

Tenofovir Disoproxil Fumarate (tenofovir disoproxil fumarate 300 mg) Dailymed
Generic: tenofovir disoproxil fumarate is used for the treatment of Hepatitis B, Chronic Acquired Immunodeficiency Syndrome

IMPRINT: LA16
SHAPE: oval
COLOR: blue
All Imprints
tenofovir disoproxil fumarate 300 mg oral tablet - la16 oval blue
Boxed Warning
Warning: Posttreatment Exacerbation Of Hepatitis

Go PRO for all pill images
Recent Major Changes Section
• Indications and Usage (1.1 ) 04/2017 • Boxed Warning, Lactic Acidosis/Severe Hepatomegaly With Steatosis Removed 04/2017 • Warnings and Precautions, Lactic Acidosis/Severe Hepatomegaly with Steatosis (5.3 ) 04/2017 • Warnings and Precautions, Coadministration with Other Products (5.4 ) 04/2017 • Warnings and Precautions, Fat Redistribution Removed 04/2017
Warning: Posttreatment Exacerbation Of Hepatitis
Severe acute exacerbations of hepatitis have been reported in HBV-infected patients who have discontinued anti-hepatitis B therapy, including Tenofovir disoproxil fumarate. Hepatic function should be monitored closely with both clinical and laboratory follow-up for at least several months in patients who discontinue anti-hepatitis B therapy, including Tenofovir disoproxil fumarate. If appropriate, resumption of anti-hepatitis B therapy may be warranted [See Warnings and Precautions (5.1)].
WARNING: POSTTREATMENT EXACERBATION OF HEPATITIS See full prescribing information for complete boxed warning. Severe acute exacerbations of hepatitis have been reported in HBV-infected patients who have discontinued anti-hepatitis B therapy, including tenofovir disoproxil fumarate. Hepatic function should be monitored closely in these patients. If appropriate, resumption of anti-hepatitis B therapy may be warranted. (5.1 )
1 Indicationsand Usage
Tenofovir disoproxil fumarate is a nucleotide analog HIV-1 reverse transcriptase inhibitor and an HBV reverse transcriptase inhibitor. • Tenofovir disoproxil fumarate tablets are indicated in combination with other antiretroviral agents for the treatment of HIV-1 infection in adults and pediatric patients 2 years of age and older. (1 )
• Tenofovir disoproxil fumarate tablets are indicated for the treatment of chronic hepatitis B in adults and pediatric patients 12 years of age and older. (1 )
1.1 HIV-1 Infection
Tenofovir disoproxil fumarate tablets are indicated in combination with other antiretroviral agents for the treatment of HIV-1 infection in adults and pediatric patients 2 years of age and older.
The following points should be considered when initiating therapy with tenofovir disoproxil fumarate tablets for the treatment of HIV-1 infection:
- Tenofovir disoproxil fumarate tablets should not be
used in combination with ATRIPLA®, COMPLERA®, DESCOVY®, GENVOYA®, ODEFSEY®, STRIBILD®, TRUVADA®, or VEMLIDY® [See Warnings and Precautions (5.4)].1.2 Chronic Hepatitis B
Tenofovir disoproxil fumarate tablets are indicated for the treatment of chronic hepatitis B in adults and pediatric patients 12 years of age and older. The following points should be considered when initiating therapy with tenofovir disoproxil fumarate tablets for the treatment of HBV infection: • The indication in adults is based on safety and efficacy data from treatment of subjects who were nucleoside-treatment-naïve and subjects who were treatment-experienced with documented resistance to lamivudine. Subjects were adults with HBeAg-positive and HBeAg-negative chronic hepatitis B with compensated liver disease [See Clinical Studies (14.2)]. • Tenofovir disoproxil fumarate was evaluated in a limited number of subjects with chronic hepatitis B and decompensated liver disease [ See Adverse Reactions (6.1) , Clinical Studies (14.2)] . • The numbers of subjects in clinical trials who had adefovir resistance-associated substitutions at baseline were too small to reach conclusions of efficacy [ See Microbiology (12.4), Clinical Studies (14.2) ].
2 Dosageand Administration
• Recommended dose for the treatment of HIV-1 or chronic hepatitis B in adults and pediatric patients 12 years of age and older (35 kg or more): 300 mg once daily taken orally without regard to food. (2.1 ) • Recommended dose for the treatment of HIV-1 in pediatric patients (2 to less than 12 years of age): o Tablets: For pediatric patients weighing greater than or equal to 17 kg who can swallow an intact tablet, one Tenofovir disoproxil fumarate tablet (300 mg based on body weight) once daily taken orally without regard to food. (2.2 ) • Dose recommended in renal impairment in adults: o Creatinine clearance 30 to 49 mL/min: 300 mg every 48 hours. (2.3 ) o Creatinine clearance 10 to 29 mL/min: 300 mg every 72 to 96 hours. (2.3 ) o Hemodialysis: 300 mg every 7 days or after approximately 12 hours of dialysis. (2.3 )
2.1 Recommended Dose in Adults and Pediatric Patients 12 Years of Age and Older (35 kg or more)
For the treatment of HIV-1 or chronic hepatitis B: The dose is one 300 mg tenofovir disoproxil fumarate tablet once daily taken orally, without regard to food. In the treatment of chronic hepatitis B, the optimal duration of treatment is unknown. Safety and efficacy in pediatric patients with chronic hepatitis B weighing less than 35 kg have not been established.
2.2 Recommended Dose in Pediatric Patients 2 Years to Less than 12 Years of Age
HIV-1 Infection For the treatment of HIV-1 in pediatric patients 2 years of age and older, the recommended oral dose of tenofovir disoproxil fumarate tablets is 8 mg of tenofovir disoproxil fumarate (tenofovir DF) per kilogram of body weight (up to a maximum of 300 mg) once daily administered as tablets. Tenofovir disoproxil fumarate is available as tablet in 300 mg strength for pediatric patients who weigh greater than or equal to 17 kg and who are able to reliably swallow intact tablets. The dose is one tablet once daily taken orally, without regard to food. Table 1 contain dosing recommendations for tenofovir disoproxil fumarate tablets based on body weight. Weight should be monitored periodically and the tenofovir disoproxil fumarate tablets dose adjusted accordingly.
Table 1 Dosing Recommendations for Pediatric Patients ≥2 Years of Age and weighing ≥17 kg using Tenofovir disoproxil fumarate tablets Body Weight Kilogram (kg) Tablets Once Daily 17 to <22 150 mg 22 to <28 200 mg 28 to <35 250 mg ≥35 300 mg
Chronic Hepatitis B Safety and efficacy of tenofovir disoproxil fumarate in patients younger than 12 years of age have not been established.
2.3 Dose Adjustment for Renal Impairment in Adults
Significantly increased drug exposures occurred when tenofovir disoproxil fumarate tablets were administered to subjects with moderate to severe renal impairment [See Clinical Pharmacology (12.3)]. Therefore, the dosing interval of tenofovir disoproxil fumarate tablets 300 mg should be adjusted in patients with baseline creatinine clearance below 50 mL/min using the recommendations in Table 2. These dosing interval recommendations are based on modeling of single-dose pharmacokinetic data in non-HIV and non-HBV infected subjects with varying degrees of renal impairment, including end-stage renal disease requiring hemodialysis. The safety and effectiveness of these dosing interval adjustment recommendations have not been clinically evaluated in patients with moderate or severe renal impairment; therefore, clinical response to treatment and renal function should be closely monitored in these patients [See Warnings and Precautions (5.2)]. No dose adjustment of tenofovir disoproxil fumarate tablets 300 mg is necessary for patients with mild renal impairment (creatinine clearance 50 to 80 mL/min). Routine monitoring of estimated creatinine clearance, serum phosphorus, urine glucose, and urine protein should be performed in patients with mild renal impairment [See Warnings and Precautions (5.2)].
Table 2 Dosage Adjustment for Patients with Altered Creatinine Clearance a Calculated using ideal (lean) body weight. b Generally once weekly assuming three hemodialysis sessions a week of approximately 4 hours duration. Tenofovir disoproxil fumarate tablets should be administered following completion of dialysis. Creatinine Clearance (mL/min)a Hemodialysis Patients ≥50 30 to 49 10 to 29 Recommended 300 mg Dosing Interval Every 24 hours Every 48 hours Every 72 to 96 hours Every 7 days or after a total of approximately 12 hours of dialysisb
The pharmacokinetics of tenofovir have not been evaluated in non-hemodialysis patients with creatinine clearance below 10 mL/min; therefore, no dosing recommendation is available for these patients. No data are available to make dose recommendations in pediatric patients with renal impairment.
3 Dosage Formsand Strengths
Tenofovir disoproxil fumarate is available as tablets. Tenofovir disoproxil fumarate tablets 300 mg contain 300 mg of tenofovir DF, which is equivalent to 245 mg of tenofovir disoproxil. The tablets are blue colored, oval shaped, film-coated, and debossed with “LA16” on one side and plain on the other.
• Tablets: 300 mg (3 )
4 Contraindications
None
None. (4 )
5 Warnings And Precautions
• New onset or worsening renal impairment: Can include acute renal failure and Fanconi syndrome. Assess estimated creatinine clearance before initiating treatment with tenofovir disoproxil fumarate. In patients at risk for renal dysfunction, assess estimated creatinine clearance, serum phosphorus, urine glucose, and urine protein before initiating treatment with tenofovir disoproxil fumarate and periodically during treatment. Avoid administering tenofovir disoproxil fumarate with concurrent or recent use of nephrotoxic drugs. (5.2 )
• Lactic acidosis/severe hepatomegaly with steatosis: Discontinue treatment in patients who develop symptoms or laboratory findings suggestive of lactic acidosis or pronounced hepatotoxicity. (5.3 )
• Coadministration with other products: Do not use with other tenofovir-containing products (e.g., ATRIPLA, COMPLERA, DESCOVY, GENVOYA, ODEFSEY, STRIBILD, TRUVADA, or VEMLIDY). Do not administer in combination with HEPSERA. (5.4 )
• HIV testing: HIV antibody testing should be offered to all HBV-infected patients before initiating therapy with tenofovir disoproxil fumarate. Tenofovir disoproxil fumarate should only be used as part of an appropriate antiretroviral combination regimen in HIV-infected patients with or without HBV coinfection. (5.5 )
• Decreases in bone mineral density (BMD): Consider assessment of BMD in patients with a history of pathologic fracture or other risk factors for osteoporosis or bone loss. (5.6 )
• Immune reconstitution syndrome: Observed in HIV-infected patients. May necessitate further evaluation and treatment. (5.7 )
• Triple nucleoside-only regimens: Early virologic failure has been reported in HIV-infected patients. Monitor carefully and consider treatment modification. (5.8 )
5.1 Exacerbation of Hepatitis after Discontinuation of Treatment
Discontinuation of anti-HBV therapy, including tenofovir disoproxil fumarate, may be associated with severe acute exacerbations of hepatitis. Patients infected with HBV who discontinue tenofovir disoproxil fumarate should be closely monitored with both clinical and laboratory follow-up for at least several months after stopping treatment. If appropriate, resumption of anti-hepatitis B therapy may be warranted.
