ibrutinib, vinblastine, everolimus
‚ÜĎAntineoplastics
LEXIVA or LEXIVA/ritonavir may increase plasma concentrations of antineoplastics metabolized by CYP3A, potentially increasing the risk of adverse events typically associated with these medications. In case of coadministration, please refer to relevant prescribing information for these medications.
Antipsychotics:
Lurasidone
‚ÜĎLurasidone
LEXIVA:
If coadministration is necessary, reduce the lurasidone dose. Refer to the lurasidone prescribing information for concomitant use with moderate CYP3A4 inhibitors.
LEXIVA/ritonavir:
Use of lurasidone is contraindicated due to potential for serious and/or life-threatening reactions [see Contraindications (4)].
Pimozide
‚ÜĎPimozide
Coadministration is contraindicated due to potential for serious and/or life-threatening reactions such as cardiac arrhythmias [see Contraindications (4)].
Quetiapine
‚ÜĎQuetiapine
LEXIVA/ritonavir:
Initiation of LEXIVA with ritonavir in
patients taking quetiapine:
Consider alternative antiretroviral therapy to avoid increases in quetiapine drug exposures. If coadministration is necessary, reduce the quetiapine dose to 1/6 of the current dose and monitor for quetiapine-associated adverse reactions. Refer to the quetiapine prescribing information for recommendations on adverse reaction monitoring.
Initiation of quetiapine in patients taking LEXIVA with ritonavir:
Refer to the quetiapine prescribing information for initial dosing and titration of quetiapine.
Benzodiazepines:
Alprazolam, clorazepate, diazepam, flurazepam
‚ÜĎBenzodiazepines
Clinical significance is unknown. A decrease in benzodiazepine dose may be needed.
Calcium channel blockers:
Diltiazem, felodipine, nifedipine, nicardipine, nimodipine, verapamil, amlodipine, nisoldipine, isradipine
‚ÜĎCalcium channel blockers
Use with caution. Clinical monitoring of patients is recommended.
Corticosteroid:
Dexamethasone
‚ÜďAmprenavir
Use with caution. LEXIVA may be less effective due to decreased amprenavir plasma concentrations.
Endothelin‚ÄĎreceptor antagonist:
Bosentan
‚ÜĎBosentan
Coadministration of bosentan in patients on LEXIVA:
In patients who have been receiving LEXIVA for at least 10 days, start bosentan at 62.5 mg once daily or every other day based upon individual tolerability.
Coadministration of LEXIVA in patients on bosentan:
Discontinue use of bosentan at least 36 hours prior to initiation of LEXIVA.
After at least 10 days following the initiation of LEXIVA, resume bosentan at 62.5 mg once daily or every other day based upon individual tolerability.
Ergot derivatives:
Dihydroergotamine, ergonovine, ergotamine, methylergonovine
‚ÜĎErgot derivatives
Coadministration is contraindicated due to potential for serious and/or life‚ÄĎthreatening reactions such as acute ergot toxicity characterized by peripheral vasospasm and ischemia of the extremities and other tissues [see Contraindications (4)].
GI motility agent:
Cisapride
‚ÜĎCisapride
Coadministration is contraindicated due to potential for serious and/or life‚ÄĎthreatening reactions such as cardiac arrhythmias [see Contraindications (4)].
Herbal product:
St. John’s wort (Hypericum perforatum)
‚ÜďAmprenavir
Coadministration is contraindicated as it may lead to loss of virologic response and possible resistance to LEXIVA or to the class of protease inhibitors [see Contraindications (4)].
Histamine H2-receptor antagonists:
Cimetidine, famotidine, nizatidine, ranitidinea
LEXIVA:
‚ÜďAmprenavir
LEXIVA/ritonavir:
Interaction not evaluated
Use with caution. LEXIVA may be less effective due to decreased amprenavir plasma concentrations.
Immunosuppressants:
Cyclosporine, tacrolimus, sirolimus
‚ÜĎImmunosuppressants
Therapeutic concentration monitoring is recommended for immunosuppressant agents.
Inhaled beta‚ÄĎagonist:
Salmeterol
‚ÜĎSalmeterol
Concurrent administration of salmeterol with LEXIVA is not recommended. The combination may result in increased risk of cardiovascular adverse events associated with salmeterol, including QT prolongation, palpitations, and sinus tachycardia.
Inhaled/nasal steroid:
Fluticasone
LEXIVA:
‚ÜĎFluticasone
LEXIVA/ritonavir:
‚ÜĎFluticasone
Use with caution. Consider alternatives to fluticasone, particularly for long‚ÄĎterm use.
May result in significantly reduced serum cortisol concentrations. Systemic corticosteroid effects including Cushing’s syndrome and adrenal suppression have been reported during postmarketing use in patients receiving ritonavir and inhaled or intranasally administered fluticasone. Coadministration of fluticasone and LEXIVA/ritonavir is not recommended unless the potential benefit to the patient outweighs the risk of systemic corticosteroid side effects.
Lipid Modifying Agents:
HMG-CoA reductase inhibitors:
Atorvastatina
‚ÜĎAtorvastatin
Titrate atorvastatin dose carefully and use the lowest necessary dose; do not exceed atorvastatin 20 mg/day.
Lovastatin, simvastatin
‚ÜĎLovastatin
‚ÜĎSimvastatin
Coadministration with lovastatin or simvastatin is contraindicated due to potential for serious reactions such as risk of myopathy including rhabdomyolysis [see Contraindications (4)].
Other lipid modifying agents:
Lomitapide
‚ÜĎLomitapide
Coadministration with lomitapide is contraindicated due to potential for markedly increased transaminases.
Narcotic analgesic:
Methadone
‚ÜďMethadone
Data suggest that the interaction is not clinically relevant; however, patients should be monitored for opiate withdrawal symptoms.
Fentanyl
‚ÜĎFentanyl
Careful monitoring of therapeutic effects and adverse effects of fentanyl (including potentially fatal respiratory depression) is recommended.
Oral contraceptives:
Ethinyl estradiol/norethindronea
LEXIVA:
‚ÜďAmprenavir
‚ÜďEthinyl estradiol
LEXIVA/ritonavir:
‚ÜďEthinyl estradiol
Alternative methods of non-hormonal contraception are recommended.
May lead to loss of virologic response.a
Increased risk of transaminase elevations. No data are available on the use of LEXIVA/ritonavir with other hormonal therapies, such as hormone replacement therapy (HRT) for postmenopausal women.
PDE5 inhibitors:
Sildenafil, tadalafil, vardenafil
‚ÜĎSildenafil
‚ÜĎTadalafil
‚ÜĎVardenafil
May result in an increase in PDE5 inhibitor‚ÄĎassociated adverse events, including hypotension, syncope, visual disturbances, and priapism.
Use of PDE5 inhibitors for pulmonary arterial hypertension (PAH):
‚ÄĘ Use of sildenafil (REVATIO) is contraindicated when used for the treatment of PAH. A safe and effective dose has not been established when used with LEXIVA. There is increased potential for sildenafil-associated adverse events (which include visual disturbances, hypotension, prolonged erection, and syncope) [see Contraindications (4)].‚ÄĘ The following dose adjustments are recommended for use of tadalafil (ADCIRCA¬ģ ) with LEXIVA: Coadministration of ADCIRCA in patients on LEXIVA: In patients receiving LEXIVA for at least one week, start ADCIRCA at 20¬†mg once daily. Increase to 40¬†mg once daily based upon individual tolerability. Coadministration of LEXIVA in patients on ADCIRCA: Avoid use of ADCIRCA during the initiation of LEXIVA. Stop ADCIRCA at least 24¬†hours prior to starting LEXIVA. After at least one week following the initiation of LEXIVA, resume ADCIRCA at 20¬†mg once daily. Increase to 40¬†mg once daily based upon individual tolerability.
Use of PDE5 inhibitors for erectile dysfunction:
LEXIVA:
Sildenafil: 25 mg every 48 hours.
Tadalafil: no more than 10 mg every 72 hours.
Vardenafil: no more than 2.5 mg every 24 hours.
LEXIVA/ritonavir:
Sildenafil: 25 mg every 48 hours.
Tadalafil: no more than 10 mg every 72 hours.
Vardenafil: no more than 2.5 mg every 72 hours.
Use with increased monitoring for adverse events.
Proton pump inhibitors:
Esomeprazolea, lansoprazole, omeprazole, pantoprazole, rabeprazole
LEXIVA:
‚ÜĒAmprenavir
‚ÜĎEsomeprazole
LEXIVA/ritonavir:
‚ÜĒAmprenavir
‚ÜĒEsomeprazole
Proton pump inhibitors can be administered at the same time as a dose of LEXIVA with no change in plasma amprenavir concentrations.
Sedative/hypnotics:
Midazolam, triazolam
‚ÜĎMidazolam
‚ÜĎTriazolam
Coadministration is contraindicated due to potential for serious and/or life‚ÄĎthreatening reactions such as prolonged or increased sedation or respiratory depression [see Contraindications (4)].
Tricyclic antidepressants:
Amitriptyline, imipramine
‚ÜĎTricyclics
Therapeutic concentration monitoring is recommended for tricyclic antidepressants.
8 Use In Specific Populations
  Lactation: Breastfeeding is not recommended due to potential for HIV transmission. (8.2 )8.1 Pregnancy
Pregnancy Exposure Registry
There is a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to LEXIVA during pregnancy. Healthcare providers are encouraged to register patients by calling the Antiretroviral Pregnancy Registry (APR) at 1-800-258-4263.
Risk Summary
Limited data are available for use of LEXIVA in pregnancy. LEXIVA 700 mg twice daily taken with ritonavir 100 mg twice daily should only be considered in pregnant patients who are already on a stable twice-daily regimen of LEXIVA/ritonavir 700 mg/100 mg prior to pregnancy, and who are virologically suppressed (HIV-1 RNA less than 50 copies per mL) (see Clinical Considerations and Data).
There are insufficient human data on the use of fosamprenavir during pregnancy to adequately assess a drug-associated risk for birth defects and miscarriage. Given the limited number of pregnancies exposed to fosamprenavir-based regimens, no conclusions can be drawn on the safety of fosamprenavir in pregnancy. The background risk for major birth defects and miscarriage for the indicated population is unknown. The background rate for major birth defects in a U.S. reference population of the Metropolitan Atlanta Congenital Defects Program (MACDP) is 2.7% (see Data). The estimated background rate of miscarriage in clinically recognized pregnancies in the U.S. general population is 15% to 20%.
In animal reproduction studies, no evidence of major adverse developmental outcomes was observed following oral administration of fosamprenavir. Systemic exposure to amprenavir (the active ingredient) was less than (rabbits) or up to 2 times (rats) those in humans at the maximum recommended human dose (MRHD) with or without ritonavir. In contrast, oral administration of amprenavir was associated with abortions in pregnant rabbits at doses that produced approximately one-twentieth the human exposure at the MRHD.
In the rat pre- and postnatal development study, toxicities to the offspring, including reduced survival and reproductive performance, were observed at maternal systemic exposures (AUC) to amprenavir that were approximately 2 times the exposure in humans at the MRHD of fosamprenavir alone or approximately the same as those seen in humans following administration of the MRHD of fosamprenavir in combination with ritonavir (see Data).
Clinical Considerations
Virologic Monitoring During Pregnancy and the Postpartum Period: Based on limited data on the use of LEXIVA during pregnancy, no dosage adjustments are required for pregnant patients who are already on a stable twice-daily regimen of LEXIVA 700 mg taken with ritonavir 100 mg prior to pregnancy, and who are virologically suppressed (HIV-1 RNA less than 50 copies per mL) [see Dosage and Administration (2.3), Clinical Pharmacology (12.3)]. In a clinical trial of 10 HIV-1‚Äďinfected pregnant women treated with LEXIVA 700 mg taken with ritonavir 100 mg twice daily through postpartum, total amprenavir exposures were lower during pregnancy compared with the postpartum period. Therefore, viral load should be monitored closely to ensure viral suppression is maintained [see Data, Dosage and Administration (2.2), Clinical Pharmacology (12.3)]. Pregnancy data with other dosage regimens of LEXIVA (with or without ritonavir) are not available.
