Xcopri Dailymed
Generic: cenobamate is used for the treatment of Epilepsies, Partial

Go PRO for all pill images
Recent Major Changes Section
Dosage and Administration, Administration Instructions (2.3 )          4/2024
1 Indications And Usage
XCOPRI is indicated for the treatment of partial-onset seizures in adult patients.
XCOPRI is indicated for the treatment of partial-onset seizures in adult patients. (1 )
2 Dosage And Administration
- The recommended initial dosage of XCOPRI is 12.5 mg once daily, titrated to the recommended maintenance dosage of 200 mg once daily. The recommended titration schedule should not be exceeded. The maximum dosage is 400 mg once daily. (
2.1 )- Hepatic impairment: For patients with mild or moderate hepatic impairment, the maximum recommended dosage is 200 mg once daily. (
2.2 ,8.7 ,12.3 )- XCOPRI can be taken whole or the tablets can be crushed. The crushed tablet can be mixed with water and either administered by mouth as an oral suspension or administered via a nasogastric tube. (
2.3 )2.1 General Dosing Recommendations
Monotherapy and Adjunctive Therapy
XCOPRI is administered orally once daily with or without food. The recommended dosage and titration, which should not be exceeded because of the potential for serious adverse reactions [see Warnings and Precautions (5.2)], is included in Table 1.
Table 1: Recommended Dosage for Partial-Onset Seizures in Adults  Initial Dosage     Week 1 and 2  12.5 mg once daily  Titration Regimen     Week 3 and 4  25 mg once daily     Week 5 and 6  50 mg once daily     Week 7 and 8  100 mg once daily     Week 9 and 10  150 mg once daily  Maintenance Dosage     Week 11 and thereafter  200 mg once daily  Maximum Dosage     If needed based on clinical response and     tolerability, dose may be increased above 200 mg     by increments of 50 mg once daily every two     weeks to 400 mg.  400 mg once daily 2.2 Dosage Modifications in Patients with Hepatic Impairment
For patients with mild to moderate (Child-Pugh Class A to B) hepatic impairment, the maximum recommended dosage is 200 mg once daily [see Use in Specific Populations (8.7)]. XCOPRI is not recommended for use in patients with severe (Child-Pugh Class C) hepatic impairment [see Clinical Pharmacology (12.3)].
2.3 Administration Instructions
XCOPRI can be taken whole or the tablets can be crushed. The crushed tablet can be mixed with water and either administered by mouth as an oral suspension or administered via a nasogastric tube, as described below [see Clinical Pharmacology (12.3)].
Administration of Crushed Tablets by Mouth as Oral Suspension
Crush the appropriate number of tablet(s) for the prescribed dose. In a cup, combine the crushed tablet(s) and 25 mL of water. Swirl to suspend the crushed tablet(s). Drink the suspension immediately. Do not store the tablet-water mixture for later use. To ensure no tablet residue is left in the container, rinse the container with 25 mL of water and drink. Visually confirm that no particles are left in the container. If particles remain, repeat step 5.
Administration of Crushed Tablets via Nasogastric (NG) Tube
Crush the appropriate number of tablet(s) for the prescribed dose. In an appropriate container, combine the crushed tablet(s) and 25 mL of water. Swirl to suspend the crushed tablet(s). Ensuring no particles are left in the container, instill the suspension with a syringe into the NG tube. Refill the catheter-tip syringe again with 10 mL of water, swirl gently, and administer. Visually confirm that no particles are left in the syringe. If particles remain, repeat step 5. 2.4 Discontinuation of XCOPRI
If XCOPRI is discontinued, the dosage should be gradually reduced over a period of at least 2 weeks, unless safety concerns require abrupt withdrawal [see Warnings and Precautions (5.1, 5.5)].
3 Dosage Forms And Strengths
XCOPRI tablets are available in the following strengths, shapes, colors, and tablet markings (Table 2).
Table 2: TRADENAME Tablet Presentations Tablet Strength Tablet Color/Shape Tablet Markings 12.5 mg Uncoated round white to off-white tablets SK on one side and 12 on the other side 25 mg Film coated round brown tablets SK on one side and 25 on the other side 50 mg Film coated round yellow tablets SK on one side and 50 on the other side 100 mg Film coated round brown tablets SK on one side and 100 on the other side 150 mg Film coated round light orange tablets SK on one side and 150 on the other side 200 mg Film coated modified oval light orange tablets SK on one side and 200 on the other side
- Tablets: 12.5 mg, 25 mg, 50 mg, 100 mg, 150 mg, and 200 mg. (
3 )
4 Contraindications
XCOPRI is contraindicated in patients with:
- Hypersensitivity to cenobamate or any of the inactive ingredients in XCOPRI. (
4 )- Familial Short QT syndrome. (
4 )
5 Warnings And Precautions
- Drug Reaction with Eosinophilia and Systemic Symptoms (DRESS)/Multi-Organ Hypersensitivity: Discontinue if no alternate etiology. (
5.1 )- QT Shortening: Use caution when administering XCOPRI with other drugs that shorten the QT interval (
5.2 )- Suicidal Behavior and Ideation: Monitor patients for suicidal behavior and ideation. (
5.3 )- Neurological Adverse Reactions: Monitor for somnolence and fatigue and advise patients not to drive or operate machinery until they have gained sufficient experience on XCOPRI. Concomitant use with other CNS depressants or alcohol may have additive effects. (
5.4 )- Withdrawal of Antiepileptic Drugs: XCOPRI should be gradually withdrawn to minimize the potential of increased seizure frequency. (
5.5 )5.1Drug Reaction with Eosinophilia and Systemic Symptoms (DRESS)/Multiorgan Hypersensitivity
Drug Reaction with Eosinophilia and Systemic Symptoms (DRESS), also known as multiorgan hypersensitivity, has been reported in patients taking XCOPRI. DRESS has occurred, including one fatality, when XCOPRI was titrated rapidly (weekly or faster titration). No cases of DRESS were reported in an open-label safety study of 1339 partial-onset seizure patients when XCOPRI was initiated at 12.5 mg once daily and titrated every two weeks. This finding does not establish that the risk of DRESS is prevented by a slower titration; however, XCOPRI should be initiated at 12.5 mg once daily and titrated every two weeks [see Dosage and Administration (2.2)]. DRESS typically, although not exclusively, presents with fever, rash, lymphadenopathy, and/or facial swelling, in association with other organ system involvement, such as hepatitis, nephritis, hematological abnormalities, myocarditis, or myositis sometimes resembling an acute viral infection. Eosinophilia is often present. This disorder is variable in its expression, and other organ systems not noted here may be involved. It is important to note that early manifestations of hypersensitivity, such as fever or lymphadenopathy, may be present even though rash is not evident. If such signs or symptoms are present, the patient should be evaluated immediately. XCOPRI should be discontinued immediately and not restarted if an alternative etiology for the signs or symptoms cannot be established [see Contraindications (4)].
5.2 QT Shortening
In a placebo-controlled study of the QT interval, a higher percentage of subjects who took XCOPRI (31% at 200 mg and 66% at 500 mg) had a QT shortening of greater than 20 msec compared to placebo (6-17%). Reductions of the QTc interval below 300 msec were not observed [see Clinical Pharmacology (12.2)]. Familial Short QT syndrome is associated with an increased risk of sudden death and ventricular arrhythmias, particularly ventricular fibrillation. Such events in this syndrome are believed to occur primarily when the corrected QT interval falls below 300 msec. Nonclinical data also indicate that QT shortening is associated with ventricular fibrillation. Patients with Familial Short QT syndrome should not be treated with XCOPRI [see Contraindications (4)]. Caution should be used when administering XCOPRI and other drugs that shorten the QT interval as there may be a synergistic effect on the QT interval that would increase the QT shortening risk.
5.3 Suicidal Behavior and Ideation
Antiepileptic drugs (AEDs), including XCOPRI, increase the risk of suicidal thoughts or behavior in patients taking these drugs for any indication. Patients treated with any AED for any indication should be monitored for the emergence or worsening of depression, suicidal thoughts or behavior, and/or any unusual changes in mood or behavior.
Pooled analyses of 199 placebo-controlled clinical trials (mono- and adjunctive therapy) of 11 different AEDs showed that patients randomized to one of the AEDs had approximately twice the risk (adjusted Relative Risk 1.8, 95% CI:1.2, 2.7) of suicidal thinking or behavior compared to patients randomized to placebo. In these trials, which had a median treatment duration of 12 weeks, the estimated incidence rate of suicidal behavior or ideation among 27,863 AED-treated patients was 0.43%, compared to 0.24% among 16,029 placebo-treated patients, representing an increase of approximately one case of suicidal thinking or behavior for every 530 patients treated. There were four suicides in drug-treated patients in the trials and none in placebo-treated patients, but the number is too small to allow any conclusion about drug effect on suicide.
The increased risk of suicidal thoughts or behavior with AEDs was observed as early as one week after starting drug treatment with AEDs and persisted for the duration of treatment assessed. Because most trials included in the analysis did not extend beyond 24 weeks, the risk of suicidal thoughts or behavior beyond 24 weeks could not be assessed.
The risk of suicidal thoughts or behavior was generally consistent among drugs in the data analyzed. The finding of increased risk with AEDs of varying mechanisms of action and across a range of indications suggests that the risk applies to all AEDs used for any indication. The risk did not vary substantially by age (5-100 years) in the clinical trials analyzed.
Table 3 shows absolute and relative risk by indication for all evaluated AEDs.
Table 3: Risk of Suicidal Thoughts or Behaviors by Indication for Antiepileptic Drugs in the Pooled Analysis Indication Placebo Patients with Events Per 1000 Patients Drug Patients with Events Per 1000 Patients Relative Risk: Incidence of Events in Drug Patients/Incidence in Placebo Patients Risk Differences: Additional Drug Patients with Events Per 1000 Patients Epilepsy 1.0 3.4 3.5 2.4 Psychiatric 5.7 8.5 1.5 2.9 Other 1.0 1.8 1.9 0.9 Total 2.4 4.3 1.8 1.9
The relative risk for suicidal thoughts or behavior was higher in clinical trials in patients with epilepsy than in clinical trials in patients with psychiatric or other conditions, but the absolute risk differences were similar for epilepsy and psychiatric indications.
Anyone considering prescribing XCOPRI or any other AED must balance this risk with the risk of untreated illness. Epilepsy and many other illnesses for which AEDs are prescribed are themselves associated with morbidity and mortality and an increased risk of suicidal thoughts and behavior. Should suicidal thoughts and behavior emerge during treatment, the prescriber needs to consider whether the emergence of these symptoms in any given patient may be related to the illness being treated.