5.2 New Onset or Worsening Renal Impairment
Tenofovir is principally eliminated by the kidney. Renal impairment, including cases of acute renal failure and Fanconi syndrome (renal tubular injury with severe hypophosphatemia), has been reported with the use of tenofovir disoproxil fumarate [See Adverse Reactions (6.2)]. It is recommended that estimated creatinine clearance be assessed in all patients prior to initiating therapy and as clinically appropriate during therapy with tenofovir disoproxil fumarate. In patients at risk of renal dysfunction, including patients who have previously experienced renal events while receiving HEPSERA®, it is recommended that estimated creatinine clearance, serum phosphorus, urine glucose, and urine protein be assessed prior to initiation of tenofovir disoproxil fumarate, and periodically during tenofovir disoproxil fumarate therapy. Dosing interval adjustment of tenofovir disoproxil fumarate and close monitoring of renal function are recommended in all patients with creatinine clearance below 50 mL/min [See Dosage and Administration (2.3)]. No safety or efficacy data are available in patients with renal impairment who received tenofovir disoproxil fumarate using these dosing guidelines, so the potential benefit of tenofovir disoproxil fumarate therapy should be assessed against the potential risk of renal toxicity. Tenofovir disoproxil fumarate should be avoided with concurrent or recent use of a nephrotoxic agent (e.g., high-dose or multiple non-steroidal anti-inflammatory drugs (NSAIDs)) [See Drug Interactions (7.4)]. Cases of acute renal failure after initiation of high dose or multiple NSAIDs have been reported in HIV-infected patients with risk factors for renal dysfunction who appeared stable on tenofovir DF. Some patients required hospitalization and renal replacement therapy. Alternatives to NSAIDs should be considered, if needed, in patients at risk for renal dysfunction. Persistent or worsening bone pain, pain in extremities, fractures and/or muscular pain or weakness may be manifestations of proximal renal tubulopathy and should prompt an evaluation of renal function in at-risk patients.
5.3 Lactic Acidosis/Severe Hepatomegaly with Steatosis
Lactic acidosis and severe hepatomegaly with steatosis, including fatal cases, have been reported with the use of nucleoside analogs, including tenofovir DF, alone or in combination with other antiretrovirals. Treatment with tenofovir disoproxil fumarateshould be suspended in any patient who develops clinical or laboratory findings suggestive of lactic acidosis or pronounced hepatotoxicity (which may include hepatomegaly and steatosis even in the absence of marked transaminase elevations).
5.4 Coadministration with Other Products
other drugs containing tenofovir DF or tenofovir alafenamide, including ATRIPLA, COMPLERA, DESCOVY, GENVOYA, ODEFSEY, STRIBILD, TRUVADA, or VEMLIDY. Tenofovir disoproxil fumarate should not be administered in combination with HEPSERA (adefovir dipivoxil) [See Drug Interactions (7.4)].
Tenofovir disoproxil fumarate should not be used in combinationwith
5.5 Patients Coinfected with HIV-1 and HBV
Due to the risk of development of HIV-1 resistance, tenofovir disoproxil fumarate should only be used in HIV-1 and HBV coinfected patients as part of an appropriate antiretroviral combination regimen. HIV-1 antibody testing should be offered to all HBV-infected patients before initiating therapy with tenofovir disoproxil fumarate. It is also recommended that all patients with HIV-1 be tested for the presence of chronic hepatitis B before initiating treatment with tenofovir disoproxil fumarate.
5.6 Bone Effects
Bone Mineral Density: In clinical trials in HIV-1 infected adults, tenofovir disoproxil fumarate was associated with slightly greater decreases in bone mineral density (BMD) and increases in biochemical markers of bone metabolism, suggesting increased bone turnover relative to comparators. Serum parathyroid hormone levels and 1,25 Vitamin D levels were also higher in subjects receiving tenofovir disoproxil fumarate [See Adverse Reactions (6.1)]. Clinical trials evaluating tenofovir disoproxil fumarate in pediatric and adolescent subjects were conducted. Under normal circumstances, BMD increases rapidly in pediatric patients. In HIV-1 infected subjects aged 2 years to less than 18 years, bone effects were similar to those observed in adult subjects and suggest increased bone turnover. Total body BMD gain was less in the tenofovir disoproxil fumarate-treated HIV-1 infected pediatric subjects as compared to the control groups. Similar trends were observed in chronic hepatitis B infected adolescent subjects aged 12 years to less than 18 years. In all pediatric trials, skeletal growth (height) appeared to be unaffected [See Adverse Reactions (6.1)]. The effects of tenofovir disoproxil fumarate-associated changes in BMD and biochemical markers on long-term bone health and future fracture risk are unknown. Assessment of BMD should be considered for adults and pediatric patients who have a history of pathologic bone fracture or other risk factors for osteoporosis or bone loss. Although the effect of supplementation with calcium and vitamin D was not studied, such supplementation may be beneficial for all patients. If bone abnormalities are suspected then appropriate consultation should be obtained. Mineralization Defects: Cases of osteomalacia associated with proximal renal tubulopathy, manifested as bone pain or pain in extremities and which may contribute to fractures, have been reported in association with the use of tenofovir disoproxil fumarate [See Adverse Reactions (6.2)]. Arthralgias and muscle pain or weakness have also been reported in cases of proximal renal tubulopathy. Hypophosphatemia and osteomalacia secondary to proximal renal tubulopathy should be considered in patients at risk of renal dysfunction who present with persistent or worsening bone or muscle symptoms while receiving products containing tenofovir DF [See Warnings and Precautions (5.2)].
5.7 Immune Reconstitution Syndrome
Immune reconstitution syndrome has been reported in HIV-infected patients treated with combination antiretroviral therapy, including tenofovir disoproxil fumarate. During the initial phase of combination antiretroviral treatment, patients whose immune system responds may develop an inflammatory response to indolent or residual opportunistic infections (such as Mycobacterium avium infection, cytomegalovirus, Pneumocystis jirovecii pneumonia [PCP], or tuberculosis), which may necessitate further evaluation and treatment. Autoimmune disorders (such as Graves’ disease, polymyositis, and Guillain-Barré syndrome) have also been reported to occur in the setting of immune reconstitution; however, the time to onset is more variable, and can occur many months after initiation of treatment.
5.8 Early Virologic Failure
Clinical trials in HIV-infected subjects have demonstrated that certain regimens that only contain three nucleoside reverse transcriptase inhibitors (NRTI) are generally less effective than triple drug regimens containing two NRTIs in combination with either a non-nucleoside reverse transcriptase inhibitor or a HIV-1 protease inhibitor. In particular, early virological failure and high rates of resistance substitutions have been reported. Triple nucleoside regimens should therefore be used with caution. Patients on a therapy utilizing a triple nucleoside-only regimen should be carefully monitored and considered for treatment modification.
6 Adverse Reactions
The following adverse reactions are discussed in other sections of the labeling: • Severe Acute Exacerbation of Hepatitis [See Boxed Warning, Warnings and Precautions (5.1)]. • New Onset or Worsening Renal Impairment [See Warnings and Precautions (5.2)]. • Lactic Acidosis/Severe Hepatomegaly with Steatosis [See Warnings and Precautions (5.3)]. • Bone Effects [See Warnings and Precautions (5.6)]. • Immune Reconstitution Syndrome [See Warnings and Precautions (5.7)].
• In HIV-infected adult subjects: Most common adverse reactions (incidence greater than or equal to 10%, Grades 2 to 4) are rash, diarrhea, headache, pain, depression, asthenia, and nausea. (6.1 )
• In HBV-infected subjects with compensated liver disease: Most common adverse reaction (all grades) was nausea (9%). (6.1 )
• In pediatric subjects: Adverse reactions in pediatric subjects were consistent with those observed in adults. (6.1 )
• In HBV-infected subjects with decompensated liver disease: Most common adverse reactions (incidence greater than or equal to 10%, all grades) were abdominal pain, nausea, insomnia, pruritus, vomiting, dizziness, and pyrexia. (6.1 ) To report SUSPECTED ADVERSE REACTIONS, contact Laurus Generics Inc. at 1-833-3-LAURUS (1-833-352-8787) or FDA at 1-800-FDA-1088 orwww.fda.gov/medwatch
6.1 Adverse Reactions from Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice. Clinical Trials in Adult Patients with HIV-1 Infection More than 12,000 subjects have been treated with tenofovir disoproxil fumarate alone or in combination with other antiretroviral medicinal products for periods of 28 days to 215 weeks in clinical trials and expanded access programs. A total of 1544 subjects have received tenofovir disoproxil fumarate 300 mg once daily in clinical trials; over 11,000 subjects have received tenofovir disoproxil fumarate in expanded access programs. The most common adverse reactions (incidence greater than or equal to 10%, Grades 2 to 4) identified from any of the 3 large controlled clinical trials include rash, diarrhea, headache, pain, depression, asthenia, and nausea. Treatment-Naïve Patients Study 903 – Treatment-Emergent Adverse Reactions: The most common adverse reactions seen in a double-blind comparative controlled trial in which 600 treatment-naïve subjects received tenofovir disoproxil fumarate (N=299) or stavudine (N=301) in combination with lamivudine and efavirenz for 144 weeks (Study 903) were mild to moderate gastrointestinal events and dizziness. Mild adverse reactions (Grade 1) were common with a similar incidence in both arms, and included dizziness, diarrhea, and nausea. Selected treatment-emergent moderate to severe adverse reactions are summarized in Table 3.
Table 3 Selected Treatment-Emergent Adverse Reactionsa (Grades 2 to 4) Reported in ≥5% in Any Treatment Group in Study 903 (0 to 144 Weeks) Tenofovir disoproxil fumarate+3TC+EFV d4T+3TC+EFV N=299 N=301 Body as a WholeHeadache Pain Fever Abdominal pain Back pain Asthenia 14%13%8%7%9%6% 17%12%7%12%8%7% Digestive System Diarrhea Nausea Dyspepsia Vomiting 11%8%4%5% 13%9%5%9% Metabolic Disorders Lipodystrophyb 1% 8% Musculoskeletal Arthralgia Myalgia 5%3% 7%5% Nervous System Depression Insomnia Dizziness Peripheral neuropathyc Anxiety 11%5%3%1%6% 10%8%6%5%6% Respiratory Pneumonia 5% 5% Skin and Appendages Rash eventd 18% 12%
a Frequencies of adverse reactions are based on all treatment-emergent adverse events, regardless of relationship to study drug. b Lipodystrophy represents a variety of investigator-described adverse events not a protocol defined syndrome. c Peripheral neuropathy includes peripheral neuritis and neuropathy. d Rash event includes rash, pruritus, maculopapular rash, urticaria, vesiculobullous rash, and pustular rash. Laboratory Abnormalities: With the exception of fasting cholesterol and fasting triglyceride elevations that were more common in the stavudine group (40% and 9%) compared with tenofovir disoproxil fumarate (19% and 1%), respectively, laboratory abnormalities observed in this trial occurred with similar frequency in the tenofovir disoproxil fumarate and stavudine treatment arms. A summary of Grades 3 to 4 laboratory abnormalities is provided in Table 4.