Data
Human Data: LEXIVA 700 mg taken with ritonavir 100 mg twice daily in combination with a background regimen was evaluated in a clinical trial of 10 HIV-1‚Äďinfected pregnant women during the second and third trimesters and postpartum. Subjects initiated LEXIVA/ritonavir during pregnancy at a median of 19 weeks‚Äô gestation; 4 had undetectable HIV-1 RNA viral load (less than 50 copies/mL) at the time of initiation. Amprenavir pharmacokinetics and placental transfer were studied during the second trimester (n¬†=¬†6) or third trimester (n¬†=¬†9) and postpartum (n¬†=¬†9). Pregnancy outcomes were available for all 10 subjects, among which 1 twin pregnancy was included. Compared to the postpartum period, geometric mean amprenavir AUC was 35% lower in the second trimester and 25% lower in the third trimester. The amprenavir geometric mean ratio (95% CI) of fetal cord to maternal peripheral plasma concentration (n¬†=¬†7) was 0.27 (0.24 to 0.30) [see Clinical Pharmacology (12.3)]. At delivery, 9 subjects had HIV-1 viral load less than 50 copies/mL, and 1 subject had a viral load of 111 copies/mL. All 11 infants born had test results that were negative for HIV-1 at the time of delivery and through 12 months postpartum. There were no new safety findings compared with the known safety profile of LEXIVA/ritonavir in HIV-1‚Äďinfected adults.
Based on prospective reports to the APR of approximately 146 live births following exposure to fosamprenavir-containing regimens, there were 2 birth defects reported in 109 first trimester exposures and 2 birth defects reported in 36 second and third trimester exposures. The background rate for major birth defects is 2.7% in a U.S. reference population of the MACDP. Prospective reports from the APR of overall major birth defects in pregnancies exposed to fosamprenavir are compared with the U.S. background major birth defect rate. Methodological limitations of the APR include the use of the MACDP as the external comparator group. Limitations of using an external comparator include differences in methodology and populations as well as confounding due to the underlying disease.
Animal Data: Fosamprenavir was administered orally to pregnant rats (300, 820, or 2,240 mg per kg per day) and rabbits (74.8, 224.3, or 672.8 mg per kg per day) on Gestation Days 6 to 17 and Days 7 to 20, respectively. No major adverse effects on embryo-fetal development were observed at these dose levels, resulting in exposures (AUC0-24 h) approximately 2 times (rats) and 0.8 times (rabbits) human exposures at the MRHD of fosamprenavir alone or 0.7 times (rats) and 0.3 times (rabbits) human exposures at the MRHD of fosamprenavir in combination with ritonavir. However, increased incidence of abortion was observed in rabbits administered a maternally toxic dose of fosamprenavir (672.8 mg per kg per day). In a study where amprenavir was administered orally to pregnant rabbits (25, 50, or 100 mg per kg per day) on Gestation Days 8 to 20, increased abortions and an increased incidence of minor skeletal variations (deficient ossification of the femur, humerus, and trochlea) were observed at doses that produced approximately one-twentieth the exposure seen at the MRHD.
In the rat pre- and postnatal development study, fosamprenavir was administered orally (300, 820, or 2,240 mg per kg per day) on Gestation Day 6 to Lactation/Postpartum Day 20. Fosamprenavir caused a reduction in pup survival and body weights. In surviving female offspring from the high-dose group, an increased time to successful mating, an increased length of gestation, a reduced number of uterine implantation sites per litter, and reduced gestational body weights were observed. Systemic exposure (AUC0-24 h) to amprenavir in rats was approximately 2 times the exposures in humans at the MRHD of fosamprenavir alone or approximately the same as those seen in humans at the MRHD of fosamprenavir in combination with ritonavir.
8.2 Lactation
Risk Summary
The Centers for Disease Control and Prevention recommends that HIV-1‚Äďinfected mothers in the United States not breastfeed their infants to avoid risking postnatal transmission of HIV-1 infection.
There is no information available on the presence of amprenavir in human milk, the effects of the drug on the breastfed infant, or the effects of the drug on milk production. When administered to lactating rats, amprenavir was present in milk (see Data). Because of the potential for (1) HIV-1 transmission (in HIV-negative infants), (2) developing viral resistance (in HIV-positive infants), and (3) adverse reactions in a breastfed infant, instruct mothers not to breastfeed if they are receiving LEXIVA.
Data
Amprenavir was excreted into the milk of lactating rats following a single dose of amprenavir (100 mg per kg); a maximal milk concentration was achieved 2 hours post-administration at a milk concentration approximately 1.2 times that of maternal plasma concentrations.
8.3 Females and Males of Reproductive Potential
Contraception
Use of LEXIVA may reduce the efficacy of combined hormonal contraceptives. Advise patients using combined hormonal contraceptives to use an effective alternative contraceptive method or an additional barrier method of contraception [see Drug Interactions (7.2)].
8.4 Pediatric Use
The safety, pharmacokinetic profile, and virologic and immunologic responses of LEXIVA with and without ritonavir were evaluated in protease inhibitor-naive and -experienced HIV-1‚Äďinfected pediatric subjects aged at least 4¬†weeks to younger than 18¬†years and weighing at least 3¬†kg in 3¬†open-label trials [see Adverse Reactions (6.1), Clinical Pharmacology (12.3), Clinical Studies (14.3)].
Treatment with LEXIVA is not recommended in protease inhibitor-experienced pediatric patients younger than 6 months. The pharmacokinetics, safety, tolerability, and efficacy of LEXIVA in pediatric patients younger than 4 weeks have not been established [see Clinical Pharmacology (12.3)]. Available pharmacokinetic and clinical data do not support once-daily dosing of LEXIVA alone or in combination with ritonavir for any pediatrics or twice-daily dosing without ritonavir in pediatric patients younger than 2 years [see Clinical Pharmacology (12.3), Clinical Studies (14.3)]. See Dosage and Administration (2.3) for dosing recommendations for pediatric patients.
8.5 Geriatric Use
Clinical studies of LEXIVA did not include sufficient numbers of patients aged 65 and over to determine whether they respond differently from younger adults. In general, dose selection for an elderly patient should be cautious, reflecting the greater frequency of decreased hepatic, renal, or cardiac function and of concomitant disease or other drug therapy.
8.6 Hepatic Impairment
Amprenavir is principally metabolized by the liver; therefore, caution should be exercised when administering LEXIVA to patients with hepatic impairment because amprenavir concentrations may be increased [see Clinical Pharmacology (12.3)]. Patients with impaired hepatic function receiving LEXIVA with or without concurrent ritonavir require dose reduction [see Dosage and Administration (2.4)].
There are no data to support dosing recommendations for pediatric subjects with hepatic impairment.
10 Overdosage
In a healthy volunteer repeat-dose pharmacokinetic trial evaluating high-dose combinations of LEXIVA plus ritonavir, an increased frequency of Grade 2/3 ALT elevations (greater than 2.5 x ULN) was observed with LEXIVA 1,400 mg twice daily plus ritonavir 200 mg twice daily (4 of 25 subjects). Concurrent Grade 1/2 elevations in AST (greater than 1.25 x ULN) were noted in 3 of these 4 subjects. These transaminase elevations resolved following discontinuation of dosing.
There is no known antidote for LEXIVA. It is not known whether amprenavir can be removed by peritoneal dialysis or hemodialysis, although it is unlikely as amprenavir is highly protein bound. If overdosage occurs, the patient should be monitored for evidence of toxicity and standard supportive treatment applied as necessary.
11 Description
LEXIVA (fosamprenavir calcium) is a prodrug of amprenavir, an inhibitor of HIV protease. The chemical name of fosamprenavir calcium is (3S)-tetrahydrofuran-3-yl (1S,2R)-3-[[(4-aminophenyl) sulfonyl](isobutyl)amino]-1-benzyl-2-(phosphonooxy) propylcarbamate monocalcium salt. Fosamprenavir calcium is a single stereoisomer with the (3S)(1S,2R) configuration. It has a molecular formula of C25H34CaN3O9PS and a molecular weight of 623.7. It has the following structural formula:
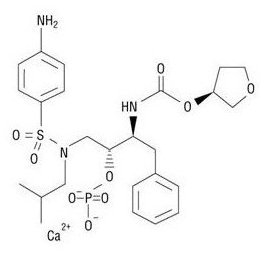
Fosamprenavir calcium is a white to cream-colored solid with a solubility of approximately 0.31¬†mg per mL in water at 25¬įC.
LEXIVA tablets are available for oral administration in a strength of 700 mg of fosamprenavir as fosamprenavir calcium (equivalent to approximately 600 mg of amprenavir). Each 700-mg tablet contains the inactive ingredients colloidal silicon dioxide, croscarmellose sodium, magnesium stearate, microcrystalline cellulose, and povidone K30. The tablet film-coating contains the inactive ingredients hypromellose, iron oxide red, titanium dioxide, and triacetin.
LEXIVA oral suspension is available in a strength of 50 mg per mL of fosamprenavir as fosamprenavir calcium equivalent to approximately 43 mg of amprenavir. LEXIVA oral suspension is a white to off-white suspension with a grape-bubblegum-peppermint flavor. Each one milliliter (1 mL) contains the inactive ingredients artificial grape-bubblegum flavor, calcium chloride dihydrate, hypromellose, methylparaben, natural peppermint flavor, polysorbate 80, propylene glycol, propylparaben, purified water, and sucralose.
12 Clinical Pharmacology
12.1 Mechanism of Action
Fosamprenavir is an HIV-1 antiretroviral agent [see Microbiology (12.4)].
12.3 Pharmacokinetics
The pharmacokinetic properties of amprenavir after administration of LEXIVA, with or without ritonavir, have been evaluated in both healthy adult volunteers and in HIV-1‚Äďinfected subjects; no substantial differences in steady-state amprenavir concentrations were observed between the 2¬†populations.
The pharmacokinetic parameters of amprenavir after administration of LEXIVA (with and without concomitant ritonavir) are shown in Table 7.
Table 7. Geometric Mean (95% CI) Steady-State Plasma Amprenavir Pharmacokinetic Parameters in Adults a Data shown are median (range). b Ctau is the concentration at the end of the dose interval. c HIV-infected adults. d AUC24 was calculated from AUC12 summary data x 2. e Healthy adults.
Regimen
Cmax
(mcg/mL)
Tmax
(hours)a
AUC24
(mcg‚ÄĘh/mL)
Ctau b
(mcg/mL)
LEXIVA 1,400 mg Twice Daily (n = 22)c
4.82
(4.06-5.72)
1.3
(0.8-4.0)
33.0d
(27.6-39.2)
0.35
(0.27-0.46)
LEXIVA 1,400 mg Once Daily plus Ritonavir 200 mg Once Daily (n = 22)e
7.24
(6.32-8.28)
2.1
(0.8-5.0)
69.4
(59.7-80.8)
1.45
(1.16-1.81)
LEXIVA 1,400 mg Once Daily plus Ritonavir 100 mg Once Daily (n = 36)e
7.93
(7.25-8.68)
1.5
(0.75-5.0)
66.4
(61.1-72.1)
0.86
(0.74-1.01)
LEXIVA 700 mg Twice Daily plus Ritonavir 100 mg Twice Daily (n = 24)e
6.08
(5.38-6.86)
1.5
(0.75-5.0)
79.2d
(69.0-90.6)
2.12
(1.77-2.54)
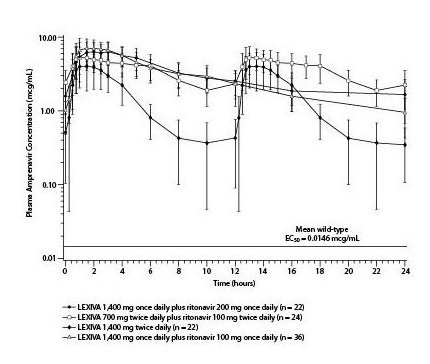
The mean plasma amprenavir concentrations of the dosing regimens over the dosing intervals are displayed in Figure 1.