5.4 Neurological Adverse Reactions
Somnolence and Fatigue
XCOPRI causes dose-dependent increases in somnolence and fatigue-related adverse reactions (somnolence, fatigue, asthenia, malaise, hypersomnia, sedation, and lethargy) [see Adverse Reactions (6.1)]. In Study 1 and Study 2, 31% of patients randomized to receive XCOPRI at 100 mg/day, 36% of patients randomized to receive XCOPRI at 200 mg/day, and 57% of patients randomized to receive XCOPRI at 400 mg/day reported at least one of these adverse reactions, compared to 19% of patients who received placebo. Somnolence and fatigue-related adverse reactions were serious in 0.4% of XCOPRI-treated patients compared to no patients who received placebo and led to discontinuation in 2% of XCOPRI-treated patients compared to 1% of patients who received placebo.
Dizziness and Disturbance in Gait and Coordination
XCOPRI causes dose-dependent adverse reactions related to dizziness and disturbance in gait and coordination (dizziness, vertigo, balance disorder, ataxia, nystagmus, gait disturbance, and abnormal coordination) [see Adverse Reactions (6.1)]. In Study 1 and Study 2, 21% of patients randomized to receive XCOPRI at 100 mg/day, 31% of patients randomized to receive XCOPRI at 200 mg/day, and 52% of patients randomized to receive XCOPRI at 400 mg/day reported at least one of these adverse reactions, compared to 18% of patients who received placebo. Dizziness and disturbance in gait and coordination adverse reactions were serious in 2% of XCOPRI-treated patients compared to no patients who received placebo and led to discontinuation in 5% of XCOPRI-treated patients compared to 1% of patients who received placebo.
Cognitive Dysfunction
XCOPRI causes adverse reactions related to cognitive dysfunction related-events (i.e., memory impairment, disturbance in attention, amnesia, confusional state, aphasia, speech disorder, slowness of thought, disorientation, and psychomotor retardation) [see Adverse Reactions (6.1)]. In Study 1 and Study 2, 6% of patients randomized to receive XCOPRI at 100 mg/day, 6% of patients randomized to receive XCOPRI at 200 mg/day, and 9% of patients randomized to receive XCOPRI at 400 mg/day reported at least one of these adverse reactions, compared to 2% of patients who received placebo. No cognitive dysfunction-related events were serious in XCOPRI-treated patients or in patients who received placebo. Cognitive dysfunction related adverse reactions led to discontinuation in 0.4% of XCOPRI-treated patients compared to no patients who received placebo.
Visual Changes
XCOPRI causes adverse reactions related to visual changes including diplopia, blurred vision, and impaired vision [see Adverse Reactions (6.1)]. In Study 1 and Study 2, 9% of patients randomized to receive XCOPRI at 100 mg/day, 9% of patients randomized to receive XCOPRI at 200 mg/day, and 18% of patients randomized to receive XCOPRI at 400 mg/day reported at least one of these adverse reactions, compared to 2% of patients who received placebo. No visual change-related events were serious in XCOPRI-treated patients or in patients who received placebo. Visual change led to discontinuation in 0.5% of XCOPRI-treated patients compared to no patients who received placebo.
Risk Amelioration
Prescribers should advise patients against engaging in hazardous activities requiring mental alertness, such as operating motor vehicles or dangerous machinery, until the effect of XCOPRI is known. Patients should be carefully observed for signs of central nervous system (CNS) depression, such as somnolence and sedation, when XCOPRI is used with other drugs with sedative properties because of potential additive effects.
5.5 Withdrawal of Antiepileptic Drugs
As with most antiepileptic drugs, XCOPRI should generally be withdrawn gradually because of the risk of increased seizure frequency and status epilepticus [see Dosage and Administration (2.4) and Clinical Studies (14)]. But if withdrawal is needed because of a serious adverse event, rapid discontinuation can be considered.
6 Adverse Reactions
The following serious adverse reactions are described in more detail in the Warnings and Precautions section of the labeling:
- Drug Reaction with Eosinophilia and Systemic Symptoms (DRESS)/Multiorgan Hypersensitivity [see Warnings and Precautions (5.1)]
- QT Shortening [see Warnings and Precautions (5.2)]
- Suicidal Behavior and Ideation [see Warnings and Precautions (5.3)]
- Neurological Adverse Reactions [see Warnings and Precautions (5.4)]
- Withdrawal of Antiepileptic Drugs [see Warnings and Precautions (5.5)]
The most common adverse reactions in patients receiving XCOPRI (at least 10% for XCOPRI and more frequently than placebo) include somnolence, dizziness, fatigue, diplopia, and headache. (6.1 )
To report SUSPECTED ADVERSE REACTIONS, contact SK Life Science, Inc. at 1-866-657-5574 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions and for varying durations, adverse reaction frequencies observed in the clinical trials of a drug cannot be directly compared with frequencies in the clinical trials of another drug and may not reflect the frequencies observed in practice.
In all controlled and uncontrolled trials performed in adult partial-onset seizure patients, XCOPRI was administered as adjunctive therapy to 1944 patients. Of these patients, 1575 were treated for at least 6 months, 710 for at least 12 months, 349 for at least 24 months, and 320 for at least 36 months. A total of 658 patients (442 patients treated with XCOPRI and 216 patients treated with placebo) constituted the safety population in the pooled analysis of placebo-controlled studies in patients with partial-onset seizures (Studies 1 and 2) [see Clinical Studies (14)]. The adverse reactions presented in Table 4 are based on this safety population; the median length of treatment in these studies was 18 weeks. Of the patients in those studies, approximately 49% were male, 76% were Caucasian, and the mean age was 39 years.
In Study 1 and Study 2, adverse events occurred in 77% of patients treated with XCOPRI and 68% treated with placebo. Table 4 gives the incidence of adverse reactions that occurred in subjects with partial-onset seizures in any XCOPRI treatment group and for which the incidence was greater than placebo during the controlled clinical trials. The most common adverse reactions that occurred in XCOPRI-treated patients (incidence at least 10% and greater than placebo) were somnolence, dizziness, fatigue, diplopia, and headache.
The discontinuation rates because of adverse events were 11%, 9%, and 21% for patients randomized to receive XCOPRI at doses of 100 mg/day, 200 mg/day, and 400 mg/day, respectively, compared to 4% in patients randomized to receive placebo. The adverse reactions most commonly (1% or greater in any XCOPRI treatment group, and greater than placebo) leading to discontinuation, in descending order of frequency, were ataxia, dizziness, somnolence, diplopia, nystagmus, and vertigo.
Table 4: Adverse Reactions in Pooled Placebo-Controlled Adjunctive Therapy Studies in Patients with Partial-Onset Seizures with XCOPRI Frequency in Any Treatment Arm Greater Than 1% Over Placebo * Reported as an adverse reaction; see Laboratory Abnormalities for ALT changes from collected laboratory values Adverse Reaction XCOPRI Placebo 100mg 200mg 400mg n = 108% n= 223% n=111% n=216% Cardiac Disorders Palpitations 0 0 2 0 Ear and Labyrinth Disorders Vertigo 1 1 6 1 Eye Disorders Diplopia 6 7 15 2 Vision Blurred 2 2 4 0 Gastrointestinal Disorders Nausea 6 6 9 3 Constipation 2 4 8 0 Diarrhea 1 3 5 0 Vomiting 2 4 5 0 Dry Mouth 1 1 3 0 Abdominal Pain 2 2 1 0 Dyspepsia 2 2 0 0 Infections and Infestations Nasopharyngitis 2 4 5 3 Pharyngitis 1 2 0 0 Urinary Tract Infection 2 5 0 2 Injury, Poisoning and Procedural Complications Head Injury 1 0 2 0 Investigations Alanine Aminotransferase Increased* 1 1 4 0 Aspartate Aminotransferase Increased 1 1 3 0 Weight Decreased 2 0 1 0 Metabolism and Nutrition Disorders Decreased Appetite 3 1 5 1 Musculoskeletal and Connective Tissue Disorders Back Pain 4 2 5 3 Musculoskeletal Chest Pain 2 1 0 0 Nervous System Disorders Somnolence 19 22 37 11 Dizziness 18 22 33 15 Fatigue 12 14 24 7 Headache 10 12 10 9 Balance Disorder 3 5 9 1 Gait Disturbance 1 3 8 1 Dysarthria 2 1 7 0 Nystagmus 3 7 6 0 Ataxia 2 3 6 2 Aphasia 2 1 4 0 Asthenia 0 1 3 1 Dysgeusia 2 0 2 0 Memory Impairment 2 1 2 0 Migraine 0 0 2 0 Sedation 1 1 2 0 Tremor 0 3 1 1 Psychiatric Disorders Confusional State 2 2 3 0 Euphoric Mood 0 0 2 0 Irritability 1 0 2 0 Suicidal Ideation 2 1 0 0 Renal and Urinary Disorders Pollakiuria 0 1 0 0 Reproductive System and Breast Disorders Dysmenorrhea 1 2 1 0 Respiratory, Thoracic and Mediastinal Disorders Hiccups 0 1 1 0 Dyspnea 0 3 0 0 Skin and Subcutaneous Tissue Disorders Pruritus 2 1 0 0 Rash Papular 2 0 0 0
Laboratory Abnormalities
Hepatic Transaminases
In Study 2, there was a post-baseline elevation of alanine aminotransferase (ALT) to greater than 3 times the upper limit of normal (ULN) in 1 (0.9%) patient treated with 100 mg XCOPRI, 2 (1.8%) patients treated with 200 mg, and 3 (2.7%) patients treated with 400 mg, compared to no patients who took placebo. The maximum ALT elevation was 7.6 times ULN in patients treated with 400 mg XCOPRI.
Potassium
In clinical studies, there was a post-baseline elevation of potassium values greater than 5 meq/L (upper reference range) in patients treated with XCOPRI. In Study 1, there were 17 (17%) patients treated with XCOPRI 200 mg compared to 8 (7%) patients who took placebo with normal baseline potassium values who had at least one post-baseline maximum value greater than 5 meq/L. In Study 2, there was a dose-related distribution where at least one post-baseline potassium value was greater than 5 meq/L, occurring in 8.3%, 9.1%, and 10.8% of the patients treated with XCOPRI 100 mg, 200 mg, and 400 mg, respectively, compared to 5.6% of patients who took placebo. Two patients had a maximum potassium value of 5.9 meq/L.
Other Adverse Reactions
Gastrointestinal disorders: There was an incidence of appendicitis in the overall clinical trial safety population of 2.9 cases of appendicitis/1000 patient-years of exposure that is in excess of the expected background rate in the general population.
Adverse Reactions Based on Gender
No significant gender differences were noted in the incidence of adverse reactions.
6.2 Postmarketing Experience
The following adverse reactions have been identified during post approval use of XCOPRI. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Psychiatric Disorders: Psychosis (hallucinations, delusions/paranoia), hostility, aggression.