Table 4 Grades 3 to 4 Laboratory Abnormalities Reported in ≥1% of Tenofovir disoproxil fumarate-Treated Subjects in Study 903 (0 to 144 Weeks) Tenofovir disoproxil fumarate+3TC+EFV d4T+3TC+EFV N=299 N=301 Any ≥ Grade 3 Laboratory Abnormality 36% 42% Fasting Cholesterol (>240 mg/dL) 19% 40% Creatine Kinase (M: >990 U/L; F: >845 U/L) 12% 12% Serum Amylase (>175 U/L) 9% 8% AST (M: >180 U/L; F: >170 U/L) 5% 7% ALT (M: >215 U/L; F: >170 U/L) 4% 5% Hematuria (>100 RBC/HPF) 7% 7% Neutrophils (<750/mm3) 3% 1% Fasting Triglycerides (>750 mg/dL) 1% 9%
Study 934 – Treatment-Emergent Adverse Reactions: In Study 934, 511 antiretroviral naïve subjects received either tenofovir disoproxil fumarate + EMTRIVA® administered in combination with efavirenz (N=257) or zidovudine/lamivudine administered in combination with efavirenz (N=254). Adverse reactions observed in this trial were generally consistent with those seen in previous studies in treatment-experienced or treatment-naïve subjects (Table 5). Changes in Bone Mineral Density In HIV-1 infected adult subjects in Study 903, there was a significantly greater mean percentage decrease from baseline in BMD at the lumbar spine in subjects receiving tenofovir disoproxil fumarate + lamivudine + efavirenz (–2.2% ± 3.9) compared with subjects receiving stavudine + lamivudine + efavirenz (–1.0% ± 4.6) through 144 weeks. Changes in BMD at the hip were similar between the two treatment groups (–2.8% ± 3.5 in the tenofovir disoproxil fumarate group vs. –2.4% ± 4.5 in the stavudine group). In both groups, the majority of the reduction in BMD occurred in the first 24 to 48 weeks of the trial and this reduction was sustained through Week 144. Twenty-eight percent of tenofovir disoproxil fumarate -treated subjects vs. 21% of the stavudine-treated subjects lost at least 5% of BMD at the spine or 7% of BMD at the hip. Clinically relevant fractures (excluding fingers and toes) were reported in 4 subjects in the tenofovir disoproxil fumarate group and 6 subjects in the stavudine group. In addition, there were significant increases in biochemical markers of bone metabolism (serum bone-specific alkaline phosphatase, serum osteocalcin, serum C telopeptide, and urinary N telopeptide) and higher serum parathyroid hormone levels and 1,25 Vitamin D levels in the tenofovir disoproxil fumarate group relative to the stavudine group; however, except for bone-specific alkaline phosphatase, these changes resulted in values that remained within the normal range [See Warnings and Precautions (5.6)].
Table 5 Selected Treatment-Emergent Adverse Reactionsa (Grades 2 to 4) Reported in ≥5% in Any Treatment Group in Study 934 (0 to 144 Weeks) Tenofovir disoproxil fumarateb+FTC+EFV AZT/3TC+EFV N=257 N=254 Gastrointestinal Disorder Diarrhea Nausea Vomiting 9% 9% 2% 5% 7% 5% General Disorders and Administration Site Condition Fatigue 9% 8% Infections and Infestations Sinusitis Upper respiratory tract infections Nasopharyngitis 8% 8% 5% 4% 5% 3% Nervous System Disorders Headache Dizziness 6% 8% 5% 7% Psychiatric Disorders DepressionInsomnia 9% 5% 7% 7% Skin and Subcutaneous Tissue Disorders Rash eventc 7% 9%
a Frequencies of adverse reactions are based on all treatment-emergent adverse events, regardless of relationship to study drug. b From Weeks 96 to 144 of the trial, subjects received TRUVADA with efavirenz in place of tenofovir disoproxil fumarate + EMTRIVA with efavirenz. c Rash event includes rash, exfoliative rash, rash generalized, rash macular, rash maculopapular, rash pruritic, and rash vesicular. Laboratory Abnormalities: Laboratory abnormalities observed in this trial were generally consistent with those seen in previous trials (Table 6).
Table 6 Significant Laboratory Abnormalities Reported in ≥1% of Subjects in Any Treatment Group in Study 934 (0 to 144 Weeks) Tenofovir disoproxil fumaratea+FTC+EFV AZT/3TC+EFV N=257 N=254 Any ≥ Grade 3 Laboratory Abnormality 30% 26% Fasting Cholesterol (>240 mg/dL) 22% 24% Creatine Kinase (M: >990 U/L; F: >845 U/L) 9% 7% Serum Amylase (>175 U/L) 8% 4% Alkaline Phosphatase (>550 U/L) 1% 0% AST (M: >180 U/L; F: >170 U/L) 3% 3% ALT (M: >215 U/L; F: >170 U/L) 2% 3% Hemoglobin (<8.0 mg/dL) 0% 4% Hyperglycemia (>250 mg/dL) 2% 1% Hematuria (>75 RBC/HPF) 3% 2% Glycosuria (≥3+) <1% 1% Neutrophils (<750/mm3) 3% 5% Fasting Triglycerides (>750 mg/dL) 4% 2%
a From Weeks 96 to 144 of the trial, subjects received TRUVADA with efavirenz in place of tenofovir disoproxil fumarate + EMTRIVA with efavirenz. Treatment-Experienced Patients Treatment-Emergent Adverse Reactions: The adverse reactions seen in treatment-experienced subjects were generally consistent with those seen in treatment-naïve subjects including mild to moderate gastrointestinal events, such as nausea, diarrhea, vomiting, and flatulence. Less than 1% of subjects discontinued participation in the clinical trials due to gastrointestinal adverse reactions (Study 907). A summary of moderate to severe treatment-emergent adverse reactions that occurred during the first 48 weeks of Study 907 is provided in Table 7.
Table 7 Selected Treatment-Emergent Adverse Reactionsa (Grades 2 to 4) Reported in ≥3% in Any Treatment Group in Study 907 (0 to 48 Weeks) Tenofovir disoproxil fumarate (N=368) (Week 0 to 24) Placebo (N=182) (Week 0 to 24) Tenofovir disoproxil fumarate (N=368) (Week 0 to 48) Placebo Crossover to Tenofovir disoproxil fumarate (N=170) (Week 24 to 48) Body as a WholeAstheniaPainHeadacheAbdominal painBack painChest painFever 7%7%5%4%3%3%2% 6%7%5%3%3%1%2% 11%12%8%7%4%3%4% 1%4%2%6%2%2%2% Digestive SystemDiarrheaNauseaVomitingAnorexiaDyspepsiaFlatulence 11%8%4%3%3%3% 10%5%1%2%2%1% 16%11%7%4%4%4% 11%7%5%1%2%1% Respiratory Pneumonia 2% 0% 3% 2% Nervous SystemDepressionInsomniaPeripheral neuropathyb Dizziness 4%3%3%1% 3%2%3%3% 8%4%5%3% 4%4%2%1% Skin and AppendageRash eventc Sweating 5%3% 4%2% 7%3% 1%1% MusculoskeletalMyalgia 3% 3% 4% 1% MetabolicWeight loss 2% 1% 4% 2%
a Frequencies of adverse reactions are based on all treatment-emergent adverse events, regardless of relationship to study drug. b Peripheral neuropathy includes peripheral neuritis and neuropathy. c Rash event includes rash, pruritus, maculopapular rash, urticaria, vesiculobullous rash, and pustular rash. Laboratory Abnormalities: Laboratory abnormalities observed in this trial occurred with similar frequency in the tenofovir disoproxil fumarate and placebo-treated groups. A summary of Grades 3 to 4 laboratory abnormalities is provided in Table 8.