Figure 1. Mean (¬ĪSD) Steady-State Plasma Amprenavir Concentrations and Mean EC50 Values against HIV from Protease Inhibitor-Naive Subjects (in the Absence of Human Serum)
Absorption
After administration of a single dose of LEXIVA to HIV-1‚Äďinfected subjects, the time to peak amprenavir concentration (Tmax) occurred between 1.5 and 4¬†hours (median 2.5¬†hours). The absolute oral bioavailability of amprenavir after administration of LEXIVA in humans has not been established.
After administration of a single 1,400-mg dose in the fasted state, LEXIVA oral suspension (50 mg per mL) and LEXIVA tablets (700 mg) provided similar amprenavir exposures (AUC); however, the Cmax of amprenavir after administration of the suspension formulation was 14.5% higher compared with the tablet.
Amprenavir is both a substrate for and inducer of P-glycoprotein.
Effects of Food on Oral Absorption
Administration of a single 1,400-mg dose of LEXIVA tablets in the fed state (standardized high-fat meal: 967¬†kcal, 67¬†grams fat, 33¬†grams protein, 58¬†grams carbohydrate) compared with the fasted state was associated with no significant changes in amprenavir Cmax, Tmax, or AUC0-‚ąě [see Dosage and Administration (2)].
Administration of a single 1,400-mg dose of LEXIVA oral suspension in the fed state (standardized high-fat meal: 967¬†kcal, 67¬†grams fat, 33¬†grams protein, 58¬†grams carbohydrate) compared with the fasted state was associated with a 46% reduction in Cmax, a 0.72-hour delay in Tmax, and a 28% reduction in amprenavir AUC0-‚ąě.
Distribution
In vitro, amprenavir is approximately 90% bound to plasma proteins, primarily to alpha1-acid glycoprotein. In vitro, concentration-dependent binding was observed over the concentration range of 1 to 10 mcg per mL, with decreased binding at higher concentrations. The partitioning of amprenavir into erythrocytes is low, but increases as amprenavir concentrations increase, reflecting the higher amount of unbound drug at higher concentrations.
Metabolism
After oral administration, fosamprenavir is rapidly and almost completely hydrolyzed to amprenavir and inorganic phosphate prior to reaching the systemic circulation. This occurs in the gut epithelium during absorption. Amprenavir is metabolized in the liver by the CYP3A4 enzyme system. The 2 major metabolites result from oxidation of the tetrahydrofuran and aniline moieties. Glucuronide conjugates of oxidized metabolites have been identified as minor metabolites in urine and feces.
Elimination
Excretion of unchanged amprenavir in urine and feces is minimal. Unchanged amprenavir in urine accounts for approximately 1% of the dose; unchanged amprenavir was not detectable in feces. Approximately 14% and 75% of an administered single dose of 14C-amprenavir can be accounted for as metabolites in urine and feces, respectively. Two metabolites accounted for greater than 90% of the radiocarbon in fecal samples. The plasma elimination half-life of amprenavir is approximately 7.7 hours.
Specific Populations
Patients with Hepatic Impairment: The pharmacokinetics of amprenavir have been studied after the administration of LEXIVA in combination with ritonavir to adult HIV-1‚Äďinfected subjects with mild, moderate, and severe hepatic impairment. Following 2¬†weeks of dosing with LEXIVA plus ritonavir, the AUC of amprenavir was increased by approximately 22% in subjects with mild hepatic impairment, by approximately 70% in subjects with moderate hepatic impairment, and by approximately 80% in subjects with severe hepatic impairment compared with HIV-1‚Äďinfected subjects with normal hepatic function. Protein binding of amprenavir was decreased in subjects with hepatic impairment. The unbound fraction at 2¬†hours (approximate Cmax) ranged between a decrease of -7% to an increase of 57% while the unbound fraction at the end of the dosing interval (Cmin) increased from 50% to 102% [see Dosage and Administration (2.4)].
The pharmacokinetics of amprenavir have been studied after administration of amprenavir given as AGENERASE capsules to adult subjects with hepatic impairment. Following administration of a single 600-mg oral dose, the AUC of amprenavir was increased by approximately 2.5-fold in subjects with moderate cirrhosis and by approximately 4.5-fold in subjects with severe cirrhosis compared with healthy volunteers [see Dosage and Administration (2.4)].
Patients with Renal Impairment: The impact of renal impairment on amprenavir elimination in adults has not been studied. The renal elimination of unchanged amprenavir represents approximately 1% of the administered dose; therefore, renal impairment is not expected to significantly impact the elimination of amprenavir.
Pregnant Women: Amprenavir pharmacokinetics were studied in pregnant women receiving LEXIVA (700 mg) plus ritonavir (100 mg) twice daily during the second trimester (n = 6) or third trimester (n = 9) and postpartum (n = 9). Compared to postpartum, geometric mean amprenavir AUC was 35% lower in the second trimester and 25% lower in the third trimester (Table 8). This decrease results in amprenavir concentrations that are within the range observed across regimens of LEXIVA in non-pregnant adults and lower concentrations compared with LEXIVA (700 mg) plus ritonavir (100 mg) twice daily in non-pregnant adults (Table 7, Table 8). This decrease is not expected to be considered clinically relevant in patients who are virologically suppressed; however, viral load should be monitored closely to ensure viral suppression is maintained [see Dosage and Administration (2.2), Use in Specific Populations (8.1)]. The amprenavir geometric mean ratio (95% CI) of fetal cord to maternal peripheral plasma concentration (n = 7) was 0.27 (0.24 to 0.30).
Table 8. Geometric Mean (95% CI) Steady-State Plasma Amprenavir Pharmacokinetic Parameters in Pregnant Women Receiving LEXIVA with Ritonavir a AUC24 was calculated from AUC12 summary data x 2. b Ctau represents the concentration at the end of the dose interval.
Pharmacokinetic Parameter
LEXIVA 700 mg Twice Daily plus Ritonavir 100 mg Twice Daily
Second Trimester
(n = 6)
Third Trimester
(n = 9)
Postpartum
(n = 9)
AUC12 (mcg‚ÄĘh/mL)
26.0
(19.5, 34.6)
30.1
(21.6, 41.9)
39.9
(31.9, 50.1)
AUC24 (mcg‚ÄĘh/mL)a
52.0
(39.0, 69.2)
60.2
(43.2, 83.8)
79.8
(63.8, 100.2)
Cmax (mcg/mL)
4.19
(3.19, 5.51)
5.36
(3.98, 7.22)
6.65
(5.24, 8.44)
Ctau (mcg/mL)b
1.31
(0.97, 1.77)
1.34
(0.95, 1.89)
2.03
(1.46, 2.83)
Pediatric Patients: The pharmacokinetics of amprenavir following administration of LEXIVA oral suspension and LEXIVA tablets, with or without ritonavir, have been studied in a total of 212¬†HIV-1‚Äďinfected pediatric subjects enrolled in 3¬†trials. LEXIVA without ritonavir was administered as 30 or 40¬†mg per kg twice daily to children aged 2 to 5¬†years. LEXIVA with ritonavir was administered as LEXIVA 30¬†mg per kg plus ritonavir 6¬†mg per kg once daily to children aged 2 to 18¬†years and as LEXIVA 18 to 60¬†mg per kg plus ritonavir 3 to 10¬†mg per kg twice daily to children aged at least 4¬†weeks to 18¬†years; body weights ranged from 3 to 103¬†kg.
Amprenavir apparent clearance decreased with increasing weight. Weight-adjusted apparent clearance was higher in children younger than 4 years, suggesting that younger children require higher mg-per-kg dosing of LEXIVA.
The pharmacokinetics of LEXIVA oral suspension in protease inhibitor-naive infants younger than 6¬†months (n¬†=¬†9) receiving LEXIVA 45¬†mg per kg plus ritonavir 10¬†mg per kg twice daily generally demonstrated lower AUC12 and Cmin than adults receiving twice-daily LEXIVA 700¬†mg plus ritonavir 100¬†mg, the dose recommended for protease-experienced adults. The mean steady-state amprenavir AUC12, Cmax, and Cmin were 26.6¬†mcg‚ÄĘhour per mL, 6.25¬†mcg per mL, and 0.86¬†mcg per mL, respectively. Because of expected low amprenavir exposure and a requirement for large volume of drug, twice-daily dosing of LEXIVA alone (without ritonavir) in pediatric subjects younger than 2¬†years was not studied.
Pharmacokinetic parameters for LEXIVA administered with food and with ritonavir in this patient population at the recommended weight-band‚Äďbased dosage regimens are provided in Table 9.
Table 9. Geometric Mean (95% CI) Steady-State Plasma Amprenavir Pharmacokinetic Parameters by Weight in Pediatric and Adolescent Subjects Aged at Least 4 Weeks to 18 Years Receiving LEXIVA with Ritonavir a Recommended dose for pediatric patients weighing 11 kg to less than 15 kg is based on population pharmacokinetic analysis.
Weight
Recommended Dosage Regimen
Cmax
AUC24
Cmin
n
(mcg/mL)
n
(mcg‚ÄĘh/mL)
n
(mcg/mL)
<11 kg
LEXIVA 45 mg/kg plus
Ritonavir 7 mg/kg twice daily
12
6.00
(3.88, 9.29)
12
57.3
(34.1, 96.2)
27
1.65
(1.22, 2.24)
11 kg - <15 kg
LEXIVA 30 mg/kg plus
Ritonavir 3 mg/kg twice daily
Not studieda
15 kg - <20 kg
LEXIVA 23 mg/kg plus
Ritonavir 3 mg/kg twice daily
5
9.54
(4.63, 19.7)
5
121
(54.2, 269)
9
3.56
(2.33, 5.43)
20 kg - <39 kg
LEXIVA 18 mg/kg plus
Ritonavir 3 mg/kg twice daily
13
6.24
(5.01, 7.77)
12
97.9
(77.0, 124)
23
2.54
(2.11, 3.06)
‚Č•39¬†kg
LEXIVA 700 mg plus
Ritonavir 100 mg twice daily
15
5.03
(4.04, 6.26)
15
72.3
(59.6, 87.6)
42
1.98
(1.72, 2.29)
Subjects aged 2 to younger than 6 years receiving LEXIVA 30 mg per kg twice daily without ritonavir achieved geometric mean (95% CI) amprenavir Cmax (n = 9), AUC12 (n = 9), and Cmin (n = 19) of 7.15 (5.05, 10.1), 22.3 (15.3, 32.6), and 0.513 (0.384, 0.686), respectively.
Geriatric Patients: The pharmacokinetics of amprenavir after administration of LEXIVA to patients older than 65 years have not been studied [see Use in Specific Populations (8.5)].
Male and Female Patients: The pharmacokinetics of amprenavir after administration of LEXIVA do not differ between males and females.
Racial Groups: The pharmacokinetics of amprenavir after administration of LEXIVA do not differ between blacks and non-blacks.
Drug Interaction Studies
[See Contraindications (4), Warnings and Precautions (5.1), Drug Interactions (7).]
Amprenavir, the active metabolite of fosamprenavir, is metabolized in the liver by the cytochrome P450 enzyme system. Amprenavir inhibits CYP3A4. Data also suggest that amprenavir induces CYP3A4. Caution should be used when coadministering medications that are substrates, inhibitors, or inducers of CYP3A4, or potentially toxic medications that are metabolized by CYP3A4. Amprenavir does not inhibit CYP2D6, CYP1A2, CYP2C9, CYP2C19, CYP2E1, or uridine glucuronosyltransferase (UDPGT). Amprenavir is both a substrate for and inducer of P-glycoprotein.
Drug interaction trials were performed with LEXIVA and other drugs likely to be coadministered or drugs commonly used as probes for pharmacokinetic interactions. The effects of coadministration on AUC, Cmax, and Cmin values are summarized in Table 10 (effect of other drugs on amprenavir) and Table 12 (effect of LEXIVA on other drugs). In addition, since LEXIVA delivers comparable amprenavir plasma concentrations as AGENERASE, drug interaction data derived from trials with AGENERASE are provided in Tables 11 and 13. For information regarding clinical recommendations, [see Drug Interactions (7)].