Hepatobiliary Disorders: Hepatic failure
7 Drug Interactions
- Phenytoin: Gradually decrease phenytoin dosage by up to 50% (
7.1 )- Phenobarbital and Clobazam: Reduce dosage as needed when used concomitantly with XCOPRI. (
7.1 )- Lamotrigine, Carbamazepine: Increase dosage as needed when used concomitantly with XCOPRI. (
7.1 )- CYP2B6 and CYP3A Substrates: Increase dosage as needed when used concomitantly with XCOPRI. (
7.1 )- CYP2C19 Substrates: Reduce dosage as needed when used concomitantly with XCOPRI. (
7.1 )- Oral Contraceptives: Effectiveness of hormonal oral contraceptives may be reduced when administered concomitantly with XCOPRI. Women should use additional or alternative non-hormonal birth control. (
7.1 )7.1Effect of XCOPRI on Other Drugs
Table 5 summarizes the effect of XCOPRI on other drugs [see Clinical Pharmacology (12.3)].
Table 5: Pharmacokinetic Drug Interactions Drug or Substrate Type Effect of XCOPRI on Drug or Substrate Clinical Recommendation Antiepileptic Drugs lamotrigine ‚Üď plasma concentrations Because of a potential for reduced efficacy of these drugs, increase the dosage of lamotrigine or carbamazepine, as needed, when used concomitantly with XCOPRI. carbamazepine ‚Üď plasma concentrations phenytoin ‚ÜĎ plasma concentrations Because of a potential 2-fold increase in phenytoin levels, gradually decrease phenytoin dosage by up to 50% as XCOPRI is being titrated. phenobarbital ‚ÜĎ plasma concentrations Because of a potential for an increase in the risk of adverse reactions from these drugs, consider a reduction in dosage of phenobarbital or clobazam, as clinically appropriate, when used concomitantly with XCOPRI. desmethylclobazam, the active metabolite of clobazam ‚ÜĎ plasma concentrations ¬† CYP2B6 Substrates ‚Üď plasma concentrations Because of a potential for reduced efficacy of these drugs, increase the dosage of CYP2B6 or CYP3A4 substrates, as needed, when used concomitantly with XCOPRI. CYP3A Substrates ‚Üď plasma concentrations Oral contraceptives ‚Üď plasma concentrations Because of the potential for reduced efficacy of oral contraceptives, women should use additional or alternative non-hormonal birth control while taking XCOPRI. ¬† CYP2C19 Substrates ‚ÜĎ plasma concentrations Because of a potential for an increase in the risk of adverse reactions from these drugs, consider a reduction in dosage of CYP2C19 substrates, as clinically appropriate, when used concomitantly with XCOPRI. 7.2Drug that Shorten the QT Interval
XCOPRI can shorten the QT interval; therefore, caution should be used when administering XCOPRI and other drugs that shorten the QT interval [see Warnings and Precautions (5.2) and Clinical Pharmacology (12.2)].
7.3 CNS Depressants and Alcohol
Concomitant use of XCOPRI with other CNS depressants, including alcohol, may increase the risk of neurological adverse reactions, including sedation and somnolence [see Warnings and Precautions (5.4)].
8 Use In Specific Populations
- Pregnancy: Based on animal data, may cause fetal harm. (
8.1 )- Renal Impairment: Use with caution and dosage reduction may be considered in patients with mild to moderate (CLcr 30 to < 90 mL/min) and severe (CLcr < 30 mL/min) renal impairment. Use not recommended in end-stage renal disease (CLcr < 15 mL/min) undergoing dialysis. (
8.6 )- Hepatic Impairment: Use with caution in patients with mild to moderate hepatic impairment; lower maximum dosage and additional dosage reduction may be considered. Use of XCOPRI in patients with severe hepatic impairment is not recommended. (
2.2 ,8.7 ,12.3 )8.1 Pregnancy
Pregnancy Exposure Registry
There is a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to antiepileptic drugs (AEDs), such as XCOPRI, during pregnancy. Encourage women who are taking XCOPRI during pregnancy to enroll in the North American Antiepileptic Drug (NAAED) Pregnancy Registry by calling the toll-free number 1-888-233-2334 or visiting http://www.aedpregnancyregistry.org/.
Risk Summary
There are no adequate data on the developmental risk associated with the use of XCOPRI in pregnant women.
In animal studies, administration of cenobamate during pregnancy or throughout pregnancy and lactation resulted in adverse effects on development (increased embryofetal mortality, decreased fetal and offspring body weights, neurobehavioral and reproductive impairment in offspring) at clinically relevant drug exposures [see Data].
In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively. The background risk of major birth defects and miscarriage for the indicated population is unknown.
Data
Animal Data
Oral administration of cenobamate (0, 10, 30, or 60 mg/kg/day) to pregnant rats during the period of organogenesis resulted in increased embryofetal mortality, reduced fetal body weights, and incomplete fetal skeletal ossification at the highest dose tested, which was associated with maternal toxicity. There was a small increase in visceral malformations at the high dose; however, teratogenic potential could not be fully evaluated because of the high rate of embryofetal deaths, which resulted in an inadequate number of fetuses examined. Maternal plasma exposure (AUC) at the no-effect dose for adverse effects on embryofetal development (30 mg/kg/day) was less than that in humans at the maximum recommended human dose (MRHD) of 400 mg.
Oral administration of cenobamate (0, 4, 12, or 36 mg/kg/day) to pregnant rabbits during the period of organogenesis resulted in increased embryofetal mortality at the highest dose tested, which was associated with maternal toxicity. Maternal plasma exposure at the no-effect dose (12 mg/kg/day) for adverse effects on embryofetal development was less than that in humans at the MRHD.
When cenobamate (0, 11, 22, or 44 mg/kg/day) was orally administered to female rats throughout pregnancy and lactation, neurobehavioral impairment (learning and memory deficit and increased auditory startle response) was observed in the offspring at all doses and decreased preweaning body weight gain and adverse effects on reproductive function (decreased numbers of corpora lutea, implantations, and live fetuses) were seen in the offspring at the high dose. Maternal plasma exposure at the lowest effect dose (11 mg/kg/day) for adverse effects on pre- and postnatal development was less than that in humans at the MRHD.
8.2 Lactation
Risk Summary
There are no data available on the presence of cenobamate in human milk, the effects on the breastfed infant, or the effects of the drug on milk production. Cenobamate was present in rat milk at concentrations similar to those in maternal plasma.
The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for XCOPRI and any potential adverse effects on the breastfed infant from XCOPRI or from the underlying maternal condition.
8.3 Females and Males of Reproductive Potential
Contraception
Women of reproductive potential concomitantly using oral contraceptives should use additional or alternative non-hormonal birth control [see Drug Interactions (7.1) and Clinical Pharmacology (12.3)].
8.4 Pediatric Use
Safety and effectiveness in pediatric patients have not been established.
Juvenile Animal Toxicity Data
Cenobamate was administered orally to juvenile rats from postnatal day (PND) 7 to 70. To maintain consistent plasma drug exposures, doses were increased during the dosing period, up to 120 and 80 mg/kg/day in males and females, respectively. Adverse effects included mortality, delayed sexual maturation, neurological (decreased grip strength) and neurobehavioral (learning and memory deficits) impairment, decreased sperm count, decreased brain weight, and ocular histopathology. Recovery from these effects was observed following discontinuation of dosing. Overall, a no-effect dose for adverse effects on postnatal development was not identified. At the lowest doses tested, plasma cenobamate exposures (AUC) were less than that in humans at the maximum recommended human dose (MRHD) of 400 mg.
8.5 Geriatric Use
Clinical studies of XCOPRI did not include sufficient numbers of patients aged 65 and over to determine the safety and efficacy of XCOPRI in the elderly population. In general, dose selection for an elderly patient should be cautious, usually starting at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy [see Clinical Pharmacology (12.3)].
8.6 Renal Impairment
XCOPRI should be used with caution and dosage reduction may be considered in patients with mild to moderate (CLcr 30 to less than 90 mL/min) and severe (CLcr less than 30 mL/min) renal impairment. Use in patients with end-stage renal disease undergoing dialysis is not recommended [see Clinical Pharmacology (12.3)].
8.7 Hepatic Impairment
XCOPRI should be used with caution in patients with mild to moderate (5-9 points on Child-Pugh assessment; Class A or B) hepatic impairment. In these patients, the maximum recommended dosage is 200 mg once daily and additional dosage reduction may be considered [see Dosage and Administration (2.2) and Clinical Pharmacology (12.3)]. Use of XCOPRI in patients with severe (10-15 points on Child-Pugh assessment; Class C) hepatic impairment is not recommended.
9 Drug Abuse And Dependence
9.1 Controlled Substance
XCOPRI contains cenobamate and is uled as a Schedule V controlled substance.
9.2 Abuse
Abuse is the intentional, non-therapeutic use of a drug, even once, for its desirable psychological or physiological effects. In a human abuse potential study conducted in recreational sedative abusers (n=39), single doses of XCOPRI (200 mg and 400 mg) were compared to placebo. XCOPRI at single doses of 400 mg produced responses on positive subjective measures such as ‚ÄúDrug Liking,‚ÄĚ ‚ÄúOverall Drug Liking,‚ÄĚ ‚ÄúTake Drug Again,‚ÄĚ and ‚ÄúGood Drug Effects‚ÄĚ that were statistically greater than the responses produced on these measures by placebo. In this study, euphoric mood occurred at greater extent with XCOPRI (400 mg) (8%) than with placebo (0%). Phase 1 multiple ascending dose studies in healthy subjects showed rates of euphoria and feeling drunk of about 3% and disturbance in attention of about 5% in subjects who received supratherapeutic doses of cenobamate, but these adverse events were absent in the placebo group. In Phase 2 and 3 studies in subjects with epilepsy, euphoric mood, confusional state, and sedation occurred at low rates in subjects who received XCOPRI (0.5-2.5%).
9.3 Dependence
Physical dependence is a state that develops as a result of physiological adaptation in response to repeated drug use, manifested by withdrawal signs and symptoms after abrupt discontinuation or a significant dose reduction of a drug. Clinical studies in healthy subjects indicate that XCOPRI may cause physical dependence and lead to a withdrawal syndrome characterized by insomnia, decreased appetite, depressed mood, tremor, and amnesia. XCOPRI should be withdrawn gradually [see Warnings and Precautions (5.5)].
10 Overdosage
There is limited clinical experience with XCOPRI overdose in humans.
There is no specific antidote for overdose with XCOPRI. In the event of overdose, standard medical practice for the management of any overdose should be used. An adequate airway, oxygenation and ventilation should be ensured; monitoring of cardiac rate and rhythm and vital signs is recommended. A certified poison control center should be contacted for updated information on the management of overdose with XCOPRI. There are no data on the removal of XCOPRI using dialysis.