Table 8 Grades 3 to 4 Laboratory Abnormalities Reported in ≥1% of tenofovir disoproxil fumarate-Treated Subjects in Study 907 (0 to 48 Weeks) Tenofovir disoproxil fumarate (N=368) (Week 0 to 24) Placebo (N=182) (Week 0 to 24) Tenofovir disoproxil fumarate (N=368) (Week 0 to 48) Placebo Crossover to Tenofovir disoproxil fumarate (N=170) (Week 24 to 48) Any ≥ Grade 3 Laboratory Abnormality 25% 38% 35% 34% Triglycerides (>750 mg/dL) 8% 13% 11% 9% Creatine Kinase (M: >990 U/L; F: >845 U/L) 7% 14% 12% 12% Serum Amylase (>175 U/L) 6% 7% 7% 6% Glycosuria (≥3+) 3% 3% 3% 2% AST (M: >180 U/L; F: >170 U/L) 3% 3% 4% 5% ALT (M: >215 U/L; F: >170 U/L) 2% 2% 4% 5% Serum Glucose (>250 U/L) 2% 4% 3% 3% Neutrophils (<750/mm3) 1% 1% 2% 1%
Clinical Trials in Pediatric Subjects 2 Years of Age and Older with HIV-1 Infection Assessment of adverse reactions is based on two randomized trials (Studies 352 and 321) in 184 HIV-1 infected pediatric subjects (2 to less than 18 years of age) who received treatment with tenofovir disoproxil fumarate (N=93) or placebo/active comparator (N=91) in combination with other antiretroviral agents for 48 weeks. The adverse reactions observed in subjects who received treatment with tenofovir disoproxil fumarate were consistent with those observed in clinical trials in adults. Eighty-nine pediatric subjects (2 to less than 12 years of age) received tenofovir disoproxil fumarate in Study 352 for a median exposure of 104 weeks. Of these, 4 subjects discontinued from the trial due to adverse reactions consistent with proximal renal tubulopathy. Three of these 4 subjects presented with hypophosphatemia and also had decreases in total body or spine BMD Z score [See Warnings and Precautions (5.6)] . Changes in Bone Mineral Density: Clinical trials in HIV-1 infected children and adolescents evaluated BMD changes. In Study 321 (12 to less than 18 years), the mean rate of BMD gain at Week 48 was less in the tenofovir disoproxil fumarate compared to the placebo treatment group. Six tenofovir disoproxil fumarate treated subjects and one placebo treated subject had significant (greater than 4%) lumbar spine BMD loss at Week 48. Changes from baseline BMD Z-scores were –0.341 for lumbar spine and–0.458 for total body in the 28 subjects who were treated with tenofovir disoproxil fumarate for 96 weeks. In Study 352 (2 to less than 12 years), the mean rate of BMD gain in lumbar spine at Week 48 was similar between the tenofovir disoproxil fumarate and the d4T or AZT treatment groups. Total body BMD gain was less in the tenofovir disoproxil fumarate compared to the d4T or AZT treatment groups. One tenofovir disoproxil fumarate -treated subject and none of the d4T or AZT-treated subjects experienced significant (greater than 4%) lumbar spine BMD loss at Week 48. Changes from baseline in BMD Z scores were –0.012 for lumbar spine and –0.338 for total body in the 64 subjects who were treated with tenofovir disoproxil fumarate for 96 weeks. In both trials, skeletal growth (height) appeared to be unaffected [See Warnings and Precautions (5.6)]. Clinical Trials in Adult Subjects with Chronic Hepatitis B and Compensated Liver Disease Treatment-Emergent Adverse Reactions: In controlled clinical trials in 641 subjects with chronic hepatitis B (0102 and 0103), more subjects treated with tenofovir disoproxil fumarate during the 48-week double-blind period experienced nausea: 9% with tenofovir disoproxil fumarate versus 2% with HEPSERA. Other treatment-emergent adverse reactions reported in more than 5% of subjects treated with tenofovir disoproxil fumarate included: abdominal pain, diarrhea, headache, dizziness, fatigue, nasopharyngitis, back pain, and skin rash. During the open-label phase of treatment with tenofovir disoproxil fumarate (weeks 48 to 384) in Studies 0102 and 0103, 2% of subjects (13/585) experienced a confirmed increase in serum creatinine of 0.5 mg/dL from baseline. No significant change in the tolerability profile was observed with continued treatment for up to 384 weeks. Laboratory Abnormalities: A summary of Grades 3 to 4 laboratory abnormalities through Week 48 is provided in Table 9. Grades 3 to 4 laboratory abnormalities were similar in subjects continuing tenofovir disoproxil fumarate treatment for up to 384 weeks in these trials.
Table 9 Grades 3 to 4 Laboratory Abnormalities Reported in ≥1% of Tenofovir disoproxil fumarate -Treated Subjects in Studies 0102 and 0103 (0 to 48 Weeks) Tenofovir disoproxil fumarate (N=426) HEPSERA (N=215) Any ≥ Grade 3 Laboratory Abnormality 19% 13% Creatine Kinase (M: >990 U/L; F: >845 U/L) 2% 3% Serum Amylase (>175 U/L) 4% 1% Glycosuria (≥3+) 3% <1% AST (M: >180 U/L; F: >170 U/L) 4% 4% ALT (M: >215 U/L; F: >170 U/L) 10% 6%
The overall incidence of on-treatment ALT flares (defined as serum ALT greater than 2 × baseline and greater than 10 × ULN, with or without associated symptoms) was similar between tenofovir disoproxil fumarate (2.6%) and HEPSERA (2%). ALT flares generally occurred within the first 4 to 8 weeks of treatment and were accompanied by decreases in HBV DNA levels. No subject had evidence of decompensation. ALT flares typically resolved within 4 to 8 weeks without changes in study medication. The adverse reactions observed in subjects with chronic hepatitis B and lamivudine resistance who received treatment with tenofovir disoproxil fumarate were consistent with those observed in other hepatitis B clinical trials in adults. Clinical Trials in Adult Subjects with Chronic Hepatitis B and Decompensated Liver Disease In a small randomized, double-blind, active-controlled trial (0108), subjects with CHB and decompensated liver disease received treatment with tenofovir disoproxil fumarate or other antiviral drugs for up to 48 weeks [See Clinical Studies (14.2) ]. Among the 45 subjects receiving tenofovir disoproxil fumarate, the most frequently reported treatment-emergent adverse reactions of any severity were abdominal pain (22%), nausea (20%), insomnia (18%), pruritus (16%), vomiting (13%), dizziness (13%), and pyrexia (11%). Two of 45 (4%) subjects died through Week 48 of the trial due to progression of liver disease. Three of 45 (7%) subjects discontinued treatment due to an adverse event. Four of 45 (9%) subjects experienced a confirmed increase in serum creatinine of 0.5 mg/dL (1 subject also had a confirmed serum phosphorus less than 2 mg/dL through Week 48). Three of these subjects (each of whom had a Child-Pugh score greater than or equal to 10 and MELD score greater than or equal to 14 at entry) developed renal failure. Because both tenofovir disoproxil fumarate and decompensated liver disease may have an impact on renal function, the contribution of tenofovir disoproxil fumarate to renal impairment in this population is difficult to ascertain. One of 45 subjects experienced an on-treatment hepatic flare during the 48-week trial. Clinical Trials in Pediatric Subjects 12 Years of Age and Older with Chronic Hepatitis B Assessment of adverse reactions is based on one randomized study (Study GS-US-174-0115) in 106 pediatric subjects (12 to less than 18 years of age) infected with chronic hepatitis B receiving treatment with tenofovir disoproxil fumarate (N=52) or placebo (N=54) for 72 weeks. The adverse reactions observed in pediatric subjects who received treatment with tenofovir disoproxil fumarate were consistent with those observed in clinical trials of tenofovir disoproxil fumarate in adults. In this study, both the tenofovir disoproxil fumarate and placebo treatment arms experienced an overall increase in mean lumbar spine BMD over 72 weeks, as expected for an adolescent population. The BMD gains from baseline to Week 72 in lumbar spine and total body BMD in tenofovir disoproxil fumarate-treated subjects (+5% and +3%, respectively) were less than the BMD gains observed in placebo-treated subjects (+8% and +5%, respectively). Three subjects in the tenofovir disoproxil fumarate group and two subjects in the placebo group had significant (greater than 4%) lumbar spine BMD loss at Week 72. At baseline, mean BMD Z-scores in subjects randomized to tenofovir disoproxil fumarate were −0.43 for lumbar spine and −0.20 for total body, and mean BMD Z-scores in subjects randomized to placebo were −0.28 for lumbar spine and −0.26 for total body. In subjects receiving tenofovir disoproxil fumarate for 72 weeks, the mean change in BMD Z-score was −0.05 for lumbar spine and −0.15 for total body compared to +0.07 and +0.06, respectively, in subjects receiving placebo. As observed in pediatric studies of HIV-infected patients, skeletal growth (height) appeared to be unaffected [See Warnings and Precautions (5.6)].
6.2 Postmarketing Experience
The following adverse reactions have been identified during postapproval use of tenofovir disoproxil fumarate. Because postmarketing reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure. Immune System Disorders allergic reaction, including angioedema Metabolism and Nutrition Disorders lactic acidosis, hypokalemia, hypophosphatemia Respiratory, Thoracic, and Mediastinal Disorders dyspnea Gastrointestinal Disorders pancreatitis, increased amylase, abdominal pain Hepatobiliary Disorders hepatic steatosis, hepatitis, increased liver enzymes (most commonly AST, ALT gamma GT) Skin and Subcutaneous Tissue Disorders rash Musculoskeletal and Connective Tissue Disorders rhabdomyolysis, osteomalacia (manifested as bone pain and which may contribute to fractures), muscular weakness, myopathy Renal and Urinary Disorders acute renal failure, renal failure, acute tubular necrosis, Fanconi syndrome, proximal renal tubulopathy, interstitial nephritis (including acute cases), nephrogenic diabetes insipidus, renal insufficiency, increased creatinine, proteinuria, polyuria General Disorders and Administration Site Conditions asthenia The following adverse reactions, uled under the body system headings above, may occur as a consequence of proximal renal tubulopathy: rhabdomyolysis, osteomalacia, hypokalemia, muscular weakness, myopathy, hypophosphatemia
7 Drug Interactions
This section describes clinically relevant drug interactions with tenofovir disoproxil fumarate. Drug interactions trials are described elsewhere in the labeling [See Clinical Pharmacology (12.3)] .
• Didanosine: Coadministration increases didanosine concentrations. Use with caution and monitor for evidence of didanosine toxicity (e.g., pancreatitis, neuropathy). Consider dose reductions or discontinuations of didanosine if warranted. (7.1 ) • HIV-1 protease inhibitors: Coadministration decreases atazanavir concentrations and increases tenofovir concentrations. When coadministered with tenofovir disoproxil fumarate, use atazanavir given with ritonavir. Coadministration of tenofovir disoproxil fumarate with atazanavir and ritonavir, darunavir and ritonavir, or lopinavir/ritonavir increases tenofovir concentrations. Monitor for evidence of tenofovir toxicity. (7.2 )
7.1 Didanosine
Coadministration of tenofovir disoproxil fumarate and didanosine should be undertaken with caution and patients receiving this combination should be monitored closely for didanosine associated adverse reactions. Didanosine should be discontinued in patients who develop didanosine-associated adverse reactions. When administered with tenofovir disoproxil fumarate, Cmax and AUC of didanosine increased significantly [See Clinical Pharmacology (12.3)]. The mechanism of this interaction is unknown. Higher didanosine concentrations could potentiate didanosine-associated adverse reactions, including pancreatitis and neuropathy. Suppression of CD4+ cell counts has been observed in patients receiving tenofovir disoproxil fumarate with didanosine 400 mg daily. In patients weighing greater than 60 kg, the didanosine dose should be reduced to 250 mg once daily when it is coadministered with tenofovir disoproxil fumarate. In patients weighing less than 60 kg, the didanosine dose should be reduced to 200 mg once daily when it is coadministered with tenofovir disoproxil fumarate. When coadministered, tenofovir disoproxil fumarate and didanosine EC may be taken under fasted conditions or with a light meal (less than 400 kcal, 20% fat). For additional information on coadministration of tenofovir disoproxil fumarate and didanosine, please refer to the full prescribing information for didanosine.
7.2 HIV-1 Protease Inhibitors
Tenofovir disoproxil fumarate decreases the AUC and Cmin of atazanavir [See Clinical Pharmacology (12.3) ]. When coadministered with tenofovir disoproxil fumarate, it is recommended that atazanavir 300 mg is given with ritonavir 100 mg. Tenofovir disoproxil fumarate should not be coadministered with atazanavir without ritonavir. Lopinavir/ritonavir, atazanavir coadministered with ritonavir, and darunavir coadministered with ritonavir have been shown to increase tenofovir concentrations [ See Clinical Pharmacology (12.3) ]. Tenofovir DF is a substrate of P-glycoprotein (Pgp) and breast cancer resistance protein (BCRP) transporters. When tenofovir DF is coadministered with an inhibitor of these transporters, an increase in absorption may be observed. Patients receiving tenofovir disoproxil fumarate concomitantly with lopinavir/ritonavir, ritonavir-boosted atazanavir, or ritonavir-boosted darunavir should be monitored for tenofovir disoproxil fumarate-associated adverse reactions. Tenofovir disoproxil fumarate should be discontinued in patients who develop tenofovir disoproxil fumarate-associated adverse reactions.