Table 10. Drug Interactions: Pharmacokinetic Parameters for Amprenavir after Administration of LEXIVA in the Presence of the Coadministered Drug(s) a Concomitant medication is also shown in this column where appropriate. b Ritonavir Cmax, AUC, and Cmin increased by 63%, 45%, and 13%, respectively, compared with historical control. c Compared with historical control. d Subjects were receiving LEXIVA/ritonavir for 10¬†days prior to the 4-day treatment period with both ketoconazole and LEXIVA/ritonavir. e Compared with LEXIVA 700¬†mg/ritonavir 100¬†mg twice daily for 2¬†weeks. f Subjects were receiving nevirapine for at least 12¬†weeks prior to trial. g Clast (C12 h or C24 h). h Doses of LEXIVA and raltegravir were given with food on pharmacokinetic sampling days and without regard to food all other days. i Compared with parallel control group.‚ÜĎ= Increase; ‚Üď= Decrease; ‚ÜĒ = No change (‚ÜĎ or ‚Üď less than or equal to 10%), NA¬†=¬†Not applicable.
Coadministered Drug(s)
and Dose(s)
Dose of LEXIVAa
n
% Change in Amprenavir Pharmacokinetic Parameters (90% CI)
Cmax
AUC
Cmin
Antacid (MAALOX TC)
    30-mL single dose
1,400-mg
single dose
30
‚Üď35
(‚Üď24 to ‚Üď42)
‚Üď18
(‚Üď9 to ‚Üď26)
‚ÜĎ14
(‚Üď7 to ‚ÜĎ39)
Atazanavir
    300 mg once daily for 10 days
700 mg twice daily
plus ritonavir 100 mg twice daily for 10 days
22
‚ÜĒ
‚ÜĒ
‚ÜĒ
Atorvastatin
    10 mg once daily for 4 days
1,400 mg twice daily
for 2 weeks
16
‚Üď18
(‚Üď34 to ‚ÜĎ1)
‚Üď27
(‚Üď41 to ‚Üď12)
‚Üď12
(‚Üď27 to ‚Üď6)
Atorvastatin
    10 mg once daily for 4 days
700 mg twice daily
plus ritonavir 100 mg twice daily for 2 weeks
16
‚ÜĒ
‚ÜĒ
‚ÜĒ
Efavirenz
    600 mg once daily for 2 weeks
1,400 mg once daily
plus ritonavir 200 mg once daily for 2 weeks
16
‚ÜĒ
‚Üď13
(‚Üď30 to ‚ÜĎ7)
‚Üď36
(‚Üď8 to ‚Üď56)
Efavirenz
    600 mg once daily plus additional
    ritonavir 100 mg once daily for 2 weeks
1,400 mg once daily
plus ritonavir 200 mg once daily for 2 weeks
16
‚ÜĎ18
(‚ÜĎ1 to ‚ÜĎ38)
‚ÜĎ11
(0 to ‚ÜĎ24)
‚ÜĒ
Efavirenz
    600 mg once daily for 2 weeks
700 mg twice daily
plus ritonavir 100 mg twice daily for 2 weeks
16
‚ÜĒ
‚ÜĒ
‚Üď17
(‚Üď4 to ‚Üď29)
Esomeprazole
    20 mg once daily for 2 weeks
1,400 mg twice daily for 2 weeks
25
‚ÜĒ
‚ÜĒ
‚ÜĒ
Esomeprazole
    20 mg once daily for 2 weeks
700 mg twice daily
plus ritonavir 100 mg twice daily for 2 weeks
23
‚ÜĒ
‚ÜĒ
‚ÜĒ
Ethinyl estradiol/norethindrone
    0.035 mg/0.5 mg once daily for 21 days
700 mg twice daily
plus ritonavirb 100 mg twice daily for 21 days
25
‚ÜĒc
‚ÜĒc
‚ÜĒc
Ketoconazoled
    200 mg once daily for 4 days
700 mg twice daily
plus ritonavir 100 mg twice daily for 4 days
15
‚ÜĒ
‚ÜĒ
‚ÜĒ
Lopinavir/ritonavir
    533 mg/133 mg twice daily
1,400 mg twice daily
for 2 weeks
18
‚Üď13e
‚Üď26e
‚Üď42e
Lopinavir/ritonavir
    400 mg/100 mg twice daily for 2 weeks
700 mg twice daily
plus ritonavir 100 mg twice daily for 2 weeks
18
‚Üď58
(‚Üď42 to ‚Üď70)
‚Üď63
(‚Üď51 to ‚Üď72)
‚Üď65
(‚Üď54 to ‚Üď73)
Maraviroc
    300 mg twice daily for 10 days
700 mg twice daily
plus ritonavir
100 mg twice daily for 20 days
14
‚Üď34
(‚Üď25 to ‚Üď41)
‚Üď35
(‚Üď29 to ‚Üď41)
‚Üď36
(‚Üď27 to ‚Üď43)
Maraviroc
    300 mg once daily for 10 days
1,400 mg once daily
plus ritonavir 100 mg once daily for 20 days
14
‚Üď29
(‚Üď20 to ‚Üď38)
‚Üď30
(‚Üď23 to ‚Üď36)
‚Üď15
(‚Üď3 to ‚Üď25)
Methadone
    70 to 120 mg once daily for 2 weeks
700 mg twice daily
plus ritonavir 100 mg twice daily for 2 weeks
19
‚ÜĒc
‚ÜĒc
‚ÜĒc
Nevirapine
    200 mg twice daily for 2 weeksf
1,400 mg twice daily for 2 weeks
17
‚Üď25
(‚Üď37 to ‚Üď10)
‚Üď33
(‚Üď45 to ‚Üď20)
‚Üď35
(‚Üď50 to ‚Üď15)
Nevirapine
    200 mg twice daily for 2 weeksf
700 mg twice daily
plus ritonavir 100 mg twice daily for 2 weeks
17
‚ÜĒ
‚Üď11
(‚Üď23 to ‚ÜĎ3)
‚Üď19
(‚Üď32 to ‚Üď4)
Phenytoin
    300 mg once daily for 10 days
700 mg twice daily
plus ritonavir 100 mg twice daily for 10 days
13
‚ÜĒ
‚ÜĎ20
(‚ÜĎ8 to ‚ÜĎ34)
‚ÜĎ19
(‚ÜĎ6 to ‚ÜĎ33)
Raltegravir
    400 mg twice daily for 14 days
1,400 mg twice daily for 14 days (fasted)
14
‚Üď27
(‚Üď46 to ‚ÜĒ)
‚Üď36
(‚Üď53 to ‚Üď13)
‚Üď43g
(‚Üď59 to ‚Üď21)
1,400 mg twice daily for 14 daysh
14
‚Üď15
(‚Üď27 to ‚Üď1)
‚Üď17
(‚Üď27 to ‚Üď6)
‚Üď32g
(‚Üď53 to ‚Üď1)
700 mg twice daily
plus ritonavir 100 mg twice daily for 14 days (fasted)
14
‚Üď14
(‚Üď39 to ‚ÜĎ20)
‚Üď17
(‚Üď38 to ‚ÜĎ12)
‚Üď20g
(‚Üď45 to ‚ÜĎ17)
700 mg twice daily
plus ritonavir 100 mg twice daily for 14 daysh
12
‚Üď25
(‚Üď42 to ‚Üď2)
‚Üď25
(‚Üď44 to ‚ÜĒ)
‚Üď33g
(‚Üď52 to ‚Üď7)
Raltegravir
    400 mg twice daily for 14 days
1,400 mg once daily
plus ritonavir
100 mg once daily for 14 days (fasted)
13
‚Üď18
(‚Üď34 to ‚ÜĒ)
‚Üď24
(‚Üď41 to ‚ÜĒ)
‚Üď50g
(‚Üď64 to ‚Üď31)
1,400 mg once daily
plus ritonavir 100 mg once daily for 14 daysh
14
‚ÜĎ27
(‚Üď1 to ‚ÜĎ62)
‚ÜĎ13
(‚Üď7 to ‚ÜĎ38)
‚Üď17g
(‚Üď45 to ‚ÜĎ26)
Ranitidine
    300-mg single dose
(administered 1 hour before fosamprenavir)
1,400-mg
single dose
30
‚Üď51
(‚Üď43 to ‚Üď58)
‚Üď30
(‚Üď22 to ‚Üď37)
‚ÜĒ
(‚Üď19 to ‚ÜĎ21)
Rifabutin
    150 mg every other day for 2 weeks
700 mg twice daily
plus ritonavir 100 mg twice daily for 2 weeks
15
‚ÜĎ36c
(‚ÜĎ18 to ‚ÜĎ55)
‚ÜĎ35c
(‚ÜĎ17 to ‚ÜĎ56)
‚ÜĎ17c
(‚Üď1 to ‚ÜĎ39)
Tenofovir
    300 mg once daily for 4 to 48 weeks
700 mg twice daily
plus ritonavir 100 mg twice daily for 4 to 48 weeks
45
NA
NA
‚ÜĒi
Tenofovir
    300 mg once daily for 4 to 48 weeks
1,400 mg once daily
plus ritonavir 200 mg once daily for 4 to 48 weeks
60
NA
NA
‚ÜĒi
Table 11. Drug Interactions: Pharmacokinetic Parameters for Amprenavir after Administration of AGENERASE in the Presence of the Coadministered Drug(s) a Compared with parallel control group. b Median percent change; confidence interval not reported. c Compared with historical data.‚ÜĎ = Increase; ‚Üď = Decrease; ‚ÜĒ = No change (‚ÜĎ or ‚Üď less than 10%); NA = Cmin not calculated for single‚ÄĎdose trial.
Coadministered Drug(s)
and Dose(s)
Dose of AGENERASEa
n
% Change in Amprenavir Pharmacokinetic Parameters
(90% CI)
Cmax
AUC
Cmin
Abacavir
    300 mg twice daily for 2 to 3 weeks
900 mg twice daily for 2 to 3 weeks
4
‚ÜĒa
‚ÜĒa
‚ÜĒa
Clarithromycin
    500 mg twice daily for 4 days
1,200 mg twice daily for 4 days
12
‚ÜĎ15
(‚ÜĎ1 to ‚ÜĎ31)
‚ÜĎ18
(‚ÜĎ8 to ‚ÜĎ29)
‚ÜĎ39
(‚ÜĎ31 to ‚ÜĎ47)
Delavirdine
    600 mg twice daily for 10 days
600 mg twice daily for 10 days
9
‚ÜĎ40b
‚ÜĎ130b
‚ÜĎ125b
Ethinyl estradiol/ norethindrone
    0.035 mg/1 mg for 1 cycle
1,200 mg twice daily for 28 days
10
‚ÜĒ
‚Üď22
(‚Üď35 to ‚Üď8)
‚Üď20
(‚Üď41 to ‚ÜĎ8)
Indinavir
    800 mg 3 times a day for 2 weeks (fasted)
750 or 800 mg
3 times a day for 2 weeks (fasted)
9
‚ÜĎ18
(‚ÜĎ13 to ‚ÜĎ58)
‚ÜĎ33
(‚ÜĎ2 to ‚ÜĎ73)
‚ÜĎ25
(‚Üď27 to ‚ÜĎ116)
Ketoconazole
    400-mg single dose
1,200-mg
single dose
12
‚Üď16
(‚Üď25 to ‚Üď6)
‚ÜĎ31
(‚ÜĎ20 to ‚ÜĎ42)
NA
Lamivudine
    150-mg single dose
600-mg
single dose
11
‚ÜĒ
‚ÜĒ
NA
Methadone
    44 to 100 mg once daily for >30 days
1,200 mg twice daily
for 10 days
16
‚Üď27c
‚Üď30c
‚Üď25c
Nelfinavir
    750 mg 3 times a day for 2 weeks (fed)
750 or 800 mg 3 times a day for 2 weeks (fed)
6
‚Üď14
(‚Üď38 to ‚ÜĎ20)
‚ÜĒ
‚ÜĎ189
(‚ÜĎ52 to ‚ÜĎ448)
Rifabutin
    300 mg once daily for 10 days
1,200 mg twice daily
for 10 days
5
‚ÜĒ
‚Üď15
(‚Üď28 to 0)
‚Üď15
(‚Üď38 to ‚ÜĎ17)
Rifampin
    300 mg once daily for 4 days
1,200 mg twice daily
for 4 days
11
‚Üď70
(‚Üď76 to ‚Üď62)
‚Üď82
(‚Üď84 to ‚Üď78)
‚Üď92
(‚Üď95 to ‚Üď89)
Saquinavir
    800 mg 3 times a day for 2 weeks (fed)
750 or 800 mg 3 times a day for 2 weeks (fed)
7
‚Üď37
(‚Üď54 to ‚Üď14)
‚Üď32
(‚Üď49 to ‚Üď9)
‚Üď14
(‚Üď52 to ‚ÜĎ54)
Zidovudine
    300-mg single dose
600-mg
single dose
12
‚ÜĒ
‚ÜĎ13
(‚Üď2 to ‚ÜĎ31)
NA
Table 12. Drug Interactions: Pharmacokinetic Parameters for Coadministered Drug in the Presence of Amprenavir after Administration of LEXIVA a Concomitant medication is also shown in this column where appropriate. b Comparison arm of atazanavir 300¬†mg once daily plus ritonavir 100¬†mg once daily for 10¬†days. c Administered as a combination oral contraceptive tablet: ethinyl estradiol 0.035¬†mg/norethindrone 0.5¬†mg. d Subjects were receiving LEXIVA/ritonavir for 10¬†days prior to the 4-day treatment period with both ketoconazole and LEXIVA/ritonavir. e Data represent lopinavir concentrations. f Compared with lopinavir 400¬†mg/ritonavir 100¬†mg twice daily for 2¬†weeks. g Dose normalized to methadone 100¬†mg. The unbound concentration of the active moiety, R‚ÄĎmethadone, was unchanged. h Subjects were receiving nevirapine for at least 12¬†weeks prior to trial. i Comparison arm of rifabutin 300¬†mg once daily for 2¬†weeks. AUC is AUC(0-48¬†h).‚ÜĎ = Increase; ‚Üď= Decrease; ‚ÜĒ = No change (‚ÜĎ or ‚Üďless than 10%); ND = Interaction cannot be determined as Cmin was below the lower limit of quantitation.