11 Description
The chemical name of XCOPRI (cenobamate) is [(1R)-1-(2-Chlorophenyl)-2-(tetrazol-2-yl) ethyl] carbamate. Its molecular formula is C10H10ClN5O2 and its molecular weight is 267.67 g/mol. The chemical structure is:
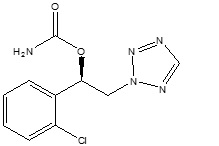
Cenobamate is a white to off-white crystalline powder. It is slightly soluble in aqueous solutions (water 1.7 mg/mL) and has higher solubility in organic solvents like ethanol (209.4 mg/mL).
XCOPRI tablets are for oral administration and contain the following inactive ingredients: colloidal silicon dioxide, lactose monohydrate, magnesium stearate, microcrystalline cellulose, and sodium starch glycolate and film coating agents specified below:
12.5 mg tablets: Not applicable, since 12.5 mg tablets are uncoated.
25 mg and 100 mg tablets: FD&C Blue# 2/indigo carmine aluminum lake, iron oxide red, iron oxide yellow, polyethylene glycol 3350, polyvinyl alcohol-part hydrolyzed, talc, and titanium dioxide.
50 mg tablets: iron oxide yellow, polyethylene glycol 3350, polyvinyl alcohol-part hydrolyzed, talc, and titanium dioxide.
150 mg and 200 mg tablets: iron oxide red, iron oxide yellow, polyethylene glycol 3350, polyvinyl alcohol-part hydrolyzed, talc, and titanium dioxide.
12 Clinical Pharmacology
12.1 Mechanism of Action
The precise mechanism by which cenobamate exerts its therapeutic effects in patients with partial-onset seizures is unknown. Cenobamate has been demonstrated to reduce repetitive neuronal firing by inhibiting voltage-gated sodium currents. It is also a positive allosteric modulator of the ő≥-aminobutyric acid (GABAA) ion channel.
12.2 Pharmacodynamics
Interactions with Alcohol
No clinically significant differences on objective attention, psychomotor performance, and memory tests, in addition to other subjective CNS tests, were observed following concomitant use of XCOPRI and ethanol (preparation of 40% ethanol in orange juice dosed at 0.7 g/kg for males and 0.57 g/kg for females) in healthy subjects.
Cardiac Electrophysiology
In a placebo-controlled QT study in healthy volunteers, dose-dependent shortening of the QTcF interval has been observed with XCOPRI [see Warnings and Precautions (5.2)]. The mean őĒőĒQTc is -11 [-13, -8] msec for 200 mg once daily and -18 [-22, -15] msec for 500 mg once daily (1.25 times the maximum recommended dosage). A higher percentage of XCOPRI-treated subjects (31% at 200 mg and 66% at 500 mg) had a QT shortening of greater than 20 msec compared to placebo (6-17%). Reductions of the QTc interval below 300 msec were not observed.
12.3 Pharmacokinetics
Cenobamate AUC increases in a greater than dose-proportional manner following single oral doses from 5 to 750 mg (0.0125 to 1.88 times the maximum recommended dosage). Cenobamate Cmax increases in a dose proportional manner. Steady-state plasma concentrations are attained after approximately two weeks of once daily dosing.
The pharmacokinetics of cenobamate are similar when used as monotherapy or as adjunctive therapy for the treatment of partial-onset seizures, except plasma cenobamate multiple-dose exposure (Cmax, AUC) decreased with co-administration of phenytoin by 27-28%.
Absorption
At least 88% of XCOPRI is absorbed following oral administration, with median Tmax ranging from 1 to 4 hours.
Plasma Cmax and AUC for XCOPRI crushed tablets mixed in water, administered either orally or through a nasogastric tube, were similar to whole tablets. The median Tmax for crushed tablets is 0.5 hours.
Effect of Food
No clinically significant differences in cenobamate pharmacokinetics were observed following administration of a high-fat meal (800 -1000 calories with 50% fat).
Distribution
The apparent volume of distribution (Vd/F) of cenobamate after oral administration of XCOPRI is approximately 40-50 L. Plasma protein binding of cenobamate is 60% and independent of concentration in vitro. Cenobamate primarily binds with human albumin protein.
Elimination
The apparent terminal half-life of cenobamate is 50-60 hours and apparent oral clearance is approximately 0.45-0.63 L/hour over a dose range from 100 mg/day to 400 mg/day.
Metabolism
Cenobamate is extensively metabolized. The primary metabolic pathways are by glucuronidation via UGT2B7 and to a lesser extent by UGT2B4, and by oxidation via CYP2E1, CYP2A6, CYP2B6, and to a lesser extent by CYP2C19 and CYP3A4/5.
Following administration of radiolabeled cenobamate, unchanged cenobamate accounted for greater than 98% of the total AUC of radioactivity in plasma. Unchanged cenobamate accounted for 6.8% of the dose which was mainly excreted in the urine (6.4%).
Excretion
Following administration of radiolabeled cenobamate, a mean of 93.0% of the total radioactive dose was recovered in urine (87.8%) and feces (5.2%). More than 50% of the radioactivity was excreted within 72 hours of dosing.
Specific Populations
No clinically significant differences in the pharmacokinetics of cenobamate were observed based on age based on data from subjects age 18 years to 77 years, sex, or race/ethnicity based on data from subjects categorized as Asian, Black, Caucasian, Hispanic, or Other.
Patients with Renal Impairment
Cenobamate plasma AUC was 1.4 fold to 1.5 fold higher in subjects with mild (CLcr 60 to less than 90 mL/min) and moderate (CLcr 30 to less than 60 mL/min) following a single oral 200 mg dose of XCOPRI compared to healthy controls. In subjects with severe (CLcr less than 30 mL/min) renal impairment, cenobamate plasma AUC did not change significantly compared to healthy controls following single oral 100 mg dose of XCOPRI [see Use in Specific Populations (8.6)]. The effect of hemodialysis on cenobamate pharmacokinetics has not been studied.
Patients with Hepatic Impairment
Cenobamate plasma AUC was 1.9-fold, 2.3-fold, and 4.2-fold higher in subjects with mild (5-6 points, Child-Pugh Class A), moderate (7-9 points, Child-Pugh Class B), and severe (10-15 points, Child-Pugh Class C) hepatic impairment, respectively, following a single oral 200 mg dose of XCOPRI in subjects with mild and moderate hepatic impairment and 100 mg dose of XCOPRI in subjects with severe hepatic impairment compared to matched healthy controls [see Dosage and Administration (2.2) and Use in Specific Populations (8.7)].
Drug Interaction Studies
Clinical Studies
Alcohol
No clinically significant pharmacokinetic differences were observed for either cenobamate or alcohol when administered concomitantly.
AEDs
Multiple doses of concomitant XCOPRI 200 mg once daily increased phenytoin mean Cmax and AUC by 70% and 84%, respectively, and phenobarbital mean Cmax and AUC by 34% and 37%, respectively [see Drug Interactions (7.1)]. Multiple doses of concomitant XCOPRI 200 mg once daily decreased carbamazepine mean Cmax and AUC each by 23% [see Drug Interactions (7.1].
No clinically significant differences in the pharmacokinetics of the following drugs were observed when used concomitantly with cenobamate: valproic acid, levetiracetam or lacosamide.
Based on population PK analyses, during treatment within the 100-400 mg/day XCOPRI dose range, lamotrigine concentrations are expected to decrease by 21-52% [see Drug Interactions (7.1)]; and levetiracetam concentrations are expected to decrease by 4-13%, which is not expected to be clinically significant.
In subjects treated with XCOPRI in Study 1 and Study 2, there was no clear relationship between efficacy and concomitant oxcarbazepine use. As such, the efficacy data from Study 1 and Study 2 do not support the existence of a clinically relevant interaction perpetrated by XCOPRI against oxcarbazepine.
CYP Substrates
Multiple doses of concomitant XCOPRI 200 mg once daily decreased total bupropion (CYP2B6 substrate) mean Cmax and AUC by 23% and 39%, respectively, and decreased midazolam (CYP3A substrate) mean Cmax and AUC by 61% and 72%, respectively [see Drug Interactions (7.1)]. Multiple doses of concomitant XCOPRI 200 mg once daily increased the omeprazole (CYP2C19 substrate) mean Cmax and AUC by 83% and 107%, respectively [see Drug Interactions (7.1)]. No clinically significant differences in the pharmacokinetics of warfarin (CYP2C9 substrate) were observed when used concomitantly with cenobamate.
The effects of concomitant AEDs on cenobamate PK
Plasma cenobamate multiple-dose exposure (Cmax, AUC) decreased with co-administration of phenytoin by 27-28%. However, repeated dosing of valproate, phenobarbital, and carbamazepine did not have any significant impact on plasma cenobamate multiple-dose exposure.
In Vitro Studies
CYP Enzymes
Cenobamate inhibits CYP2B6, CYP2C19, and CYP3A, but cenobamate does not inhibit CYP1A2, CYP2C8, CYP2C9, or CYP2D6.
Cenobamate induces CYP2B6, CYP2C8, and CYP3A4, but cenobamate does not induce CYP1A2, CYP2C9, or CYP2C19.
Transporters Systems
Cenobamate was not a substrate of P-gp, BCRP, OAT1, OAT3, OCT2, MATE1, or MATE2-K, and cenobamate did not inhibit P-gp, OAT1, OCT1, OCT2, OATP1B3, BSEP, OAT3, or OATP1B1.
13 Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Oral administration of cenobamate (0, 5, 15, or 35 mg/kg/day) to Tg.rasH2 mice for up to 26 weeks did not result in an increase in tumors. Oral administration of cenobamate (0, 4, 8, or 20 mg/kg/day) to male and female rats for up to 87 or 90 weeks, respectively, did not result in an increase in tumors. Plasma exposure at the highest dose tested in rats was less than that in humans at the maximum recommended human dose (MRHD) of 400 mg/day.
Mutagenesis
Cenobamate was negative for genotoxicity in in vitro (Ames, mouse lymphoma) and in vivo (rat bone marrow micronucleus) assays.
Impairment of Fertility
Oral administration of cenobamate (0, 11, 22, or 44 mg/kg/day) to male and female rats prior to and throughout mating and continuing in females to Gestation Day 6 did not produce adverse effects on fertility, general reproductive performance, or early embryonic development. Plasma exposure (AUC) at the highest dose tested in rats was less than that in humans at the MRHD.
14 Clinical Studies
The efficacy of XCOPRI for the treatment of partial-onset seizures was established in two multicenter, randomized, double-blind, placebo-controlled studies in adult patients (Study 1 and Study 2). Patients enrolled in the studies had partial-onset seizures with or without secondary generalization and were not adequately controlled with 1 to 3 concomitant AEDs. During an 8-week baseline period, patients were required to have at least 3 or 4 partial-onset seizures per 28 days on average with no seizure-free period exceeding 3 to 4 weeks. In these studies, patients had a mean duration of epilepsy of approximately 24 years and median baseline seizure frequency of 8.5 seizures per 28 days. More than 80% of patients were taking 2 or more concomitant AEDs.