7.3 Hepatitis C Antiviral Agents
Coadministration of tenofovir disoproxil fumarate and EPCLUSA® (sofosbuvir/velpatasvir) or HARVONI® (ledipasvir/sofosbuvir) has been shown to increase tenofovir exposure [See Clinical Pharmacology (12.3)]. In patients receiving tenofovir disoproxil fumarate concomitantly with EPCLUSA, monitor for adverse reactions associated with tenofovir DF. In patients receiving tenofovir disoproxil fumarate concomitantly with HARVONI without an HIV-1 protease inhibitor/ritonavir or an HIV-1 protease inhibitor/cobicistat combination, monitor for adverse reactions associated with tenofovir DF. In patients receiving tenofovir disoproxil fumarate concomitantly with HARVONI and an HIV-1 protease inhibitor/ritonavir or an HIV-1 protease inhibitor/cobicistat combination, consider an alternative HCV or antiretroviral therapy, as the safety of increased tenofovir concentrations in this setting has not been established. If coadministration is necessary, monitor for adverse reactions associated with tenofovir DF.
7.4 Drugs Affecting Renal Function
Since tenofovir is primarily eliminated by the kidneys [See Clinical Pharmacology (12.3)] , coadministration of tenofovir disoproxil fumarate with drugs that reduce renal function or compete for active tubular secretion may increase serum concentrations of tenofovir and/or increase the concentrations of other renally eliminated drugs. Some examples include, but are not limited to, cidofovir, acyclovir, valacyclovir, ganciclovir, valganciclovir, aminoglycosides (e.g., gentamicin), and high-dose or multiple NSAIDs [ See Warnings and Precautions (5.2) ]. In the treatment of chronic hepatitis B, tenofovir disoproxil fumarate should not be administered in combination with HEPSERA (adefovir dipivoxil).
8 Use In Specific Populations
Nursing mothers: Women infected with HIV should be instructed not to breastfeed. (8.3 )
8.1 Pregnancy
Pregnancy Category B There are no adequate and well-controlled studies in pregnant women. Because animal reproduction studies are not always predictive of human response, tenofovir disoproxil fumarate should be used during pregnancy only if clearly needed. Antiretroviral Pregnancy Registry: To monitor fetal outcomes of pregnant women exposed to tenofovir disoproxil fumarate, an Antiretroviral Pregnancy Registry has been established. Healthcare providers are encouraged to register patients by calling 1-800-258-4263. Risk Summary Animal Data Reproduction studies have been performed in rats and rabbits at doses up to 14 and 19 times the human dose based on body surface area comparisons and revealed no evidence of impaired fertility or harm to the fetus due to tenofovir.
8.3 Nursing Mothers
Nursing Mothers: The Centers for Disease Control and Prevention recommend that HIV-1 infected mothers not breastfeed their infants to avoid risking postnatal transmission of HIV-1. Samples of breast milk obtained from five HIV-1 infected mothers in the first post-partum week show that tenofovir is secreted in human milk. The impact of this exposure in breastfed infants is unknown. Because of both the potential for HIV-1 transmission and the potential for serious adverse reactions in nursing infants, mothers should be instructed not to breastfeed if they are receiving tenofovir disoproxil fumarate.
8.4 Pediatric Use
Pediatric Patients 2 Years of Age and Older with HIV-1 infection The safety of tenofovir disoproxil fumarate in pediatric patients aged 2 to less than 18 years is supported by data from two randomized trials in which tenofovir disoproxil fumarate was administered to HIV-1 infected treatment-experienced subjects. In addition, the pharmacokinetic profile of tenofovir in patients 2 to less than 18 years of age at the recommended doses was similar to that found to be safe and effective in adult clinical trials [See Clinical Pharmacology (12.3)]. In Study 352, 92 treatment-experienced subjects 2 to less than 12 years of age with stable, virologic suppression on stavudine-or zidovudine-containing regimen were randomized to either replace stavudine or zidovudine with tenofovir disoproxil fumarate (N=44) or continue their original regimen (N=48) for 48 weeks. Five additional subjects over the age of 12 were enrolled and randomized (tenofovir disoproxil fumarate N=4, original regimen N=1) but are not included in the efficacy analysis. After 48 weeks, all eligible subjects were allowed to continue in the study receiving open-label tenofovir disoproxil fumarate. At Week 48, 89% of subjects in the tenofovir disoproxil fumarate treatment group and 90% of subjects in the stavudine or zidovudine treatment group had HIV-1 RNA concentrations less than 400 copies/mL. During the 48 week randomized phase of the study, 1 subject in the tenofovir disoproxil fumarate group discontinued the study prematurely because of virologic failure/lack of efficacy and 3 subjects (2 subjects in the tenofovir disoproxil fumarate group and 1 subject in the stavudine or zidovudine group) discontinued for other reasons. In Study 321, 87 treatment-experienced subjects 12 to less than 18 years of age were treated with tenofovir disoproxil fumarate (N=45) or placebo (N=42) in combination with an optimized background regimen (OBR) for 48 weeks. The mean baseline CD4 cell count was 374 cells/mm3 and the mean baseline plasma HIV-1 RNA was 4.6 log10 copies/mL. At baseline, 90% of subjects harbored NRTI resistance-associated substitutions in their HIV-1 isolates. Overall, the trial failed to show a difference in virologic response between the tenofovir disoproxil fumarate and placebo treatment groups. Subgroup analyses suggest the lack of difference in virologic response may be attributable to imbalances between treatment arms in baseline viral susceptibility to tenofovir disoproxil fumarate and OBR. Although changes in HIV-1 RNA in these highly treatment-experienced subjects were less than anticipated, the comparability of the pharmacokinetic and safety data to that observed in adults supports the use of tenofovir disoproxil fumarate in pediatric patients 12 years of age and older who weigh greater than or equal to 35 kg and whose HIV-1 isolate is expected to be sensitive to tenofovir disoproxil fumarate. [See Warnings and Precautions (5.6), Adverse Reactions (6.1), and Clinical Pharmacology (12.3)]. Safety and effectiveness of tenofovir disoproxil fumarate in pediatric patients younger than 2 years of age with HIV-1 infection have not been established. Pediatric Patients 12 Years of Age and Older with Chronic Hepatitis B In Study 115, 106 HBeAg negative (9%) and positive (91%) subjects aged 12 to less than 18 years with chronic HBV infection were randomized to receive blinded treatment with tenofovir disoproxil fumarate 300 mg (N=52) or placebo (N=54) for 72 weeks. At study entry, the mean HBV DNA was 8.1 log10 copies/mL and mean ALT was 101 U/L. Of 52 subjects treated with tenofovir disoproxil fumarate, 20 subjects were nucleos(t)ide-naïve and 32 subjects were nucleos(t)ide-experienced. Thirty-one of the 32 nucleos(t)ide-experienced subjects had prior lamivudine experience. At Week 72, 88% (46/52) of subjects in the tenofovir disoproxil fumarate group and 0% (0/54) of subjects in the placebo group had HBV DNA <400 copies/mL (69 IU/mL). Among subjects with abnormal ALT at baseline, 74% (26/35) of subjects receiving tenofovir disoproxil fumarate had normalized ALT at Week 72 compared to 31% (13/42) in the placebo group. One tenofovir disoproxil fumarate -treated subject experienced sustained HBsAg-loss and seroconversion to anti-HBs during the first 72 weeks of study participation. Safety and effectiveness of tenofovir disoproxil fumarate in pediatric patients younger than 12 years of age or less than 35 kg with chronic hepatitis B have not been established.
8.5 Geriatric Use
Clinical trials of tenofovir disoproxil fumarate did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects. In general, dose selection for the elderly patient should be cautious, keeping in mind the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy.
8.6 Patients with Impaired Renal Function
It is recommended that the dosing interval for tenofovir disoproxil fumarate be modified in patients with estimated creatinine clearance below 50 mL/min or in patients with ESRD who require dialysis [See Dosage and Administration (2.3), Clinical Pharmacology (12.3)].
10 Overdosage
Limited clinical experience at doses higher than the therapeutic dose of tenofovir disoproxil fumarate 300 mg is available. In Study 901, 600 mg tenofovir DF was administered to 8 subjects orally for 28 days. No severe adverse reactions were reported. The effects of higher doses are not known. If overdose occurs the patient must be monitored for evidence of toxicity and standard supportive treatment applied as necessary. Tenofovir is efficiently removed by hemodialysis with an extraction coefficient of approximately 54%. Following a single 300 mg dose of tenofovir disoproxil fumarate, a four-hour hemodialysis session removed approximately 10% of the administered tenofovir dose.
11 Description
Tenofovir disoproxil fumarate (a prodrug of tenofovir) which is a fumaric acid salt of bis-isopropoxycarbonyloxymethyl ester derivative of tenofovir. In vivo tenofovir DF is converted to tenofovir, an acyclic nucleoside phosphonate (nucleotide) analog of adenosine 5’-monophosphate. Tenofovir exhibits activity against HIV-1 reverse transcriptase. The chemical name of tenofovir DF is 9-[(R)-2 [[bis[[(isopropoxycarbonyl)oxy]methoxy]phosphinyl]methoxy]propyl]adenine fumarate (1:1). It has a molecular formula of C19H30N5O10P • C4H4O4 and a molecular weight of 635.52. It has the following structural formula: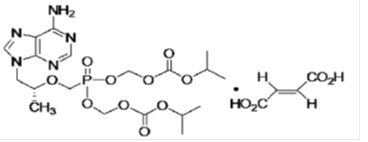
Tenofovir DF is a white to off-white crystalline powder with a solubility of 13.4 mg/mL in distilled water at 25°C. It has an octanol/phosphate buffer (pH 6.5) partition coefficient (log p) of 1.25 at 25°C. Tenofovir disoproxil fumarate is available as tablets. Tenofovir disoproxil fumarate tablets are for oral administration in strength 300 mg of tenofovir DF, which are equivalent to 245 mg of tenofovir disoproxil. Each tablet contains the following inactive ingredients: lactose monohydrate, microcrystalline cellulose, pregelatinized starch, croscarmellose sodium and magnesium stearate. The 300 mg tablets are coated with Opadry II Blue 32K505023, which contains FD&C Blue 2/Indigo Carmine Aluminium Lake, Hypromellose 2910, Lactose monohydrate, Titanium dioxide and Triacetin. In this insert, all dosages are expressed in terms of tenofovir DF except where otherwise noted.