Coadministered Drug(s)
and Dose(s)
Dose of LEXIVAa
n
% Change in Pharmacokinetic Parameters of Coadministered Drug (90% CI)
Cmax
AUC
Cmin
Atazanavir
    300 mg once daily for 10 daysb
700 mg twice daily
plus ritonavir 100 mg twice daily
for 10 days
21
‚Üď24
(‚Üď39 to ‚Üď6)
‚Üď22
(‚Üď34 to ‚Üď9)
‚ÜĒ
Atorvastatin
    10 mg once daily for 4 days
1,400 mg twice daily for 2 weeks
16
‚ÜĎ304
(‚ÜĎ205 to ‚ÜĎ437)
‚ÜĎ130
(‚ÜĎ100 to ‚ÜĎ164)
‚Üď10
(‚Üď27 to ‚ÜĎ12)
Atorvastatin
    10 mg once daily for 4 days
700 mg twice daily
plus ritonavir 100 mg twice daily
for 2 weeks
16
‚ÜĎ184
(‚ÜĎ126 to ‚ÜĎ257)
‚ÜĎ153
(‚ÜĎ115 to ‚ÜĎ199)
‚ÜĎ73
(‚ÜĎ45 to ‚ÜĎ108)
Esomeprazole
    20 mg once daily for 2 weeks
1,400 mg twice daily for 2 weeks
25
‚ÜĒ
‚ÜĎ55
(‚ÜĎ39 to ‚ÜĎ73)
ND
Esomeprazole
    20 mg once daily for 2 weeks
700 mg twice daily
plus ritonavir
100 mg twice daily for 2 weeks
23
‚ÜĒ
‚ÜĒ
ND
Ethinyl estradiolc
    0.035 mg once daily for 21 days
700 mg twice daily
plus ritonavir 100 mg twice daily
for 21 days
25
‚Üď28
(‚Üď21 to ‚Üď35)
‚Üď37
(‚Üď30 to ‚Üď42)
ND
Dolutegravir
    50 mg once daily
700 mg twice daily
plus ritonavir 100 mg twice daily
12
‚Üď24
(‚Üď8 to ‚Üď37)
‚Üď35
(‚Üď22 to ‚Üď46)
‚Üď49
(‚Üď37 to ‚Üď59)
Ketoconazoled
    200 mg once daily for 4 days
700 mg twice daily
plus ritonavir 100 mg twice daily for 4 days
15
‚ÜĎ25
(‚ÜĎ0 to ‚ÜĎ56)
‚ÜĎ169
(‚ÜĎ108 to ‚ÜĎ248)
ND
Lopinavir/ritonavire
    533 mg/133 mg twice daily for 2 weeks
1,400 mg twice daily for 2 weeks
18
‚ÜĒf
‚ÜĒf
‚ÜĒf
Lopinavir/ritonavire
    400 mg/100 mg twice daily for 2 weeks
700 mg twice daily
plus ritonavir 100 mg twice daily for 2 weeks
18
‚ÜĎ30
(‚Üď15 to ‚ÜĎ47)
‚ÜĎ37
(‚Üď20 to ‚ÜĎ55)
‚ÜĎ52
(‚Üď28 to ‚ÜĎ82)
Maraviroc
    300 mg twice daily for 10 days
700 mg twice daily
plus ritonavir 100 mg twice daily for 20 days
14
‚ÜĎ52
(‚ÜĎ27 to ‚ÜĎ82)
‚ÜĎ149
(‚ÜĎ119 to ‚ÜĎ182)
‚ÜĎ374
(‚ÜĎ303 to ‚ÜĎ457)
Maraviroc
    300 mg once daily for 10 days
1,400 mg once daily plus ritonavir 100 mg once daily for 20 days
14
‚ÜĎ45
(‚ÜĎ20 to ‚ÜĎ74)
‚ÜĎ126
(‚ÜĎ99 to ‚ÜĎ158)
‚ÜĎ80
(‚ÜĎ53 to ‚ÜĎ113)
Methadone
    70 to 120 mg once daily for 2 weeks
700 mg twice daily
plus ritonavir 100 mg twice daily for 2 weeks
19
R-Methadone (active)
‚Üď21g
(‚Üď30 to ‚Üď12)
‚Üď18g
(‚Üď27 to ‚Üď8)
‚Üď11g
(‚Üď21 to ‚ÜĎ1)
S-Methadone (inactive)
‚Üď43g
(‚Üď49 to ‚Üď37)
‚Üď43g
(‚Üď50 to ‚Üď36)
‚Üď41g
(‚Üď49 to ‚Üď31)
Nevirapine
    200 mg twice daily for 2 weeksh
1,400 mg twice daily for 2 weeks
17
‚ÜĎ25
(‚ÜĎ14 to ‚ÜĎ37)
‚ÜĎ29
(‚ÜĎ19 to ‚ÜĎ40)
‚ÜĎ34
(‚ÜĎ20 to ‚ÜĎ49)
Nevirapine
    200 mg twice daily for 2 weeksh
700 mg twice daily plus ritonavir 100 mg twice daily for 2 weeks
17
‚ÜĎ13
(‚ÜĎ3 to ‚ÜĎ24)
‚ÜĎ14
(‚ÜĎ5 to ‚ÜĎ24)
‚ÜĎ22
(‚ÜĎ9 to ‚ÜĎ35)
Norethindronec
    0.5 mg once daily for 21 days
700 mg twice daily
plus ritonavir 100 mg twice daily
for 21 days
25
‚Üď38
(‚Üď32 to ‚Üď44)
‚Üď34
(‚Üď30 to ‚Üď37)
‚Üď26
(‚Üď20 to ‚Üď32)
Phenytoin
    300 mg once daily for 10 days
700 mg twice daily
plus ritonavir 100 mg twice daily for 10 days
14
‚Üď20
(‚Üď12 to ‚Üď27)
‚Üď22
(‚Üď17 to ‚Üď27)
‚Üď29
(‚Üď23 to ‚Üď34)
Rifabutin
    150 mg every other day for 2 weeks i
700 mg twice daily
plus ritonavir 100 mg twice daily for 2 weeks
15
‚Üď14
(‚Üď28 to ‚ÜĎ4)
‚ÜĒ
‚ÜĎ28
(‚ÜĎ12 to ‚ÜĎ46)
     (25-O-desacetylrifabutin
metabolite)
‚ÜĎ579
(‚ÜĎ479 to ‚ÜĎ698)
‚ÜĎ1,120
(‚ÜĎ965 to ‚ÜĎ1,300)
‚ÜĎ2,510
(‚ÜĎ1,910 to ‚ÜĎ3,300)
    Rifabutin + 25-O-
desacetylrifabutin
metabolite
NA
‚ÜĎ64
(‚ÜĎ46 to ‚ÜĎ84)
NA
Rosuvastatin
    10-mg single dose
700 mg twice daily
plus ritonavir 100 mg twice daily for 7 days
(‚ÜĎ45)
(‚ÜĎ8)
NA
Table 13. Drug Interactions: Pharmacokinetic Parameters for Coadministered Drug in the Presence of Amprenavir after Administration of AGENERASE a Compared with historical data. b Median percent change; confidence interval not reported.‚ÜĎ = Increase; ‚Üď = Decrease; ‚ÜĒ= No change (‚ÜĎ or ‚Üď less than 10%); NA = Cmin not calculated for single-dose trial; ND = Interaction cannot be determined as Cmin was below the lower limit of quantitation.
Coadministered Drug(s) and Dose(s)
Dose of AGENERASE
n
% Change in Pharmacokinetic Parameters of Coadministered Drug (90% CI)
Cmax
AUC
Cmin
Abacavir
    300 mg twice daily for 2 to 3 weeks
900 mg twice daily for 2 to 3 weeks
4
‚ÜĒa
‚ÜĒa
‚ÜĒa
Clarithromycin
    500 mg twice daily for 4 days
1,200 mg twice daily for 4 days
12
‚Üď10
(‚Üď24 to ‚ÜĎ7)
‚ÜĒ
‚ÜĒ
Delavirdine
    600 mg twice daily for 10 days
600 mg twice daily for 10 days
9
‚Üď47b
‚Üď61b
‚Üď88b
Ethinyl estradiol
    0.035 mg for 1 cycle
1,200 mg twice daily for 28 days
10
‚ÜĒ
‚ÜĒ
‚ÜĎ32
(‚Üď3 to ‚ÜĎ79)
Indinavir
    800 mg 3 times a day for 2 weeks (fasted)
750 mg or 800 mg 3 times a day for 2 weeks (fasted)
9
‚Üď22a
‚Üď38a
‚Üď27a
Ketoconazole
    400-mg single dose
1,200-mg
single dose
12
‚ÜĎ19
(‚ÜĎ8 to ‚ÜĎ33)
‚ÜĎ44
(‚ÜĎ31 to ‚ÜĎ59)
NA
Lamivudine
    150-mg single dose
600-mg
single dose
11
‚ÜĒ
‚ÜĒ
NA
Methadone
    44 to 100 mg once daily for >30 days
1,200 mg twice daily for 10 days
16
R-Methadone (active)
‚Üď25
(‚Üď32 to ‚Üď18)
‚Üď13
(‚Üď21 to ‚Üď5)
‚Üď21
(‚Üď32 to ‚Üď9)
S-Methadone (inactive)
‚Üď48
(‚Üď55 to ‚Üď40)
‚Üď40
(‚Üď46 to ‚Üď32)
‚Üď53
(‚Üď60 to ‚Üď43)
Nelfinavir
    750 mg 3 times a day for 2 weeks (fed)
750 mg or 800 mg 3 times a day for 2 weeks (fed)
6
‚ÜĎ12a
‚ÜĎ15a
‚ÜĎ14a
Norethindrone
    1 mg for 1 cycle
1,200 mg twice daily for 28 days
10
‚ÜĒ
‚ÜĎ18
(‚ÜĎ1 to ‚ÜĎ38)
‚ÜĎ45
(‚ÜĎ13 to ‚ÜĎ88)
Rifabutin
    300 mg once daily for 10 days
1,200 mg twice daily for 10 days
5
‚ÜĎ119
(‚ÜĎ82 to ‚ÜĎ164)
‚ÜĎ193
(‚ÜĎ156 to ‚ÜĎ235)
‚ÜĎ271
(‚ÜĎ171 to ‚ÜĎ409)
Rifampin
    300 mg once daily for 4 days
1,200 mg twice daily
for 4 days
11
‚ÜĒ
‚ÜĒ
ND
Saquinavir
    800 mg 3 times a day for 2 weeks (fed)
750 mg or 800 mg 3 times a day for 2 weeks (fed)
7
‚ÜĎ21a
‚Üď19a
‚Üď48a
Zidovudine
    300-mg single dose
600-mg
single dose
12
‚ÜĎ40
(‚ÜĎ14 to ‚ÜĎ71)
‚ÜĎ31
(‚ÜĎ19 to ‚ÜĎ45)
NA
12.4 Microbiology
Mechanism of Action
Fosamprenavir is a prodrug that is rapidly hydrolyzed to amprenavir by cellular phosphatases in the gut epithelium as it is absorbed. Amprenavir is an inhibitor of HIV-1 protease. Amprenavir binds to the active site of HIV-1 protease and thereby prevents the processing of viral Gag and Gag-Pol polyprotein precursors, resulting in the formation of immature non-infectious viral particles.