Study 1 (NCT01397968) compared doses of XCOPRI 200 mg/day with placebo. Study 2 (NCT01866111) compared doses of XCOPRI 100 mg/day, 200 mg/day, and 400 mg/day with placebo. Both studies had an 8-week baseline period to establish a baseline seizure frequency, following which patients were randomized to a treatment arm. Patients entered a treatment period consisting of an initial titration phase (6 weeks), and a subsequent maintenance phase (6 weeks for Study 1 and 12 weeks for Study 2). In Study 1, patients were started on a daily dose of 50 mg (a higher starting dose than currently recommended) and subsequently increased by 50 mg/day every two weeks, until the final daily target dose of 200 mg/day was achieved. In Study 2, patients were started on a daily dose of 50 mg (a higher starting dose than currently recommended) and subsequently increased by 50 mg/day every week (a faster titration than currently recommended) until 100 mg/day or 200 mg/day was reached and then increased by 100 mg/day every week in patients randomized to 400 mg/day [see Dosage and Administration (2.2), Warnings and Precautions (5.1), and Adverse Reactions (6.1)].
The primary efficacy outcome in Study 1 and Study 2 was the percent change from baseline in seizure frequency per 28 days in the treatment period. Table 6 summarizes the results of the primary endpoint for XCOPRI in each study.
Table 6: Percent Change from Baseline in Seizure Frequency per 28 Days in the Treatment Period (Study 1 and Study 2) * A negative percent change from baseline in seizure frequency indicates reduction in seizure frequency from baseline. ** Statistically significant compared to placebo N Median Percent Change from Baseline in Seizure Frequency per 28 Days (%)* p-value(Compared to Placebo) Study 1 Placebo 108 -21.5 -- XCOPRI 200 mg/day 113 -55.6 <0.0001** Study 2 Placebo 106 -24.3 -- XCOPRI 100 mg/day 108 -36.3 0.006** XCOPRI 200 mg/day 109 -55.2 <0.001** XCOPRI 400 mg/day 111 -55.3 <0.001**
Figure 1 and Figure 2 show the proportion of patients with different percent reductions during the maintenance phase over baseline in Study 1 and Study 2, respectively. Patients in whom the seizure frequency increased are shown in the left-most column as ‚Äúworse.‚ÄĚ Patients in whom the seizure frequency decreased are shown in the remaining four categories.
Figure 1: Proportion of Patients Exhibiting Different Percent Reductions During the Maintenance Phase over Baseline in Study 1
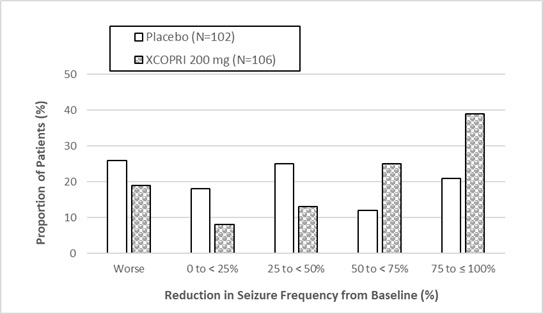
Figure 2: Proportion of Patients Exhibiting Different Percent Reductions During the Maintenance Phase over Baseline in Study 2
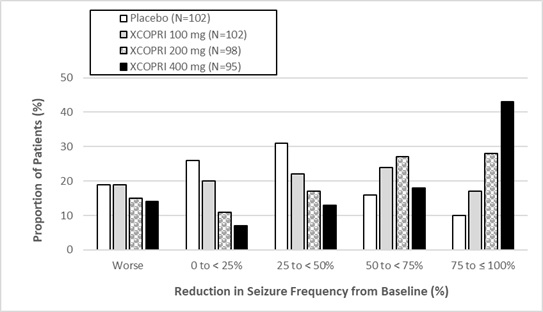
In Study 2, 4 of 102 (4%) patients in the XCOPRI 100 mg/day group, 11 of 98 (11%) patients in the XCOPRI 200 mg/day group, and 20 of 95 (21%) patients in the XCOPRI 400 mg/day group and 1 of 102 (1%) of patients in the placebo group reported no partial seizures during the maintenance phase.
16 How Supplied/storage And Handling
16.1 How Supplied
XCOPRI tablets are supplied in the following configurations:
Bottles; 30 count
Strength NDC Number Tablet Description (Color, Shape, Markings) 25 mg 71699-025-30 Film coated round brown tablets with SK on one side and 25 on the other side 50 mg 71699-050-30 Film coated round yellow tablets with SK on one side and 50 on the other side 100 mg 71699-100-30 Film coated round brown tablets with SK on one side and 100 on the other side 150 mg 71699-150-30 Film coated round light orange tablets with SK on one side and 150 on the other side 200 mg 71699-200-30 Film coated modified oval light orange tablets with SK on one side and 200 on the other side Titration Buler Packs; 14-Day
Daily Dose NDC Number Supplied As [strength(quantity)] Tablet Description (Color, Shape, Markings) 12.5 mg per day for 14 days 71699-204-14 12.5 mg (14-count) Uncoated round white to off-white tablets with SK on one side and 12 on the other side
Titration Buler Packs; 28-Day
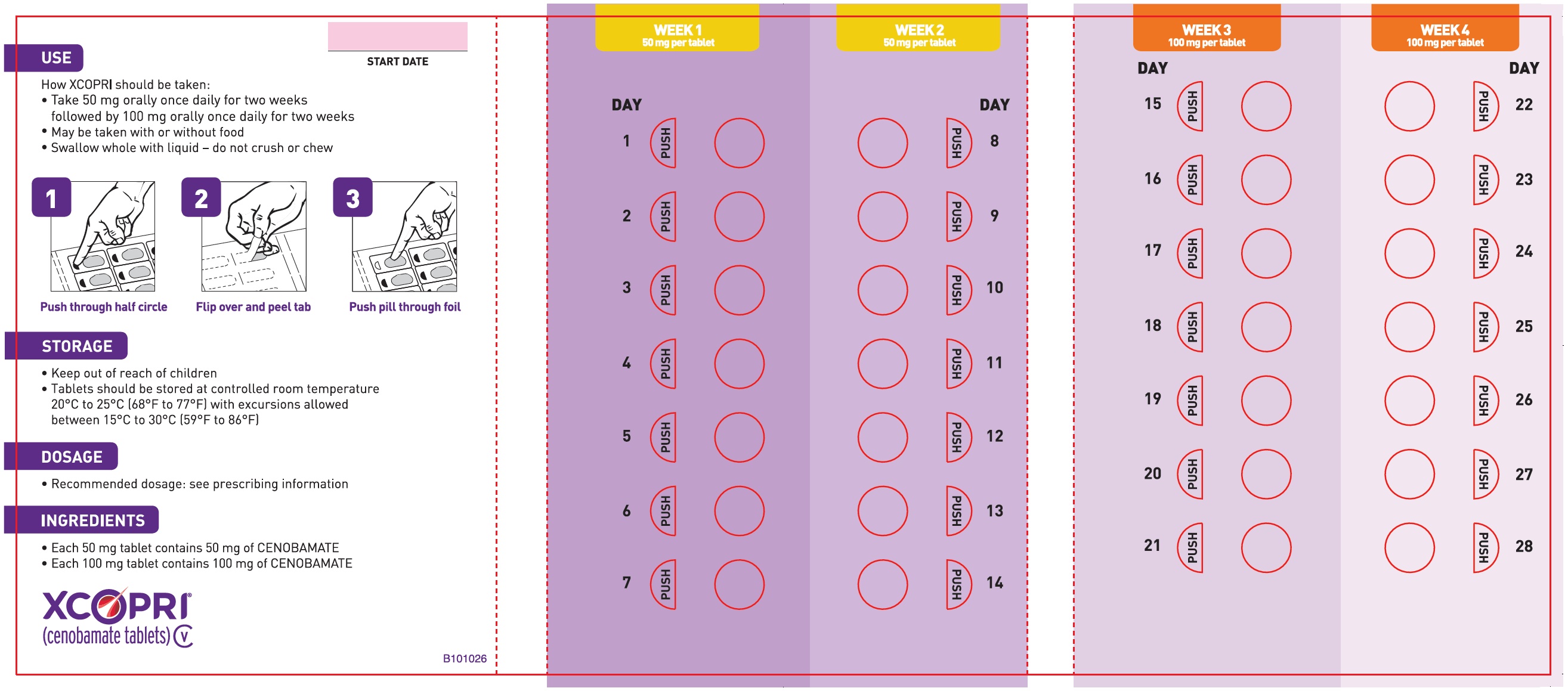
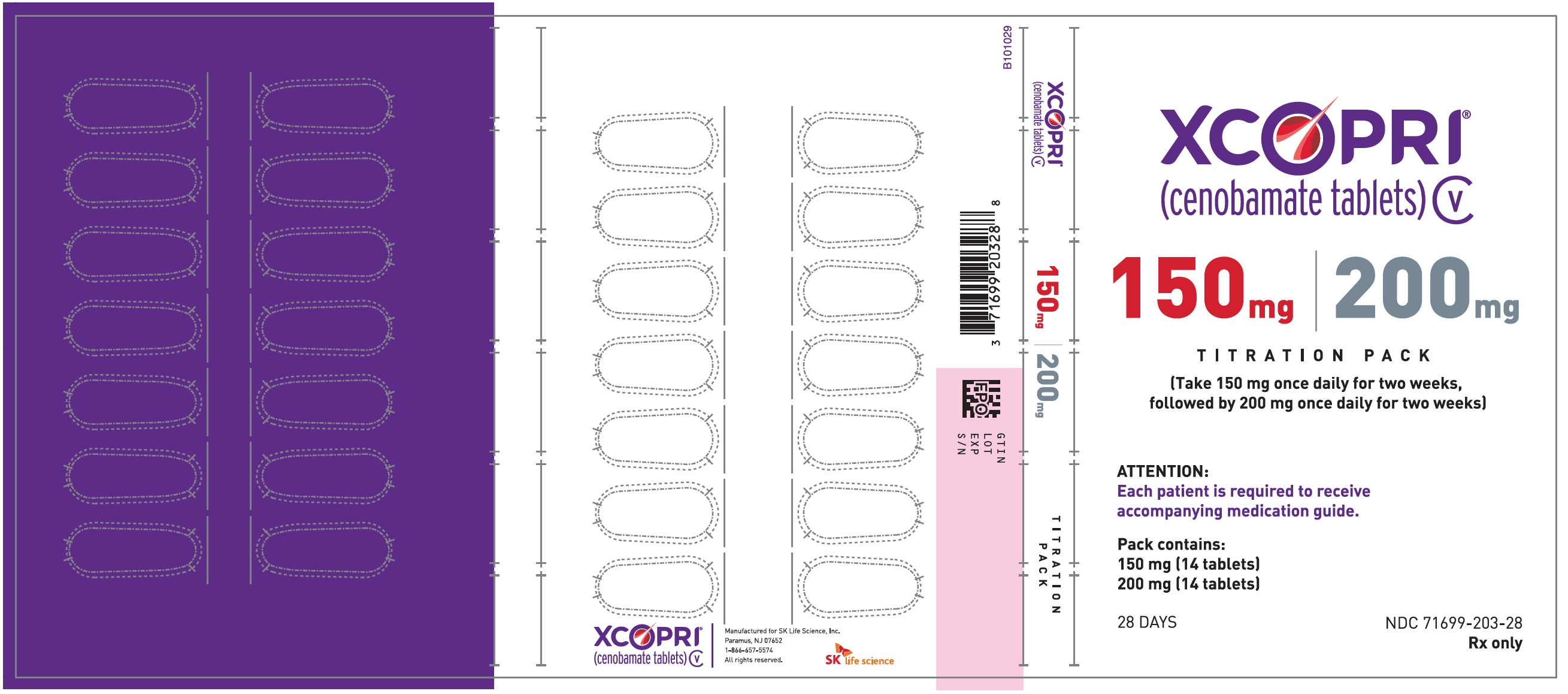
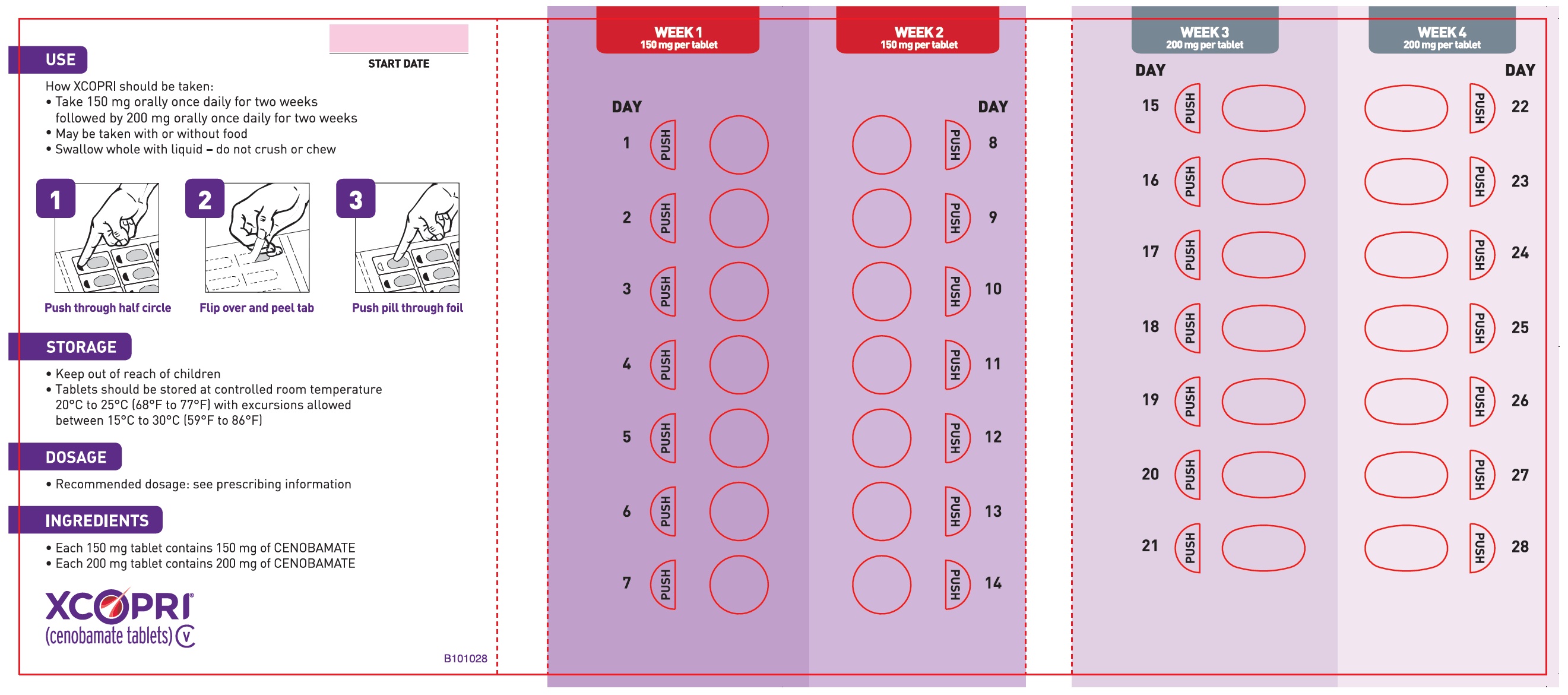
Daily Dose NDC Number Supplied As [strength(quantity)] Tablet Description (Color, Shape, Markings) 12.5 mg per day for 14 days, then 25 mg per day for 14 days 71699-201-28 12.5 mg (14-count) Uncoated round white to off-white tablets with SK on one side and 12 on the other side 25 mg (14-count) Film coated round brown tablets with SK on one side and 25 on the other side 50 mg per day for 14 days, then 100 mg per day for 14 days 71699-202-28 50 mg (14-count) Film coated round yellow tablets with SK on one side and 50 on the other side 100 mg (14-count) Film coated round brown tablets with SK on one side and 100 on the other side 150 mg per day for 14 days, then 200 mg per day for 14 days 71699-203-28 150 mg (14-count) Film coated round light orange tablets with SK on one side and 150 on the other side 200 mg (14-count) Film coated modified oval light orange tablets with SK on one side and 200 on the other side
Maintenance Buler Packs; 28-Day
Daily Dose NDC Number Supplied As [strength(quantity)] Tablet Description (Color, Shape, Markings) 250 mg per day 71699-104-56 100 mg (28-count) Film coated round brown tablets with SK on one side and 100 on the other side 150 mg (28-count) Film coated round light orange tablets with SK on one side and 150 on the other side 350 mg per day 71699-103-56 150 mg (28-count) Film coated round light orange tablets with SK on one side and 150 on the other side 200 mg (28-count) Film coated modified oval light orange tablets with SK on one side and 200 on the other side 16.2 Storage and Handling