12 Clinical Pharmacology
12.1 Mechanism of Action
Tenofovir DF is an antiviral drug [See Microbiology (12.4)].
12.3 Pharmacokinetics
The pharmacokinetics of tenofovir DF have been evaluated in healthy volunteers and HIV-1 infected individuals. Tenofovir pharmacokinetics are similar between these populations.
Absorption
Tenofovir disoproxil fumarate is a water soluble diester prodrug of the active ingredient tenofovir. The oral bioavailability of tenofovir from tenofovir disoproxil fumarate in fasted subjects is approximately 25%. Following oral administration of a single dose of tenofovir disoproxil fumarate 300 mg to HIV-1 infected subjects in the fasted state, maximum serum concentrations (Cmax) are achieved in 1.0 ± 0.4 hrs. Cmax and AUC values are 0.30 ± 0.09 mcg/mL and 2.29 ± 0.69 mcg•hr/mL, respectively.
The pharmacokinetics of tenofovir are dose proportional over a tenofovir disoproxil fumarate dose range of 75 to 600 mg and are not affected by repeated dosing.
Distribution
In vitro binding of tenofovir to human plasma or serum proteins is less than 0.7 and 7.2%, respectively, over the tenofovir concentration range 0.01 to 25 mcg/mL. The volume of distribution at steady-state is 1.3 ± 0.6 L/kg and 1.2 ± 0.4 L/kg, following intravenous administration of tenofovir 1.0 mg/kg and 3.0 mg/kg.
Metabolism and Elimination
In vitro studies indicate that neither tenofovir disoproxil nor tenofovir are substrates of CYP enzymes.
Following IV administration of tenofovir, approximately 70 to 80% of the dose is recovered in the urine as unchanged tenofovir within 72 hours of dosing. Following single dose, oral administration of tenofovir disoproxil fumarate, the terminal elimination half-life of tenofovir is approximately 17 hours. After multiple oral doses of tenofovir disoproxil fumarate 300 mg once daily (under fed conditions), 32 ± 10% of the administered dose is recovered in urine over 24 hours.
Tenofovir is eliminated by a combination of glomerular filtration and active tubular secretion. There may be competition for elimination with other compounds that are also renally eliminated.
Effects of Food on Oral Absorption
Administration of tenofovir disoproxil fumarate 300 mg tablets following a high-fat meal (~700 to 1000 kcal containing 40 to 50% fat) increases the oral bioavailability, with an increase in tenofovir AUC0-∞ of approximately 40% and an increase in Cmax of approximately 14%. However, administration of tenofovir disoproxil fumarate with a light meal did not have a significant effect on the pharmacokinetics of tenofovir when compared to fasted administration of the drug. Food delays the time to tenofovir Cmax by approximately 1 hour. Cmax and AUC of tenofovir are 0.33 ± 0.12 mcg/mL and 3.32 ± 1.37 mcg•hr/mL following multiple doses of tenofovir disoproxil fumarate 300 mg once daily in the fed state, when meal content was not controlled.
Special Populations
Race: There were insufficient numbers from racial and ethnic groups other than Caucasian to adequately determine potential pharmacokinetic differences among these populations.
Gender: Tenofovir pharmacokinetics are similar in male and female subjects.
Pediatric Patients 2 Years of Age and Older: Steady-state pharmacokinetics of tenofovir were evaluated in 31 HIV-1 infected pediatric subjects 2 to less than 18 years (Table 10). Tenofovir exposure achieved in these pediatric subjects receiving oral once daily doses of tenofovir disoproxil fumarate 300 mg (tablet) or 8 mg/kg of body weight (powder) up to a maximum dose of 300 mg was similar to exposures achieved in adults receiving once-daily doses of tenofovir disoproxil fumarate 300 mg.
Table 10 Mean (± SD) Tenofovir Pharmacokinetic Parameters by Age Groups for HIV-1-infected Pediatric Patients Dose and Formulation 300 mg Tablet 8 mg/kg Oral Powder 12 to <18 Years (N=8) 2 to <12 Years (N=23) Cmax (mcg/mL) 0.38 ± 0.13 0.24 ± 0.13 AUCtau (mcg•hr/mL) 3.39 ± 1.22 2.59 ± 1.06
Tenofovir exposures in 52 HBV-infected pediatric subjects (12 to less than 18 years of age) receiving oral once-daily doses of tenofovir disoproxil fumarate 300 mg tablet were comparable to exposures achieved in HIV-1 infected adults and adolescents receiving once-daily doses of 300 mg.
Geriatric Patients: Pharmacokinetic trials have not been performed in the elderly (65 years and older).
Patients with Impaired Renal Function: The pharmacokinetics of tenofovir are altered in subjects with renal impairment [See Warnings and Precautions (5.2)]. In subjects with creatinine clearance below 50 mL/min or with end-stage renal disease (ESRD) requiring dialysis, Cmax, and AUC0-∞ of tenofovir were increased (Table 11). It is recommended that the dosing interval for tenofovir disoproxil fumarate be modified in patients with estimated creatinine clearance below 50 mL/min or in patients with ESRD who require dialysis [See Dosage and Administration (2.3)].
Table 11 Pharmacokinetic Parameters (Mean ± SD) of Tenofovira in Subjects with Varying Degrees of Renal Function a 300 mg, single dose of tenofovir disoproxil fumarate. Baseline Creatinine Clearance (mL/min) >80 (N=3) 50 to 80 (N=10) 30 to 49 (N=8) 12 to 29 (N=11) Cmax (mcg/mL) 0.34 ± 0.03 0.33 ± 0.06 0.37 ± 0.16 0.60 ± 0.19 AUC0–∞ (mcg•hr/mL) 2.18 ± 0.26 3.06 ± 0.93 6.01 ± 2.50 15.98 ± 7.22 CL/F (mL/min) 1043.7 ± 115.4 807.7 ± 279.2 444.4 ± 209.8 177.0 ± 97.1 CLrenal (mL/min) 243.5 ± 33.3 168.6 ± 27.5 100.6 ± 27.5 43.0 ± 31.2
Tenofovir is efficiently removed by hemodialysis with an extraction coefficient of approximately 54%. Following a single 300 mg dose of tenofovir disoproxil fumarate, a four-hour hemodialysis session removed approximately 10% of the administered tenofovir dose.
Patients with Hepatic Impairment: The pharmacokinetics of tenofovir following a 300 mg single dose of tenofovir disoproxil fumarate have been studied in non-HIV infected subjects with moderate to severe hepatic impairment. There were no substantial alterations in tenofovir pharmacokinetics in subjects with hepatic impairment compared with unimpaired subjects. No change in tenofovir disoproxil fumarate dosing is required in patients with hepatic impairment.
Assessment of Drug Interactions
At concentrations substantially higher (~300-fold) than those observed in vivo, tenofovir did not inhibit in vitro drug metabolism mediated by any of the following human CYP isoforms: CYP3A4, CYP2D6, CYP2C9, or CYP2E1. However, a small (6%) but statistically significant reduction in metabolism of CYP1A substrate was observed. Based on the results of in vitro experiments and the known elimination pathway of tenofovir, the potential for CYP-mediated interactions involving tenofovir with other medicinal products is low.
Tenofovir disoproxil fumarate has been evaluated in healthy volunteers in combination with other antiretroviral and potential concomitant drugs. Tables 12 and 13 summarize pharmacokinetic effects of coadministered drug on tenofovir pharmacokinetics and effects of tenofovir disoproxil fumarate on the pharmacokinetics of coadministered drug. Coadministration of tenofovir disoproxil fumarate with didanosine results in changes in the pharmacokinetics of didanosine that may be of clinical significance. Concomitant dosing of tenofovir disoproxil fumarate with didanosine significantly increases the Cmax and AUC of didanosine. When didanosine 250 mg enteric-coated capsules were administered with tenofovir disoproxil fumarate, systemic exposures of didanosine were similar to those seen with the 400 mg enteric-coated capsules alone under fasted conditions (Table 13). The mechanism of this interaction is unknown.
No clinically significant drug interactions have been observed between tenofovir disoproxil fumarate and efavirenz, methadone, nelfinavir, oral contraceptives, ribavirin, or sofosbuvir.