Antiviral Activity
Fosamprenavir has little or no antiviral activity in cell culture. The antiviral activity of amprenavir was evaluated against HIV-1 IIIB in both acutely and chronically infected lymphoblastic cell lines (MT-4, CEM-CCRF, H9) and in peripheral blood lymphocytes in cell culture. The 50% effective concentration (EC50) of amprenavir ranged from 0.012 to 0.08 microM in acutely infected cells and was 0.41 microM in chronically infected cells (1 microM = 0.50 mcg per mL). The median EC50 value of amprenavir against HIV-1 isolates from clades A to G was 0.00095 microM in peripheral blood mononuclear cells (PBMCs). Similarly, the EC50 values for amprenavir against monocytes/macrophage tropic HIV-1 isolates (clade B) ranged from 0.003 to 0.075 microM in monocyte/macrophage cultures. The EC50 values of amprenavir against HIV-2 isolates grown in PBMCs were higher than those for HIV-1 isolates, and ranged from 0.003 to 0.11 microM. The anti-HIV-1 activity of amprenavir was not antagonistic in combination with the nucleoside reverse transcriptase inhibitors (NRTIs); abacavir, didanosine, lamivudine, stavudine, tenofovir, and zidovudine; the non-nucleoside reverse transcriptase inhibitors (NNRTIs) delavirdine, efavirenz, and nevirapine; the protease inhibitors (PIs) atazanavir, indinavir, lopinavir, nelfinavir, ritonavir, and saquinavir; and the gp41 fusion inhibitor enfuvirtide. These drug combinations have not been adequately studied in humans.
Resistance
HIV-1 isolates with decreased susceptibility to amprenavir have been selected in cell culture and obtained from subjects treated with fosamprenavir. Genotypic analysis of isolates from treatment-naive subjects failing amprenavir-containing regimens showed substitutions in the HIV-1 protease resulting in amino acid substitutions primarily at positions V32I, M46I/L, I47V, I50V, I54L/M, and I84V, as well as substitutions in the p7/p1 and p1/p6 Gag and Gag-Pol polyprotein precursor cleavage sites. Some of these amprenavir resistance-associated substitutions have also been detected in HIV-1 isolates from antiretroviral-naive subjects treated with LEXIVA. Of the 488 antiretroviral-naive subjects treated with LEXIVA 1,400 mg twice daily or LEXIVA 1,400 mg plus ritonavir 200 mg once daily in Trials APV30001 and APV30002, respectively, isolates from 61 subjects (29 receiving LEXIVA and 32 receiving LEXIVA/ritonavir) with virologic failure (plasma HIV-1 RNA greater than 1,000 copies per mL on 2 occasions on or after Week 12) were genotyped. Isolates from 5 of the 29 antiretroviral-naive subjects (17%) receiving LEXIVA without ritonavir in Trial APV30001 had evidence of genotypic resistance to amprenavir: I54L/M (n = 2), I54L + L33F (n = 1), V32I + I47V (n = 1), and M46I + I47V (n = 1). No amprenavir resistance-associated substitutions were detected in isolates from antiretroviral-naive subjects treated with LEXIVA/ritonavir for 48 weeks in Trial APV30002. However, the M46I and I50V substitutions were detected in isolates from 1 virologic failure subject receiving LEXIVA/ritonavir once daily at Week 160 (HIV-1 RNA greater than 500 copies per mL). Upon retrospective analysis of stored samples using an ultrasensitive assay, these resistant substitutions were traced back to Week 84 (76 weeks prior to clinical virologic failure).
Cross-Resistance
Varying degrees of cross-resistance among HIV-1 protease inhibitors have been observed. An association between virologic response at 48 weeks (HIV-1 RNA level less than 400 copies per mL) and protease inhibitor-resistance substitutions detected in baseline HIV-1 isolates from protease inhibitor-experienced subjects receiving LEXIVA/ritonavir twice daily (n = 88), or lopinavir/ritonavir twice daily (n = 85) in Trial APV30003 is shown in Table 14. The majority of subjects had previously received either one (47%) or 2 protease inhibitors (36%), most commonly nelfinavir (57%) and indinavir (53%). Out of 102 subjects with baseline phenotypes receiving twice-daily LEXIVA/ritonavir, 54% (n = 55) had resistance to at least one protease inhibitor, with 98% (n = 54) of those having resistance to nelfinavir. Out of 97 subjects with baseline phenotypes in the lopinavir/ritonavir arm, 60% (n = 58) had resistance to at least one protease inhibitor, with 97% (n = 56) of those having resistance to nelfinavir.
Table 14. Responders at Trial Week 48 by Presence of Baseline Protease Inhibitor Resistance-Associated Substitutionsa a Results should be interpreted with caution because the subgroups were small. b Most subjects had greater than 1 protease inhibitor resistance-associated substitution at baseline.
Protease Inhibitor Resistance-Associated Substitutionsb
LEXIVA/Ritonavir Twice Daily
(n = 88)
Lopinavir/Ritonavir Twice Daily
(n = 85)
D30N
21/22
95%
17/19
89%
N88D/S
20/22
91%
12/12
100%
L90M
16/31
52%
17/29
59%
M46I/L
11/22
50%
12/24
50%
V82A/F/T/S
2/9
22%
6/17
35%
I54V
2/11
18%
6/11
55%
I84V
1/6
17%
2/5
40%
The virologic response based upon baseline phenotype was assessed. Baseline isolates from protease inhibitor-experienced subjects responding to LEXIVA/ritonavir twice daily had a median shift in susceptibility to amprenavir relative to a standard wild-type reference strain of 0.7 (range: 0.1 to 5.4, n = 62), and baseline isolates from individuals failing therapy had a median shift in susceptibility of 1.9 (range: 0.2 to 14, n = 29). Because this was a select patient population, these data do not constitute definitive clinical susceptibility break points. Additional data are needed to determine clinically relevant break points for LEXIVA.
Isolates from 15 of the 20 subjects receiving twice-daily LEXIVA/ritonavir up to Week 48 and experiencing virologic failure/ongoing replication were subjected to genotypic analysis. The following amprenavir resistance-associated substitutions were found either alone or in combination: V32I, M46I/L, I47V, I50V, I54L/M, and I84V. Isolates from 4 of the 16 subjects continuing to receive twice-daily LEXIVA/ritonavir up to Week 96 who experienced virologic failure underwent genotypic analysis. Isolates from 2 subjects contained amprenavir resistance-associated substitutions: V32I, M46I, and I47V in 1 isolate and I84V in the other.
13 Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
In long-term carcinogenicity studies, fosamprenavir was administered orally for up to 104 weeks at doses of 250, 400, or 600 mg per kg per day in mice and at doses of 300, 825, or 2,250 mg per kg per day in rats. Exposures at these doses were 0.3- to 0.7-fold (mice) and 0.7- to 1.4-fold (rats) those in humans given 1,400 mg twice daily of fosamprenavir alone, and 0.2- to 0.3-fold (mice) and 0.3- to 0.7-fold (rats) those in humans given 1,400 mg once daily of fosamprenavir plus 200 mg ritonavir once daily. Exposures in the carcinogenicity studies were 0.1- to 0.3-fold (mice) and 0.3- to 0.6-fold (rats) those in humans given 700 mg of fosamprenavir plus 100 mg ritonavir twice daily. There was an increase in hepatocellular adenomas and hepatocellular carcinomas at all doses in male mice and at 600 mg per kg per day in female mice, and in hepatocellular adenomas and thyroid follicular cell adenomas at all doses in male rats, and at 825 mg per kg per day and 2,250 mg per kg per day in female rats. The relevance of the hepatocellular findings in the rodents for humans is uncertain. Repeat-dose studies with fosamprenavir in rats produced effects consistent with enzyme induction, which predisposes rats, but not humans, to thyroid neoplasms. In addition, in rats only there was an increase in interstitial cell hyperplasia at 825 mg per kg per day and 2,250 mg per kg per day, and an increase in uterine endometrial adenocarcinoma at 2,250 mg per kg per day. The incidence of endometrial findings was slightly increased over concurrent controls, but was within background range for female rats. The relevance of the uterine endometrial adenocarcinoma findings in rats for humans is uncertain.
Fosamprenavir was not mutagenic or genotoxic in a battery of in vitro and in vivo assays. These assays included bacterial reverse mutation (Ames), mouse lymphoma, rat micronucleus, and chromosome aberrations in human lymphocytes.
The effects of fosamprenavir on fertility and general reproductive performance were investigated in male (treated for 4 weeks before mating) and female rats (treated for 2 weeks before mating through Postpartum Day 6) that received doses of 300, 820, or 2,240 mg per kg per day. Systemic exposures (AUC0-24 h) to amprenavir in these studies were 3 (males) to 4 (females) times higher than exposures in humans following administration of the MRHD of fosamprenavir alone or similar to those seen in humans following administration of fosamprenavir in combination with ritonavir. Fosamprenavir did not impair mating or fertility of male or female rats and did not affect the development and maturation of sperm from treated rats.
14 Clinical Studies
14.1 Therapy-Naive Adult Trials
APV30001
A randomized, open-label trial evaluated treatment with LEXIVA tablets (1,400 mg twice daily) versus nelfinavir (1,250 mg twice daily) in 249 antiretroviral treatment-naive subjects. Both groups of subjects also received abacavir (300 mg twice daily) and lamivudine (150 mg twice daily).
The mean age of the subjects in this trial was 37 years (range: 17 to 70 years); 69% of the subjects were male, 20% were CDC Class C (AIDS), 24% were white, 32% were black, and 44% were Hispanic. At baseline, the median CD4+ cell count was 212 cells per mm3 (range: 2 to 1,136 cells per mm3; 18% of subjects had a CD4+ cell count of less than 50 cells per mm3 and 30% were in the range of 50 to less than 200 cells per mm3). Baseline median HIV-1 RNA was 4.83 log10 copies per mL (range: 1.69 to 7.41 log10 copies per mL; 45% of subjects had greater than 100,000 copies per mL).
The outcomes of randomized treatment are provided in Table 15.
Table 15. Outcomes of Randomized Treatment through Week 48 (APV30001) a Subjects achieved and maintained confirmed HIV-1 RNA less than 400 copies per mL (less than 50 copies per mL) through Week 48 (Roche AMPLICOR HIV-1 MONITOR Assay Version 1.5). b Includes consent withdrawn, lost to follow up, protocol violations, those with missing data, and other reasons.
Outcome
(Rebound or discontinuation = failure)
LEXIVA
1,400 mg Twice Daily
(n = 166)
Nelfinavir
1,250 mg Twice Daily
(n = 83)
Respondera
66% (57%)
52% (42%)
Virologic failure
19%
32%
    Rebound
16%
19%
    Never suppressed through Week 48
3%
13%
Clinical progression
1%
1%
Death
0%
1%
Discontinued due to adverse reactions
4%
2%
Discontinued due to other reasonsb
10%
10%
Treatment response by viral load strata is shown in Table 16.