Store XCOPRI tablets at 20¬įC to 25¬įC (68¬įF to 77¬įF) with excursions permitted to 15¬įC to 30¬įC (59¬įF to 86¬įF) (See USP Controlled Room Temperature).
17 Patient Counseling Information
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
DRESS/Multi-organ Hypersensitivity
Instruct patients and caregivers that a fever or rash associated with signs of other organ system involvement (e.g., lymphadenopathy, hepatic dysfunction) may be drug-related and should be reported to their healthcare provider immediately. XCOPRI should be discontinued immediately if a serious hypersensitivity reaction is suspected [see Warnings and Precautions (5.1)].
QT Shortening
Instruct patients to inform their healthcare provider of all of the medications, over-the-counter medications, and herbal supplements that they are taking. Instruct patients to notify their healthcare provider if they have any symptoms of shortening of the QT interval, including prolonged heart palpitations or a loss of consciousness [see Warnings and Precautions (5.2)].
Suicidal Behavior and Ideation
Counsel patients, their caregivers, and/or families that antiepileptic drugs, including XCOPRI, may increase the risk of suicidal thoughts and behavior, and advise patients to be alert for the emergence or worsening of symptoms of depression; unusual changes in mood or behavior; or suicidal thoughts, behavior, or thoughts about self-harm. Advise patients, their caregivers, and/or families to report behaviors of concern immediately to a healthcare provider [see Warnings and Precautions (5.3)].
Neurological Adverse Reactions
Counsel patients that XCOPRI causes somnolence, fatigue, dizziness, and gait disturbance. These adverse reactions, if observed, are more likely to occur early in treatment but can occur at any time. Advise patients not to drive or operate machinery until they have gained sufficient experience on XCOPRI to gauge whether it adversely affects their ability to drive or operate machinery and that other CNS depressants or alcohol may have additive effects [see Warnings and Precautions (5.4)].
Withdrawal of XCOPRI
Advise patients not to discontinue use of XCOPRI without consulting with their healthcare provider. XCOPRI should normally be gradually withdrawn to reduce the potential for increased seizure frequency and status epilepticus [see Warnings and Precautions (5.5)].
Contraceptives
Counsel females of reproductive potential that XCOPRI may decrease the efficacy of oral contraceptives and advise them to use additional or alternative non-hormonal birth control [see Drug Interactions (7.1)].
Pregnancy
Advise patients to notify their healthcare provider if they become pregnant or intend to become pregnant during XCOPRI therapy. Encourage patients to enroll in the North American Antiepileptic Drug Pregnancy Registry if they become pregnant. This registry is collecting information about the safety of antiepileptic drugs during pregnancy [see Use in Specific Populations (8.1)].
Dosing Instructions
Counsel patients that XCOPRI may be taken any time with or without food. Instruct patients that XCOPRI tablets can be taken whole or crushed. The crushed tablet can be mixed with water and either administered by mouth as an oral suspension or administered via a nasogastric (NG) tube. Counsel patients administering XCOPRI as an oral suspension or via NG tube on appropriate preparation and administration instructions [see Dosage and Administration (2.3)].
Abuse and Dependence
Advise patients that XCOPRI is a federally controlled substance (CV) because it can be abused or lead to dependence [see Drug Abuse and Dependence (9)]. Advise patients to keep their medication in a safe place to prevent misuse and abuse.
Manufactured for: SK Life Science, Inc. Paramus, NJ 07652 Phone: 1-866-657-5574 © 2019 SK Life Science, Inc.
XCOPRI¬ģ is a registered trademark of SK Life Science, Inc. or its affiliates and protected by U.S. Patent 7,598,279
Spl Medguide Section
This Medication Guide has been approved by the U.S. Food and Drug Administration. Revised: 4/2024
MEDICATION GUIDE
XCOPRI¬ģ (ex-koh-pree)
(cenobamate)
tablets, for oral use, CV
What is the most important information I should know about XCOPRI?
XCOPRI can cause serious side effects, including:
- Serious or life threatening allergic reactions which may affect organs and other parts of your body like the liver or blood cells. You may or may not have a rash with these types of reactions. Call your healthcare provider right away and go to the nearest emergency room if you have any of the following:
- swelling of your face, eyes, lips, or tongue
- trouble swallowing or breathing
- a skin rash
- hives
- fever, swollen glands, or sore throat that does not go away or comes and goes
- painful sores in the mouth or around your eyes
- yellowing of your skin or eyes
- unusual bruising or bleeding
- severe fatigue or weakness
- severe muscle pain
- frequent infections that do not go away
These symptoms may be the first signs of a serious reaction. A healthcare provider should examine you to decide if you should continue taking XCOPRI.