Table 12 Drug Interactions: Changes in Pharmacokinetic Parameters for Tenofovira in the Presence of the Coadministered Drug a. Subjects received tenofovir disoproxil fumarate 300 mg once daily.b. Increase = ↑; Decrease = ↓; No Effect = ⇔c. Reyataz Prescribing Information.d. Prezista Prescribing Information. e Data generated from simultaneous dosing with HARVONI (ledipasvir/sofosbuvir). Staggered administration (12 hours apart) provide similar results.f. Comparison based on exposures when administered as atazanavir/ritonavir + emtricitabine/tenofovir DF.g. Comparison based on exposures when administered as darunavir/ritonavir + emtricitabine/tenofovir DF.h Study conducted with ATRIPLA (efavirenz/emtricitabine/tenofovir DF) coadministered with HARVONI.i. Study conducted with COMPLERA (emtricitabine/rilpivirine/tenofovir DF) coadministered with HARVONI. j. Study conducted with TRUVADA (emtricitabine/tenofovir DF) + dolutegravir coadministered with HARVONI. k. Study conducted with ATRIPLA coadministered with SOVALDI® (sofosbuvir). l. Comparison based on exposures when administered as atazanavir/ritonavir + emtricitabine/tenofovir DF. m. Comparison based on exposures when administered as darunavir/ritonavir + emtricitabine/tenofovir DF. n. Study conducted with ATRIPLA coadministered with EPCLUSA (sofosbuvir/velpatasvir). o. Study conducted with STRIBILD (elvitegravir/cobicistat/emtricitabine/tenofovir DF) coadministered with EPCLUSA. p. Study conducted with COMPLERA coadministered with EPCLUSA. q. Administered as raltegravir + emtricitabine/tenofovir DF. r. Aptivus Prescribing Information. Coadministered Drug Dose of Coadministered Drug (mg) N % Change of Tenofovir Pharmacokinetic Parametersb (90% CI) Cmax AUC Cmin Abacavirc 400 once daily × 14 days 33 ↑ 14 (↑ 8 to ↑ 20) ↑ 24 (↑ 21 to ↑ 28) ↑ 22 (↑ 15 to ↑ 30) Atazanavir/ Ritonavirc 300/100 once daily 12 ↑ 34 (↑ 20 to ↑ 51) ↑ 37 (↑ 30 to ↑ 45) ↑ 29 (↑ 21 to ↑ 36) Darunavir/Ritonavird 300/100 twice daily 12 ↑24 (↑ 8 to ↑ 42) ↑ 22 (↑ 10 to ↑ 35) ↑ 37 (↑ 19 to ↑ 57) Indinavir 800 three times daily × 7 days 13 ↑14 (↓ 3 to ↑ 33) ⇔ ⇔ Ledipasvir/ Sofosbuvire,f 90/400 once daily x 10 days 24 ↑ 47 (↑ 37 to ↑ 58) ↑ 35 (↑ 29 to ↑ 42) ↑ 47 (↑ 38 to ↑ 57) Ledipasvir/ Sofosbuvire,g 23 ↑ 64 (↑ 54 to ↑ 74) ↑ 50 (↑ 42 to ↑ 59) ↑ 59 (↑ 49 to ↑ 70) Ledipasvir/ Sofosbuvirh 90/400 once daily x 14 days 15 ↑ 79 (↑ 56 to ↑ 104) ↑ 98 (↑ 77 to ↑ 123) ↑ 163 (↑ 132 to ↑ 197) Ledipasvir/ Sofosbuviri 90/400 once daily x 10 days 14 ↑ 32 (↑ 25 to ↑ 39) ↑ 40 (↑ 31 to ↑ 50) ↑ 91 (↑74 to ↑ 110) Ledipasvir/ Sofosbuvirj 90/400 once daily x 10 days 29 ↑ 61 (↑ 51 to ↑ 72) ↑ 65 (↑ 59 to ↑ 71) ↑ 115 (↑ 105 to ↑ 126) Lopinavir/ Ritonavir 400/100 twice daily × 14 days 24 ⇔ ↑ 32 (↑ 25 to ↑ 38) ↑ 51 (↑ 37 to ↑ 66) Saquinavir/ Ritonavir 1000/100 twice daily × 14 days 35 ⇔ ⇔ ↑ 23 (↑ 16 to ↑ 30) Sofosbuvirk 400 single dose 16 ↑ 25 (↑8 to ↑ 45) ⇔ ⇔ Sofosbuvir/ Velpatasvirl 400/100 once daily 24 ↑ 55 (↑ 43 to ↑ 68) ↑ 30 (↑ 24 to ↑ 36) ↑ 39 (↑ 31 to ↑ 48) Sofosbuvir/ Velpatasvirm 400/100 once daily 29 ↑ 55 (↑ 45 to ↑ 66) ↑ 39 (↑ 33 to ↑ 44) ↑ 52 (↑ 45 to ↑ 59) Sofosbuvir/ Velpatasvirn 400/100 once daily 15 ↑ 77 (↑ 53 to ↑ 104) ↑ 81 (↑ 68 to ↑ 94) ↑ 121 (↑ 100 to ↑ 143) Sofosbuvir/ Velpatasviro 400/100 once daily 24 ↑ 36 (↑ 25 to ↑ 47) ↑ 35 (↑ 29 to ↑ 42) ↑ 45 (↑ 39 to ↑ 51) Sofosbuvir/ Velpatasvirp 400/100 once daily 24 ↑ 44 (↑ 33 to ↑ 55) ↑ 40 (↑ 34 to ↑ 46) ↑ 84 (↑ 76 to ↑ 92) Sofosbuvir/ Velpatasvirq 400/100 once daily 30 ↑ 46 (↑ 39 to ↑ 54) ↑ 40 (↑ 34 to ↑ 45) ↑ 70 (↑ 61 to ↑ 79) Tacrolimus 0.05 mg/kg twice daily x 7 days 21 ↑ 13 (↑1 to ↑ 27) ⇔ ⇔ Tipranavir/ Ritonavirr 500/100 twice daily 22 ↓ 23 (↓ 32 to ↓ 13) ↓ 2 (↓ 9 to ↑ 5) ↑ 7 (↓ 2 to ↑ 17) 750/200 twice daily (23 doses) 20 ↓ 38 (↓ 46 to ↓ 29) ↑ 2 (↓ 6 to ↑ 10) ↑ 14 (↑ 1 to ↑ 27)
No effect on the pharmacokinetic parameters of the following coadministered drugs was observed with tenofovir disoproxil fumarate: abacavir, didanosine (buffered tablets), emtricitabine, entecavir, and lamivudine.
Table 13 Drug Interactions: Changes in Pharmacokinetic Parameters for Coadministered Drug in the Presence of tenofovir disoproxil fumarate a. Increase = ↑; Decrease = ↓; No Effect = ⇔; NA = Not Applicableb. Reyataz Prescribing Information.c. In HIV-infected subjects, addition of tenofovir DF to atazanavir 300 mg plus ritonavir 100 mg, resulted in AUC and Cmin values of atazanavir that were 2.3-and 4-fold higher than the respective values observed for atazanavir 400 mg when given alone.d. Prezista Prescribing Information.e. Videx EC Prescribing Information. Subjects received didanosine enteric-coated capsules.f. 373 kcal, 8.2 g fatg. Compared with didanosine (enteric-coated) 400 mg administered alone under fasting conditions.h. Increases in AUC and Cmin are not expected to be clinically relevant; hence no dose adjustments are required when tenofovir DF and ritonavir-boosted saquinavir are coadministered.i. Aptivus Prescribing Information. Coadministered Drug Dose of Coadministered Drug (mg) N % Change of Coadministered Drug Pharmacokinetic Parametersa (90% CI) Cmax AUC Cmin Abacavir 300 once 8 ↑ 12 (↓1 to ↑ 16) ⇔ NA Atazanavirb 400 once daily × 14 days 34 ↓ 21 (↓27 to ↓ 14) ↓ 25 (↓30 to ↓ 19) ↓ 40 (↓48 to↓32) Atazanavirb Atazanavir/ Ritonavir 300/100 once daily × 42 days 10 ↓ 28 (↓50 to ↑ 5) ↓ 25c (↓42 to ↓ 3) ↓ 23c (↓46 to ↑ 10) Darunavird Darunavir/Ritonavir 300/100 once daily 12 ↑ 16 (↓6 to ↑ 42) ↑ 21 (↓5 to ↑ 54) ↑ 24 (↓10 to ↑ 69) Didanosinee 250 once, simultaneously with tenofovir disoproxil fumarate and a light mealf 33 ↓ 20g (↓32 to ↓ 7) ⇔g NA Emtricitabine 200 once daily × 7 days 17 ⇔ ⇔ ↑ 20 (↑12 to ↑ 29) Entecavir 1 mg once daily × 10 days 28 ⇔ ↑ 13 (↑11 to ↑ 15) ⇔ Indinavir 800 three times daily × 7 days 12 ↓ 11 (↓30 to ↑12) ⇔ ⇔ Lamivudine 150 twice daily × 7 days 15 ↓ 24 (↓34 to ↓ 12) ⇔ ⇔ Lopinavir Ritonavir Lopinavir/Ritonavir 400/100 twice daily × 14 days 24 ⇔ ⇔ ⇔ Saquinavir Ritonavir Saquinavir/Ritonavir 1000/100 twice daily × 14 days 32 ↑ 22 (↑6 to ↑ 41) ↑ 29h (↑12 to ↑ 48) ↑ 47h (↑23 to ↑ 76) ⇔ ⇔ ↑ 23 (↑3 to ↑ 46) Tacrolimus 0.05 mg/kg twice daily x 7 days 21 ⇔ ⇔ ⇔ Tipranaviri Tipranavir/Ritonavir 500/100 twice daily 22 ↓ 17 (↓26 to ↓ 6) ↓18 (↓25 to ↓ 9) ↓ 21 (↓30 to ↓ 10) Tipranavir/Ritonavir 750/200 twice daily (23 doses) 20 ↓ 11 (↓16 to ↓ 4) ↓ 9 (↓15 to ↓ 3) ↓ 12 (↓22 to ↓0) 12.4 Microbiology
Mechanism of Action
Tenofovir DF is an acyclic nucleoside phosphonate diester analog of adenosine monophosphate. Tenofovir DF requires initial diester hydrolysis for conversion to tenofovir and subsequent phosphorylations by cellular enzymes to form tenofovir diphosphate, an obligate chain terminator. Tenofovir diphosphate inhibits the activity of HIV-1 reverse transcriptase and HBV reverse transcriptase by competing with the natural substrate deoxyadenosine 5’-triphosphate and, after incorporation into DNA, by DNA chain termination. Tenofovir diphosphate is a weak inhibitor of mammalian DNA polymerases α, β, and mitochondrial DNA polymerase γ.
Activity against HIV
Antiviral Activity
The antiviral activity of tenofovir against laboratory and clinical isolates of HIV-1 was assessed in lymphoblastoid cell lines, primary monocyte/macrophage cells and peripheral blood lymphocytes. The EC50 (50% effective concentration) values for tenofovir were in the range of 0.04 mcM to 8.5 mcM. In drug combination studies, tenofovir was not antagonistic with nucleoside reverse transcriptase inhibitors (abacavir, didanosine, lamivudine, stavudine, zalcitabine, zidovudine), non-nucleoside reverse transcriptase inhibitors (delavirdine, efavirenz, nevirapine), and protease inhibitors (amprenavir, indinavir, nelfinavir, ritonavir, saquinavir). Tenofovir displayed antiviral activity in cell culture against HIV-1 clades A, B, C, D, E, F, G, and O (EC50 values ranged from 0.5 mcM to 2.2 mcM) and strain-specific activity against HIV-2 (EC50 values ranged from 1.6 mcM to 5.5 mcM).
Resistance
HIV-1 isolates with reduced susceptibility to tenofovir have been selected in cell culture. These viruses expressed a K65R substitution in reverse transcriptase and showed a 2 to 4-fold reduction in susceptibility to tenofovir. In addition, a K70E substitution in HIV-1 reverse transcriptase has been selected by tenofovir and results in low-level reduced susceptibility to tenofovir.
In Study 903 of treatment-naïve subjects (Tenofovir disoproxil fumarate + lamivudine + efavirenz versus stavudine + lamivudine + efavirenz) [See Clinical Studies (14.1)], genotypic analyses of isolates from subjects with virologic failure through Week 144 showed development of efavirenz and lamivudine resistance-associated substitutions to occur most frequently and with no difference between the treatment arms. The K65R substitution occurred in 8/47 (17%) of analyzed patient isolates in the tenofovir disoproxil fumarate arm and in 2/49 (4%) of analyzed patient isolates in the stavudine arm. Of the 8 subjects whose virus developed K65R in the tenofovir disoproxil fumarate arm through 144 weeks, 7 occurred in the first 48 weeks of treatment and one at Week 96. One patient in the tenofovir disoproxil fumarate arm developed the K70E substitution in the virus. Other substitutions resulting in resistance to tenofovir disoproxil fumarate were not identified in this trial.