Table 16. Proportions of Responders through Week 48 by Screening Viral Load (APV30001)
Screening Viral Load HIV-1 RNA (copies/mL)
LEXIVA 1,400 mg
Twice Daily
Nelfinavir 1,250 mg
Twice Daily
<400 copies/mL
n
<400 copies/mL
n
‚ȧ100,000
65%
93
65%
46
>100,000
67%
73
36%
37
Through 48 weeks of therapy, the median increases from baseline in CD4+ cell counts were 201 cells per mm3 in the group receiving LEXIVA and 216 cells per mm3 in the nelfinavir group.
APV30002
A randomized, open-label trial evaluated treatment with LEXIVA tablets (1,400 mg once daily) plus ritonavir (200 mg once daily) versus nelfinavir (1,250 mg twice daily) in 649 treatment-naive subjects. Both treatment groups also received abacavir (300 mg twice daily) and lamivudine (150 mg twice daily).
The mean age of the subjects in this trial was 37 years (range: 18 to 69 years); 73% of the subjects were male, 22% were CDC Class C, 53% were white, 36% were black, and 8% were Hispanic. At baseline, the median CD4+ cell count was 170 cells per mm3 (range: 1 to 1,055 cells per mm3; 20% of subjects had a CD4+ cell count of less than 50 cells per mm3 and 35% were in the range of 50 to less than 200 cells per mm3). Baseline median HIV-1 RNA was 4.81 log10 copies per mL (range: 2.65 to 7.29 log10 copies per mL; 43% of subjects had greater than 100,000 copies per mL).
The outcomes of randomized treatment are provided in Table 17.
Table 17. Outcomes of Randomized Treatment through Week 48 (APV30002) a Subjects achieved and maintained confirmed HIV-1 RNA less than 400 copies per mL (less than 50 copies per mL) through Week 48 (Roche AMPLICOR HIV-1 MONITOR Assay Version 1.5). b Includes consent withdrawn, lost to follow up, protocol violations, those with missing data, and other reasons.
Outcome
(Rebound or discontinuation = failure)
LEXIVA 1,400 mg/
Ritonavir 200 mg
Once Daily
(n = 322)
Nelfinavir 1,250 mg
Twice Daily
(n = 327)
Respondera
69% (58%)
68% (55%)
Virologic failure
6%
16%
    Rebound
5%
8%
    Never suppressed through Week 48
1%
8%
Death
1%
0%
Discontinued due to adverse reactions
9%
6%
Discontinued due to other reasonsb
15%
10%
Treatment response by viral load strata is shown in Table 18.
Table 18. Proportions of Responders through Week 48 by Screening Viral Load (APV30002)
Screening Viral Load HIV-1 RNA
LEXIVA 1,400 mg/Ritonavir 200 mg
Once Daily
Nelfinavir 1,250 mg
Twice Daily
(copies/mL)
<400 copies/mL
n
<400 copies/mL
n
‚ȧ100,000
72%
197
73%
194
>100,000
66%
125
64%
133
Through 48 weeks of therapy, the median increases from baseline in CD4+ cell counts were 203 cells per mm3 in the group receiving LEXIVA and 207 cells per mm3 in the nelfinavir group.
14.2 Protease Inhibitor-Experienced Adult Trials
APV30003
A randomized, open-label, multicenter trial evaluated 2 different regimens of LEXIVA plus ritonavir (LEXIVA tablets 700 mg twice daily plus ritonavir 100 mg twice daily or LEXIVA tablets 1,400 mg once daily plus ritonavir 200 mg once daily) versus lopinavir/ritonavir (400 mg/100 mg twice daily) in 315 subjects who had experienced virologic failure to 1 or 2 prior protease inhibitor-containing regimens.
The mean age of the subjects in this trial was 42 years (range: 24 to 72 years); 85% were male, 33% were CDC Class C, 67% were white, 24% were black, and 9% were Hispanic. The median CD4+ cell count at baseline was 263 cells per mm3 (range: 2 to 1,171 cells per mm3). Baseline median plasma HIV-1 RNA level was 4.14 log10 copies per mL (range: 1.69 to 6.41 log10 copies per mL).
The median durations of prior exposure to NRTIs were 257 weeks for subjects receiving LEXIVA/ritonavir twice daily (79% had greater than or equal to 3 prior NRTIs) and 210 weeks for subjects receiving lopinavir/ritonavir (64% had greater than or equal to 3 prior NRTIs). The median durations of prior exposure to protease inhibitors were 149 weeks for subjects receiving LEXIVA/ritonavir twice daily (49% received greater than or equal to 2 prior protease inhibitors) and 130 weeks for subjects receiving lopinavir/ritonavir (40% received greater than or equal to 2 prior protease inhibitors).
The time-averaged changes in plasma HIV-1 RNA from baseline (AAUCMB) at 48 weeks (the endpoint on which the trial was powered) were -1.4 log10 copies per mL for twice-daily LEXIVA/ritonavir and -1.67 log10 copies per mL for the lopinavir/ritonavir group.
The proportions of subjects who achieved and maintained confirmed HIV-1 RNA less than 400 copies per mL (secondary efficacy endpoint) were 58% with twice-daily LEXIVA/ritonavir and 61% with lopinavir/ritonavir (95% CI for the difference: -16.6, 10.1). The proportions of subjects with HIV-1 RNA less than 50 copies per mL with twice-daily LEXIVA/ritonavir and with lopinavir/ritonavir were 46% and 50%, respectively (95% CI for the difference: -18.3, 8.9). The proportions of subjects who were virologic failures were 29% with twice-daily LEXIVA/ritonavir and 27% with lopinavir/ritonavir.
The frequency of discontinuations due to adverse events and other reasons, and deaths were similar between treatment arms.
Through 48 weeks of therapy, the median increases from baseline in CD4+ cell counts were 81 cells per mm3 with twice-daily LEXIVA/ritonavir and 91 cells per mm3 with lopinavir/ritonavir.
This trial was not large enough to reach a definitive conclusion that LEXIVA/ritonavir and lopinavir/ritonavir are clinically equivalent.
Once-daily administration of LEXIVA plus ritonavir is not recommended for protease inhibitor-experienced patients. Through Week 48, 50% and 37% of subjects receiving LEXIVA 1,400 mg plus ritonavir 200 mg once daily had plasma HIV-1 RNA less than 400 copies per mL and less than 50 copies per mL, respectively.
14.3 Pediatric Trials
Three open-label trials in pediatric subjects aged at least 4 weeks to 18 years were conducted. In one trial (APV29005), twice-daily dosing regimens (LEXIVA with or without ritonavir) were evaluated in combination with other antiretroviral agents in pediatric subjects aged 2 to 18 years. In a second trial (APV20002), twice-daily dosing regimens (LEXIVA with ritonavir) were evaluated in combination with other antiretroviral agents in pediatric subjects aged at least 4 weeks to younger than 2 years. A third trial (APV20003) evaluated once-daily dosing of LEXIVA with ritonavir; the pharmacokinetic data from this trial did not support a once-daily dosing regimen in any pediatric patient population.
APV29005
LEXIVA: Twenty (18 therapy-naive and 2 therapy-experienced) pediatric subjects received LEXIVA oral suspension without ritonavir twice daily. At Week 24, 65% (13 of 20) achieved HIV-1 RNA less than 400 copies per mL, and the median increase from baseline in CD4+ cell count was 350 cells per mm3.
LEXIVA plus Ritonavir: Forty-nine protease inhibitor-naive and 40 protease inhibitor-experienced pediatric subjects received LEXIVA oral suspension or tablets with ritonavir twice daily. At Week 24, 71% of protease inhibitor-naive (35 of 49) and 55% of protease inhibitor-experienced (22 of 40) subjects achieved HIV-1 RNA less than 400 copies per mL; median increases from baseline in CD4+ cell counts were 184 cells per mm3 and 150 cells per mm3 in protease inhibitor-naive and experienced subjects, respectively.
APV20002
Fifty-four pediatric subjects (49 protease inhibitor-naive and 5 protease inhibitor-experienced) received LEXIVA oral suspension with ritonavir twice daily. At Week 24, 72% of subjects achieved HIV-1 RNA less than 400 copies per mL. The median increases from baseline in CD4+ cell counts were 400 cells per mm3 in subjects aged at least 4 weeks to younger than 6 months and 278 cells per mm3 in subjects aged 6 months to 2 years.
16 How Supplied/storage And Handling
LEXIVA tablets, 700¬†mg, are pink, film-coated, capsule-shaped, biconvex tablets, with ‚ÄúGX¬†LL7‚ÄĚ debossed on one face.
Bottle of 60 with child-resistant closure (NDC 49702-207-18).
Store at controlled room temperature of 25¬įC (77¬įF); excursions permitted to 15¬į to 30¬įC (59¬į to 86¬įF) (see USP Controlled Room Temperature). Keep container tightly closed.
LEXIVA oral suspension, a white to off-white grape-bubblegum-peppermint‚Äďflavored suspension, contains 50¬†mg of fosamprenavir as fosamprenavir calcium equivalent to approximately 43¬†mg of amprenavir in each 1¬†mL.
Bottle of 225 mL with child-resistant closure (NDC 49702-208-53).
This product does not require reconstitution.
Store in refrigerator or at room temperature (5¬į to 30¬įC; 41¬į to 86¬įF). Shake vigorously before using. Do not freeze.
17 Patient Counseling Information
Advise the patient to read the FDA-approved patient labeling (Patient Information).
Drug Interactions
A statement to patients and healthcare providers is included on the product’s bottle label: ALERT: Find out about medicines that should NOT be taken with LEXIVA.
LEXIVA may interact with many drugs; therefore, advise patients to report to their healthcare provider the use of any other prescription or nonprescription medication or herbal products, particularly St. John’s wort.
Advise patients receiving PDE5 inhibitors that they may be at an increased risk of PDE5 inhibitor‚ÄĎassociated adverse events, including hypotension, visual changes, and priapism, and should promptly report any symptoms to their healthcare provider [see Contraindications (4), Warnings and Precautions (5.1), Drug Interactions (7)].
Contraception
Instruct patients receiving combined hormonal contraception to use an effective alternative contraceptive method or an additional barrier method during therapy with LEXIVA because hormonal levels may decrease, and if used in combination with LEXIVA and ritonavir, liver enzyme elevations may occur [see Drug Interactions (7.2), Use in Specific Populations (8.3)].
Severe Skin Reactions
Advise patients that skin reactions ranging from mild to severe, including Stevens-Johnson Syndrome, have been reported with LEXIVA. Advise patients to discontinue LEXIVA immediately for severe or life-threatening skin reactions or for moderate rashes accompanied by systemic symptoms [see Warnings and Precautions (5.2), Adverse Reactions (6)].
Sulfa Allergy
Advise patients to inform their healthcare provider if they have a sulfa allergy. The potential for cross‚ÄĎsensitivity between drugs in the sulfonamide class and fosamprenavir is unknown [see Warnings and Precautions (5.3)].
Hepatic Toxicity
Advise patients that it is recommended to have laboratory testing before and during therapy as patients with underlying hepatitis B or C or marked elevations of transaminases prior to treatment may be at increased risk for developing or worsening transaminase elevations with use of LEXIVA, particularly at higher than recommended doses which should not be used. [see Warnings and Precautions (5.4)].
Immune Reconstitution Syndrome
Advise patients to inform their healthcare provider immediately of any signs or symptoms of infection as inflammation from previous infection may occur soon after combination antiretroviral therapy, including when LEXIVA is started [see Warnings and Precautions (5.6)].
Increase in Body Fat
Inform patients that an increase of body fat may occur in patients receiving protease inhibitors, including LEXIVA, and that the cause and long-term health effects of these conditions are not known at this time [see Warnings and Precautions (5.7)].
Lipid Elevations
Advise patients that it is recommended to have laboratory testing before and during therapy as increases in the concentration of triglycerides and cholesterol have been reported with use of LEXIVA [see Warnings and Precautions (5.8), Adverse Reactions (6)].
Pregnancy Registry
Advise patients that there is a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to LEXIVA during pregnancy [see Use in Specific Populations (8.1)].
Lactation
Instruct women with HIV-1 infection not to breastfeed because HIV-1 can be passed to the baby in the breast milk [see Use in Specific Populations (8.2)].
Missed Dose
Instruct patients that if they miss a dose of LEXIVA, to take it as soon as they remember. Advise patients not to double their next dose or take more than the prescribed dose [see Dosage and Administration (2)].