- Like other antiepileptic drugs, XCOPRI may cause suicidal thoughts or actions in a very small number of people, about 1 in 500. Call a healthcare provider right away if you have any of these symptoms, especially if they are new, worse, or worry you:
- thoughts about suicide or dying
- attempts to commit suicide
- new or worse depression
- new or worse anxiety
- feeling agitated or restless
- panic attacks
- trouble sleeping (insomnia)
- new or worse irritability
- acting aggressive, being angry, or violent
- acting on dangerous impulses
- an extreme increase in activity and talking (mania)
- other unusual changes in behavior or mood
- Suicidal thoughts or actions may be caused by things other than medicines. If you have suicidal thoughts or actions, your healthcare provider may check for other causes.
How can I watch for early symptoms of suicidal thoughts and actions?
- Pay attention to any changes, especially sudden changes, in mood, behaviors, thoughts, or feelings.
- Keep all follow-up visits with your healthcare provider as scheduled.
Call your healthcare provider between visits as needed, especially if you are worried about symptoms.
- Do not stop taking XCOPRI without first talking to your healthcare provider.
- Stopping XCOPRI suddenly can cause serious problems.
- Stopping a seizure medicine suddenly can cause seizures that will not stop (status epilepticus).
- XCOPRI is a federally controlled substance (CV) because it can be abused or lead to dependence. Keep XCOPRI in a safe place to prevent misuse and abuse. Tell your healthcare provider if you have ever abused or been dependent on alcohol, prescription medicines, or street drugs. Selling or giving away XCOPRI may harm others and is against the law.
What is XCOPRI? XCOPRI is a prescription medicine used to treat partial-onset seizures in adults. It is not known if XCOPRI is safe and effective in children. Do not take XCOPRI if you:
- are allergic to cenobamate, any of the other ingredients in XCOPRI. See the end of this Medication Guide for a complete ul of ingredients in XCOPRI.
- have a genetic problem (called familial short QT syndrome) that affects the electrical system of the heart.
Before taking XCOPRI, tell your healthcare provider about all your medical conditions, including if you:
- have or had depression, mood problems, or suicidal thoughts or actions.
- have liver, kidney, or blood problems.
- have had an allergic reaction to a medicine which caused a rash or affected internal organs, like the liver or blood cells.
- use birth control medicine. XCOPRI may cause your birth control medicine to be less effective. Talk to your healthcare provider about the best birth control method to use while taking XCOPRI.
- are pregnant or plan to become pregnant. It is not known if XCOPRI will harm your unborn baby. Tell your healthcare provider right away if you become pregnant while taking XCOPRI. You and your healthcare provider will decide if you should take XCOPRI while you are pregnant.
- If you become pregnant while taking XCOPRI, talk to your healthcare provider about registering with the North American Antiepileptic Drug (NAAED) Pregnancy Registry. The purpose of this registry is to collect information about the safety of antiepileptic medicine during pregnancy. You can enroll in this registry by calling 1-888-233-2334 or go to www.aedpregnancyregistry.org
- are breastfeeding or plan to breastfeed. It is not known if XCOPRI passes into breastmilk. Talk to your healthcare provider about the best way to feed your baby while taking XCOPRI.
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements.
XCOPRI may affect the way other medicines work, and other medicines may affect how XCOPRI works. Do not start a new medicine without first talking with your healthcare provider.
Ask your healthcare provider or pharmacist for a ul of medicines you are taking, if you are not sure. Know the medicines you take. Keep a ul of them and show it to your healthcare provider and pharmacist when you get a new medicine.
How should I take XCOPRI?
- Take XCOPRI exactly as your healthcare provider tells you to take it.
- It is very important to increase your dose of XCOPRI slowly, as instructed by your healthcare provider.
- Do not stop taking XCOPRI without talking to your healthcare provider. Stopping XCOPRI suddenly can cause serious problems, including seizures that will not stop (status epilepticus).
- Your healthcare provider may change your dose, if needed.
- Your healthcare provider will tell you how much XCOPRI to take.
- XCOPRI can be taken at any time of day, with or without food.
- XCOPRI tablets can be taken whole or crushed.
- If you crush the tablet, it can be mixed with water and either taken by mouth as an oral suspension or given via a nasogastric (NG) feeding tube. Talk with your healthcare provider about how to take crushed tablets.
- Talk with your healthcare provider about what you should do if you miss a dose.
- If you take too much XCOPRI, call your healthcare provider or go to the nearest hospital emergency room right away.
What should I avoid while taking XCOPRI?
- Do not drive, operate machinery, or do other dangerous activities until you know how XCOPRI affects you. XCOPRI may slow your thinking and motor skills and may affect your vision.
- Do not drink alcohol or take other medicines that can make you sleepy or dizzy while taking XCOPRI without first talking to your healthcare provider.
What are the possible side effects of XCOPRI?XCOPRI may cause serious side effects, including:
- See ‚ÄúWhat is the most important information I should know about XCOPRI?‚ÄĚ
- problems with the electrical system of the heart (QT shortening). Call your healthcare provider if you have symptoms of QT shortening including fast heartbeat (heart palpitations) that last a long time or fainting.
- nervous system problems. XCOPRI may cause problems that can affect your nervous system. Symptoms of nervous system problems include:
- dizziness
- trouble walking or with coordination
- feeling sleepy and tired
- trouble concentrating, remembering, and thinking clearly
- vision problems
The most common side effects of XCOPRI include:
- feeling sleepy and tired
- dizziness
- double vision
- headache
Tell your healthcare provider if you have any side effect that bothers you or that does not go away. These are not all the possible side effects of XCOPRI.
For more information, ask your healthcare provider or pharmacist.Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store XCOPRI?
- Store XCOPRI at room temperature between 68‚ĄČ to 77‚ĄČ (20‚ĄÉ to 25‚ĄÉ).
- Safely throw away medicine that is out of date or no longer needed.
- Keep XCOPRI and all medicines out of the reach of children.
General information about the safe and effective use of XCOPRI. Medicines are sometimes prescribed for purposes other than those uled in a Medication Guide. Do not use XCOPRI for a condition for which it was not prescribed. Do not give XCOPRI to other people, even if they have the same symptoms that you have. It may harm them. You can ask your pharmacist or healthcare provider for information about XCOPRI that is written for health professionals. What are the ingredients in XCOPRI?
Active ingredient: cenobamate
Inactive ingredients: colloidal silicon dioxide, lactose monohydrate, magnesium stearate,
microcrystalline cellulose, and sodium starch glycolate.
25 mg and 100 mg tablets: FD&C Blue #2/indigo carmine aluminum lake, iron oxide red, iron oxide yellow, polyethylene glycol 3350, polyvinyl alcohol-part hydrolyzed, talc, and titanium dioxide.
50 mg tablets: iron oxide yellow, polyethylene glycol 3350, polyvinyl alcohol-part hydrolyzed, talc, and titanium dioxide.
150 mg and 200 mg tablets: iron oxide red, iron oxide yellow, polyethylene glycol 3350, polyvinyl alcohol-part hydrolyzed, talc, and titanium dioxide.
Manufactured for SK Life Science, Inc., Paramus, NJ 07652© 2019 SK Life Science, Inc.
XCOPRI¬ģ is a registered trademark of SK Life Science, Inc. or its affiliates and protected by U.S. Patent 7,598,279
For more information, go to www.XCOPRI.com or call 1-866-657-5574.
Principal Display Panel - Ndc: 71699-050-30 - 50 Mg 30-count Bottle Label

Principal Display Panel - Ndc: 71699-050-99 - 50 Mg 30-count Bottle Label

Principal Display Panel - Ndc: 71699-100-30 - 100 Mg 30-count Bottle Label

Principal Display Panel - Ndc: 71699-150-30 - 150 Mg 30-count Bottle Label

Principal Display Panel - Ndc: 71699-200-30 - 200 Mg 30-count Bottle Label

Principal Display Panel - Ndc: 71699-201-28 - 12.5 Mg 14-count And 25 Mg 14-count Titration Pack Label (front)

Principal Display Panel - Ndc: 71699-201-28 - 12.5 Mg 14-count And 25 Mg 14-count Titration Pack Label (back)
Principal Display Panel - Ndc: 71699-202-28 - 50 Mg 14-count And 100 Mg 14-count Titration Pack Label (front)

Principal Display Panel - Ndc: 71699-202-28 - 50 Mg 14-count And 100 Mg 14-count Titration Pack Label (back)
Principal Display Panel - Ndc: 71699-203-28 - 150 Mg 14-count And 200 Mg 14-count Titration Pack Label (front)
Principal Display Panel - Ndc: 71699-203-28 - 150 Mg 14-count And 200 Mg 14-count Titration Pack Label (back)
Principal Display Panel - Ndc: 71699-103-56 - 200 Mg 28-count And 150 Mg 28-count Maintenance Pack (front)

Principal Display Panel - Ndc: 71699-103-56 - 200 Mg 28-count And 150 Mg 28-count Maintenance Pack (back)
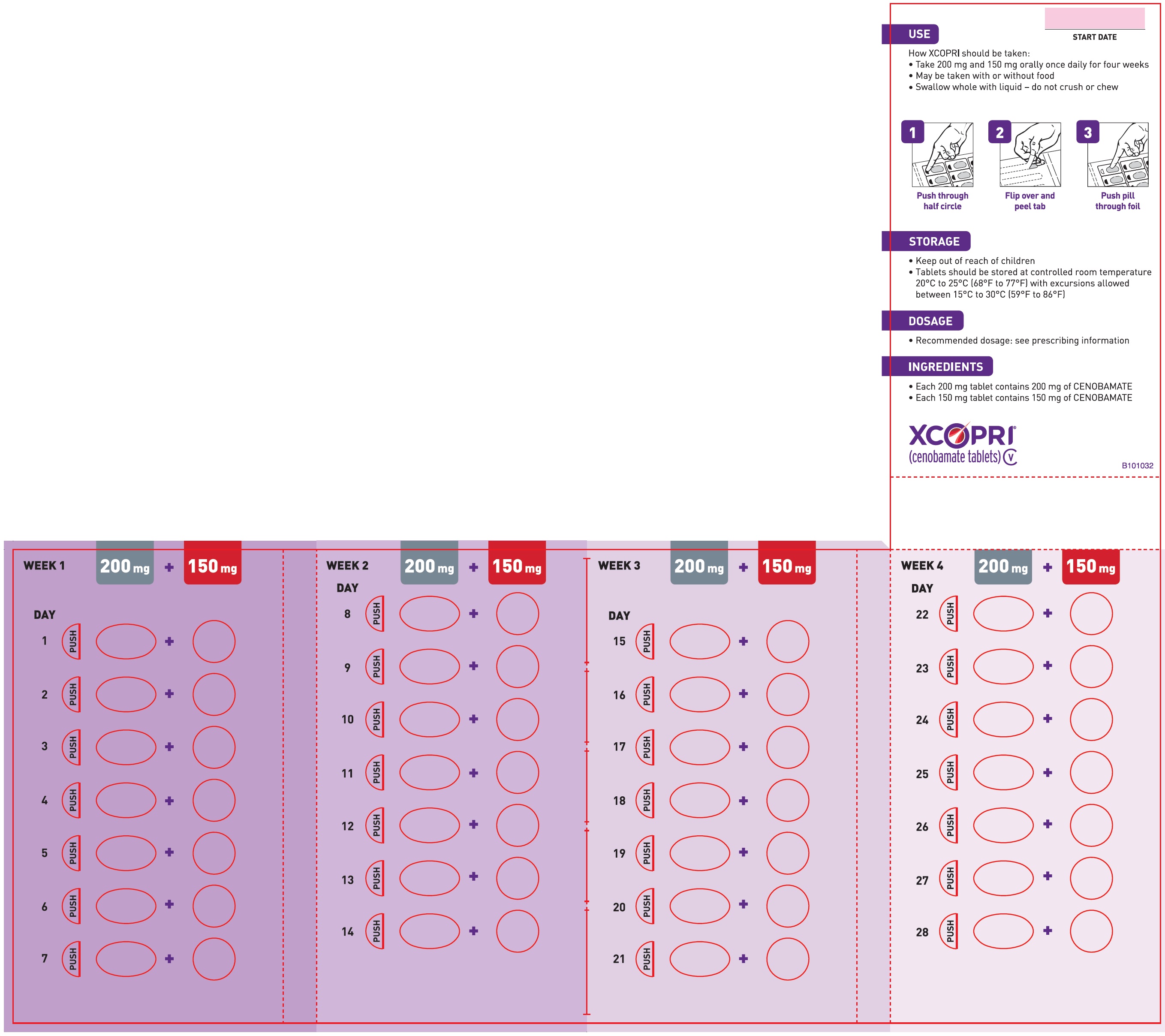
Principal Display Panel - Ndc: 71699-104-56 - 150 Mg 28-count And 100 Mg 28-count Maintenance Pack (front)

Principal Display Panel - Ndc: 71699-104-56 - 150 Mg 28-count And 100 Mg 28-count Maintenance Pack (back)
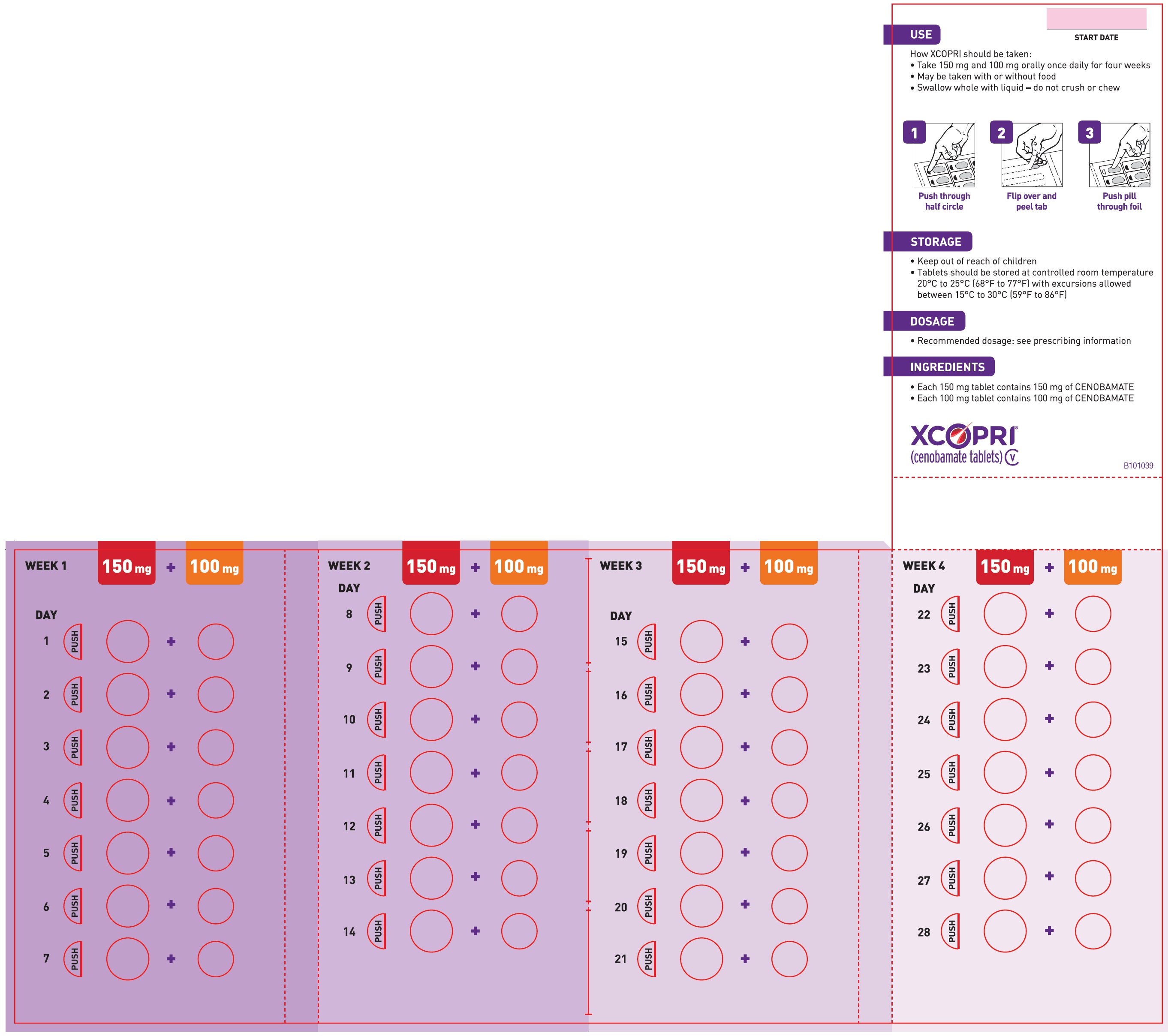
Principal Display Panel - Ndc: 71699-999-99 - 12.5 Mg 14-count And 25 Mg 14-count Titration Pack Label (front)

Principal Display Panel - Ndc: 71699-999-99 - 12.5 Mg 14-count And 25 Mg 14-count Titration Pack Label (back)
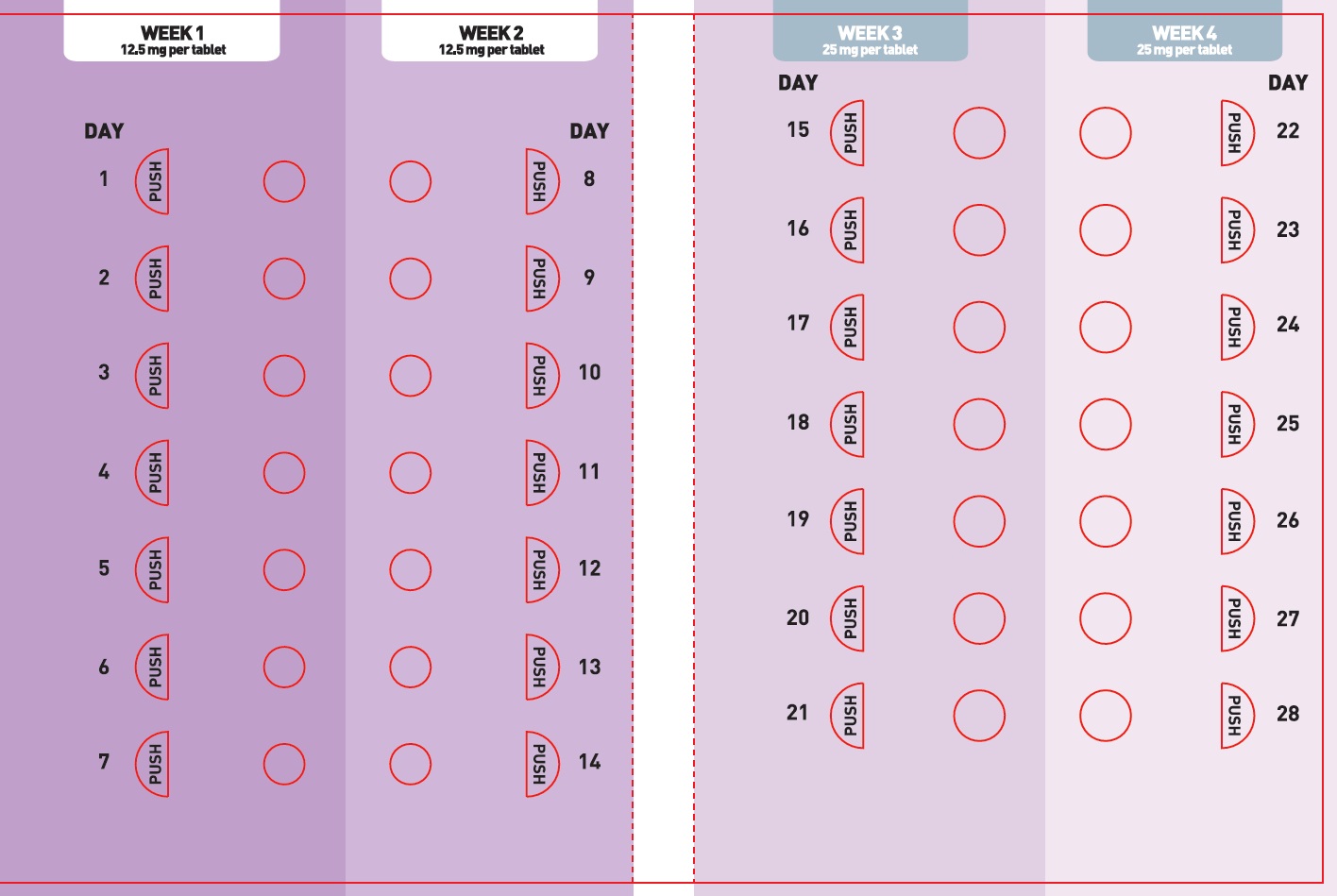
Principal Display Panel - Ndc: 71699-025-30 - 25 Mg 30-count Bottle Label

Principal Display Panel - Ndc: 71699-204-14 - 12.5 Mg 14-count Titration Pack Label (front)

Principal Display Panel - Ndc: 71699-204-14 - 12.5 Mg 14-count Titration Pack Label (back)

DISCLAIMER:
"This tool does not provide medical advice, and is for informational and educational purposes only, and is not a substitute for professional medical advice, treatment or diagnosis. Call your doctor to receive medical advice. If you think you may have a medical emergency, please dial 911."
"Do not rely on openFDA to make decisions regarding medical care. While we make every effort to ensure that data is accurate, you should assume all results are unvalidated. We may limit or otherwise restrict your access to the API in line with our Terms of Service."
"This product uses publicly available data from the U.S. National Library of Medicine (NLM), National Institutes of Health, Department of Health and Human Services; NLM is not responsible for the product and does not endorse or recommend this or any other product."
PillSync may earn a commission via links on our site