Oral Suspension
Instruct patients to shake the bottle vigorously before each use and inform them that refrigeration of the oral suspension may improve the taste for some patients [see How Supplied/Storage and Handling (16)].
LEXIVA and AGENERASE are trademarks owned by or licensed to the ViiV Healthcare group of companies.
ADCIRCA is a registered trademark owned by Eli Lilly and Company.
The other brands uled are trademarks owned by or licensed to their respective owners and are not owned by or licensed to the ViiV Healthcare group of companies. The makers of these brands are not affiliated with and do not endorse the ViiV Healthcare group of companies or its products.
Manufactured for:
ViiV Healthcare
Research Triangle Park, NC 27709
©2020 ViiV Healthcare group of companies or its licensor.
LXV:26PI
PHARMACIST-DETACH HERE AND GIVE INSTRUCTIONS TO PATIENT
_ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ __ _ _ _ _ _ _ _
Spl Patient Package Insert Section
PATIENT INFORMATION
LEXIVA (lex-EE-vah)
(fosamprenavir calcium)
tablets
LEXIVA (lex-EE-vah)
(fosamprenavir calcium)
oral suspension
What is the most important information I should know about LEXIVA?
LEXIVA can interact with other medicines and cause serious side effects. It is important to know the medicines that should not be taken with LEXIVA. See the section ‚ÄúWho should not take LEXIVA?‚ÄĚ
LEXIVA can cause serious side effects, including:
‚ÄĘ Severe skin reactions. LEXIVA may cause severe or life-threatening skin reactions or rash. If you get a rash with any of the following symptoms, stop taking LEXIVA and call your healthcare provider or get medical help right away:
o hives or sores in your mouth, or your skin bulers and peelso trouble swallowing or breathing
o swelling of your face, eyes, lips, tongue, or throat
For more information about side effects, see ‚ÄúWhat are the possible side effects of LEXIVA?‚ÄĚ
What is LEXIVA?
LEXIVA is a prescription medicine that is used together with other antiretroviral medicines to treat human immunodeficiency virus 1 (HIV-1) infection.
HIV-1 is the virus that causes Acquired Immune Deficiency Syndrome (AIDS).
It is not known if LEXIVA is safe and effective in children younger than 4 weeks of age.
Do not take LEXIVA if you:
‚ÄĘ are allergic to amprenavir, fosamprenavir calcium, or any of the ingredients in LEXIVA. See the end of this leaflet for a complete ul of ingredients in LEXIVA.‚ÄĘ take any of the following medicines:
o alfuzosino rifampino ergot including:
‚Ė™ dihydroergotamine mesylate‚Ė™ ergonovine‚Ė™ ergotamine tartrate‚Ė™ methylergonovineo cisaprideo St. John‚Äôs wort (Hypericum perforatum)o lomitapideo lovastatino simvastatino pimozideo delavirdine mesylateo sildenafil (REVATIO), for treatment of pulmonary arterial hypertensiono triazolamo midazolam, when taken by mouth¬† Serious problems can happen if you or your child take any of the medicines uled above with LEXIVA.
If you are taking LEXIVA with ritonavir, do not take the following medicines:
 
‚ÄĘ flecainide‚ÄĘ propafenone‚ÄĘ lurasidone
Before taking LEXIVA, tell your healthcare provider about all of your medical conditions, including if you:
‚ÄĘ are allergic to medicines that contain sulfa.‚ÄĘ have or have had liver problems, including hepatitis B or C virus infection.‚ÄĘ have kidney problems.‚ÄĘ have high blood sugar (diabetes).‚ÄĘ have hemophilia.‚ÄĘ have high cholesterol.‚ÄĘ are pregnant or plan to become pregnant. It is not known if LEXIVA will harm your unborn baby. Talk to your healthcare provider if you are pregnant or plan to become pregnant during treatment with LEXIVA.
‚ÄĘ LEXIVA may reduce how well hormonal contraceptives (birth control pills) work. Females who may become pregnant should use a different form of birth control or an additional barrier method of birth control during treatment with LEXIVA.¬† Pregnancy Registry. There is a pregnancy registry for women who take LEXIVA during pregnancy. The purpose of the registry is to collect information about the health of you and your baby. Talk to your healthcare provider about how you can take part in this registry.‚ÄĘ are breastfeeding or plan to breastfeed. Do not breastfeed if you take LEXIVA.
o You should not breastfeed if you have HIV-1 because of the risk of passing HIV-1 to your baby.o It is not known if LEXIVA can pass to your baby in your breast milk.o Talk with your healthcare provider about the best way to feed your baby.
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements.
Some medicines interact with LEXIVA. Keep a ul of your medicines to show your healthcare provider and pharmacist.
‚ÄĘ You can ask your healthcare provider or pharmacist for a ul of medicines that interact with LEXIVA.‚ÄĘ Do not start taking a new medicine without telling your healthcare provider. Your healthcare provider can tell you if it is safe to take LEXIVA with other medicines.
How should I take LEXIVA?
‚ÄĘ Take LEXIVA exactly as your healthcare provider tells you to take it.‚ÄĘ If you miss a dose of LEXIVA, take it as soon as you remember. Do not take 2 doses at the same time or take more than your healthcare provider tells you to take.‚ÄĘ Stay under the care of a healthcare provider during treatment with LEXIVA.‚ÄĘ If your child is taking LEXIVA, your child‚Äôs healthcare provider will decide the right dose based on your child‚Äôs weight.‚ÄĘ LEXIVA tablets may be taken with or without food.‚ÄĘ Adults should take LEXIVA oral suspension without food.‚ÄĘ Children should take LEXIVA oral suspension with food. If your child vomits within 30 minutes after taking a dose of LEXIVA, the dose should be repeated.‚ÄĘ Shake LEXIVA oral suspension well before each use.‚ÄĘ Do not run out of LEXIVA. The virus in your blood may increase and the virus may become harder to treat. When your supply starts to run low, get more from your healthcare provider or pharmacy.‚ÄĘ If you take too much LEXIVA, call your healthcare provider or go to the nearest hospital emergency room right away.
What are the possible side effects of LEXIVA?
LEXIVA may cause serious side effects including:
‚ÄĘ See ‚ÄúWhat is the most important information I should know about LEXIVA?‚ÄĚ‚ÄĘ Liver problems. Your healthcare provider should do blood tests before and during your treatment with LEXIVA to check your liver function. Some people with liver problems, including hepatitis B or C, may have an increased risk of developing worsening liver problems during treatment with LEXIVA.‚ÄĘ Diabetes and high blood sugar (hyperglycemia). Some people who take protease inhibitors, including LEXIVA, can get high blood sugar, develop diabetes, or your diabetes can get worse. Tell your healthcare provider if you notice an increase in thirst or urinate often during treatment with LEXIVA.‚ÄĘ Changes in your immune system (Immune Reconstitution Syndrome) can happen when you start taking HIV-1 medicines. Your immune system may get stronger and begin to fight infections that have been hidden in your body for a long time. Call your healthcare provider right away if you start having new symptoms after starting your HIV-1 medicine.‚ÄĘ Increase in body fat. An increase in body fat can happen in people who take protease inhibitors, including LEXIVA. The exact cause and long-term health effects of these conditions are not known.‚ÄĘ Changes in blood tests. Some people have changes in blood tests while taking LEXIVA. These include an increase in liver function tests, blood fat levels (cholesterol and triglycerides) and decrease in red blood cells. Your healthcare provider should do regular blood tests before and during your treatment with LEXIVA.‚ÄĘ Increased bleeding problems in some people with hemophilia. Some people with hemophilia have increased bleeding with protease inhibitors, including LEXIVA.‚ÄĘ Kidney stones. Some people have developed kidney stones during treatment with LEXIVA. Tell your healthcare provider right away if you develop any of the following signs or symptoms of kidney stones:
o pain in your sideo blood in your urineo pain when you urinate
The most common side effects of LEXIVA in adults include:
‚ÄĘ nausea‚ÄĘ vomiting‚ÄĘ rash
‚ÄĘ diarrhea‚ÄĘ headache
The most common side effects of LEXIVA in children include vomiting and decrease in white blood cells.
These are not all the possible side effects of LEXIVA.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store LEXIVA?
‚ÄĘ Store LEXIVA tablets at room temperature between 68¬įF to 77¬įF (20¬įC to 25¬įC).‚ÄĘ Keep the bottle of LEXIVA tablets tightly closed.‚ÄĘ Store LEXIVA oral suspension at room temperature or in the refrigerator between 41¬įF to 86¬įF (5¬įC to 30¬įC). Refrigeration of LEXIVA oral suspension may improve taste for some people.‚ÄĘ Do not freeze.‚ÄĘ LEXIVA comes in a child-resistant package.
Keep LEXIVA and all medicines out of the reach of children.
General information about the safe and effective use of LEXIVA.
Medicines are sometimes prescribed for purposes other than those uled in a Patient Information leaflet. Do not use LEXIVA for a condition for which it was not prescribed. Do not give LEXIVA to other people, even if they have the same symptoms you have. It may harm them. You can ask your pharmacist or healthcare provider for information about LEXIVA that is written for health professionals.
What are the ingredients in LEXIVA?
Active ingredient: fosamprenavir calcium
Inactive ingredients:
Tablets: colloidal silicon dioxide, croscarmellose sodium, magnesium stearate, microcrystalline cellulose, and povidone K30. The tablet film-coating contains the inactive ingredients hypromellose, iron oxide red, titanium dioxide, and triacetin.
Oral Suspension: artificial grape-bubblegum flavor, calcium chloride dihydrate, hypromellose, methylparaben, natural peppermint flavor, polysorbate 80, propylene glycol, propylparaben, purified water, and sucralose.
Manufactured for:
ViiV Healthcare
Research Triangle Park, NC 27709
Trademark is owned by or licensed to the ViiV Healthcare group of companies.
©2020 ViiV Healthcare group of companies or its licensor.
LXV:23PIL
For more information call 877-844-8872.
This Patient Information has been approved by the U.S. Food and Drug Administration.
Revised: 10/2020
Package Label.principal Display Panel
PRINCIPAL DISPLAY PANEL
NDC 49702-208-53
LEXIVA
(fosamprenavir calcium)
Oral Suspension
50 mg/mL
ViiV Healthcare
SHAKE VIGOROUSLY BEFORE USING.
ALERT: Find out about medicines that should NOT be taken with LEXIVA.
225 mL
Rx only
Made in Canada
©2020 ViiV Healthcare group of companies or its licensor.
  62000000056125 Rev. 10/20
Package Label.principal Display Panel
PRINCIPAL DISPLAY PANEL
NDC 49702-207-18
LEXIVA
(fosamprenavir calcium)
TABLETS
700 mg
60 Tablets
ALERT: Find out about medicines that should NOT be taken with LEXIVA.
Note to Pharmacist: Do not cover ALERT box with pharmacy label.
Rx only
For use in combination regimens with or without ritonavir.
Each tablet contains 700 mg of fosamprenavir as fosamprenavir calcium. 
This package is child-resistant. Keep out of reach of children.
Store at controlled room temperature of 25ňöC (77ňöF); excursions permitted to 15ňö to 30ňöC (59ňö to 86ňöF) (see USP Controlled Room Temperature).
Keep bottle tightly closed. 
See prescribing information for dosage information. 
Trademarks owned or licensed by ViiV Healthcare.
©2020 ViiV Healthcare or licensor.
Manufactured for:  
ViiV Healthcare  
Research Triangle Park, NC 27709 
Made in Belgium
Rev. 11/20
62000000055505
DISCLAIMER:
"This tool does not provide medical advice, and is for informational and educational purposes only, and is not a substitute for professional medical advice, treatment or diagnosis. Call your doctor to receive medical advice. If you think you may have a medical emergency, please dial 911."
"Do not rely on openFDA to make decisions regarding medical care. While we make every effort to ensure that data is accurate, you should assume all results are unvalidated. We may limit or otherwise restrict your access to the API in line with our Terms of Service."
"This product uses publicly available data from the U.S. National Library of Medicine (NLM), National Institutes of Health, Department of Health and Human Services; NLM is not responsible for the product and does not endorse or recommend this or any other product."
PillSync may earn a commission via links on our site