In Study 934 of treatment-naïve subjects (Tenofovir disoproxil fumarate + EMTRIVA + efavirenz versus zidovudine (AZT)/lamivudine (3TC) + efavirenz) [See Clinical Studies (14.1)], genotypic analysis performed on HIV-1 isolates from all confirmed virologic failure subjects with greater than 400 copies/mL of HIV-1 RNA at Week 144 or early discontinuation showed development of efavirenz resistance-associated substitutions occurred most frequently and was similar between the two treatment arms. The M184V substitution, associated with resistance to EMTRIVA and lamivudine, was observed in 2/19 of analyzed subject isolates in the tenofovir disoproxil fumarate + EMTRIVA group and in 10/29 of analyzed subject isolates in the zidovudine/lamivudine group. Through 144 weeks of Study 934, no subjects have developed a detectable K65R substitution in their HIV-1 as analyzed through standard genotypic analysis.
Cross Resistance
Cross resistance among certain reverse transcriptase inhibitors has been recognized. The K65R and K70E substitutions selected by tenofovir are also selected in some HIV-1 infected subjects treated with abacavir or didanosine. HIV-1 isolates with this substitution also show reduced susceptibility to emtricitabine and lamivudine. Therefore, cross resistance among these drugs may occur in patients whose virus harbors the K65R or K70E substitution. HIV-1 isolates from subjects (N=20) whose HIV-1 expressed a mean of three zidovudine-associated reverse transcriptase substitutions (M41L, D67N, K70R, L210W, T215Y/F, or K219Q/E/N), showed a 3.1-fold decrease in the susceptibility to tenofovir.
In Studies 902 and 907 conducted in treatment-experienced subjects (Tenofovir disoproxil fumarate + Standard Background Therapy (SBT) compared to Placebo + SBT) [See Clinical Studies (14.1)], 14/304 (5%) of the tenofovir disoproxil fumarate -treated subjects with virologic failure through Week 96 had greater than 1.4-fold (median 2.7-fold) reduced susceptibility to tenofovir. Genotypic analysis of the baseline and failure isolates showed the development of the K65R substitution in the HIV-1 reverse transcriptase gene.
The virologic response to tenofovir disoproxil fumarate therapy has been evaluated with respect to baseline viral genotype (N=222) in treatment-experienced subjects participating in Studies 902 and 907. In these clinical trials, 94% of the participants evaluated had baseline HIV-1 isolates expressing at least one NRTI substitution. Virologic responses for subjects in the genotype substudy were similar to the overall trial results.
Several exploratory analyses were conducted to evaluate the effect of specific substitutions and substitutional patterns on virologic outcome. Because of the large number of potential comparisons, statistical testing was not conducted. Varying degrees of cross resistance of tenofovir disoproxil fumarate to pre-existing zidovudine resistance-associated substitutions (M41L, D67N, K70R, L210W, T215Y/F, or K219Q/E/N) were observed and appeared to depend on the type and number of specific substitutions. Tenofovir disoproxil fumarate -treated subjects whose HIV-1 expressed 3 or more zidovudine resistance-associated substitutions that included either the M41L or L210W reverse transcriptase substitution showed reduced responses to tenofovir disoproxil fumarate therapy; however, these responses were still improved compared with placebo. The presence of the D67N, K70R, T215Y/F, or K219Q/E/N substitution did not appear to affect responses to tenofovir disoproxil fumarate therapy. Subjects whose virus expressed an L74V substitution without zidovudine resistance associated substitutions (N=8) had reduced response to tenofovir disoproxil fumarate. Limited data are available for subjects whose virus expressed a Y115F substitution (N=3), Q151M substitution (N=2), or T69 insertion (N=4), all of whom had a reduced response.
In the protocol defined analyses, virologic response to tenofovir disoproxil fumarate was not reduced in subjects with HIV-1 that expressed the abacavir/emtricitabine/lamivudine resistance-associated M184V substitution. HIV-1 RNA responses among these subjects were durable through Week 48.
Studies 902 and 907 Phenotypic Analyses
Phenotypic analysis of baseline HIV-1 from treatment-experienced subjects (N=100) demonstrated a correlation between baseline susceptibility to tenofovir disoproxil fumarate and response to tenofovir disoproxil fumarate therapy. Table 14 summarizes the HIV-1 RNA response by baseline tenofovir disoproxil fumarate susceptibility.
Table 14 HIV-1 RNA Response at Week 24 by Baseline tenofovir disoproxil fumarate Susceptibility (Intent-To-Treat)a a. Tenofovir susceptibility was determined by recombinant phenotypic Antivirogram assay (Virco). b. Fold change in susceptibility from wild-type. c. Average HIV-1 RNA change from baseline through Week 24 (DAVG24) in log10 copies/mL. Baseline tenofovir disoproxil fumarate Susceptibilityb Change in HIV-1 RNAc (N) <1 –0.74 (35) >1 and ≤3 –0.56 (49) >3 and ≤4 –0.3 (7) >4 –0.12 (9)
Activity against HBV
Antiviral Activity
The antiviral activity of tenofovir against HBV was assessed in the HepG2 2.2.15 cell line. The EC50 values for tenofovir ranged from 0.14 to 1.5 mcM, with CC50 (50% cytotoxicity concentration) values greater than 100 mcM. In cell culture combination antiviral activity studies of tenofovir with the nucleoside HBV reverse transcriptase inhibitors entecavir, lamivudine, and telbivudine, and with the nucleoside HIV-1 reverse transcriptase inhibitor emtricitabine, no antagonistic activity was observed.
Resistance
Cumulative tenofovir disoproxil fumarate genotypic resistance has been evaluated annually for up to 384 weeks in Studies 0102, 0103, 0106, 0108, and 0121 with the paired HBV reverse transcriptase amino acid sequences of the pretreatment and on-treatment isolates from subjects who received at least 24 weeks of tenofovir disoproxil fumarate monotherapy and remained viremic with HBV DNA greater than or equal to 400 copies/mL (69 IU/mL) at the end of each study year (or at discontinuation of tenofovir disoproxil fumarate monotherapy) using an as-treated analysis. In the nucleotide-naïve population from Studies 0102 and 0103, HBeAg-positive subjects had a higher baseline viral load than HBeAg-negative subjects and a significantly higher proportion of the subjects remained viremic at their last time point on tenofovir disoproxil fumarate monotherapy (15% versus 5%, respectively).
HBV isolates from these subjects who remained viremic showed treatment-emergent substitutions (Table 15); however, no specific substitutions occurred at a sufficient frequency to be associated with resistance to tenofovir disoproxil fumarate (genotypic and phenotypic analyses).
Table 15 Amino Acid Substitutions in Viremic Subjects across HBV Trials of tenofovir disoproxil fumarate a. Nucleotide-naïve subjects from Studies 0102 (N=246) and 0103 (N=171) receiving up to 384 weeks of treatment with tenofovir disoproxil fumarate.b. HEPSERA-experienced subjects from Studies 0102/0103 (N=195) and 0106 (N=52) receiving up to 336 weeks of treatment with tenofovir disoproxil fumarate after switching to tenofovir disoproxil fumarate from HEPSERA. Study 0106, a randomized, double-blind, 168-week Phase 2 trial, has been completed.c. Lamivudine-resistant subjects from Study 0121 (N=136) receiving up to 96 weeks of treatment with tenofovir disoproxil fumarate after switching to tenofovir disoproxil fumarate from lamivudine.d. Subjects with decompensated liver disease from Study 0108 (N=39) receiving up to 48 weeks of treatment with tenofovir disoproxil fumarate.e. Denominator includes those subjects who were viremic at last time point on tenofovir disoproxil fumarate monotherapy and had evaluable paired genotypic data.f. Of the 18 subjects with treatment-emergent amino acid substitutions during Studies 0102 and 0103, 5 subjects had substitutions at conserved sites and 13 subjects had substitutions only at polymorphic sites, and 8 subjects had only transient substitutions that were not detected at the last time point on tenofovir disoproxil fumarate.g. Of the 11 HEPSERA-experienced subjects with treatment-emergent amino acid substitutions, 2 subjects had substitutions at conserved sites and 9 had substitutions only at polymorphic sites.h. Of the 6 lamivudine-resistant subjects with treatment-emergent substitutions during Study 0121, 3 subjects had substitutions at conserved sites and 3 had substitutions only at polymorphic sites. Compensated Liver Disease Decompensated Liver Disease (N=39)d Nucleotide-Naïve (N=417)a HEPSERA-Experienced (N=247)b Lamivudine- Resistant (N=136)c Viremic at Last Time Point on tenofovir disoproxil fumarate 38/417 (9%) 37/247 (15%) 9/136 (7%) 7/39 (18%) Treatment-Emergent Amino Acid Substitutionse 18f/32 (56%) 11g/31 (35%) 6h/8 (75%) 3/5 (60%)
Cross Resistance
Cross resistance has been observed between HBV nucleoside/nucleotide analogue reverse transcriptase inhibitors.
In cell based assays, HBV strains expressing the rtV173L, rtL180M, and rtM204I/V substitutions associated with resistance to lamivudine and telbivudine showed a susceptibility to tenofovir ranging from 0.7-to 3.4-fold that of wild type virus. The rtL180M and rtM204I/V double substitutions conferred 3.4-fold reduced susceptibility to tenofovir.
HBV strains expressing the rtL180M, rtT184G, rtS202G/I, rtM204V, and rtM250V substitutions associated with resistance to entecavir showed a susceptibility to tenofovir ranging from 0.6-to 6.9-fold that of wild type virus.
HBV strains expressing the adefovir resistance-associated substitutions rtA181V and/or rtN236T showed reductions in susceptibility to tenofovir ranging from 2.9-to 10-fold that of wild type virus. Strains containing the rtA181T substitution showed changes in susceptibility to tenofovir ranging from 0.9-to 1.5-fold that of wild type virus.
One hundred fifty-two subjects initiating tenofovir disoproxil fumarate therapy in Studies 0102, 0103, 0106, 0108, and 0121 harbored HBV with known resistance substitutions to HBV nucleos(t)ide analogue reverse transcriptase inhibitors: 14 with adefovir resistance-associated substitutions (rtA181S/T/V and/or rtN236T), 135 with lamivudine resistance-associated substitutions (rtM204I/V), and 3 with both adefovir and lamivudine resistance-associated substitutions. Following up to 384 weeks of tenofovir disoproxil fumarate treatment, 10 of the 14 subjects with adefovir-resistant HBV, 124 of the 135 subjects with lamivudine-resistant HBV, and 2 of the 3 subjects with both adefovir-and lamivudine-resistant HBV achieved and maintained virologic suppression (HBV DNA less than 400 copies/mL [69 IU/mL]). Three of the 5 subjects whose virus harbored both the rtA181T/V and rtN236T substitutions remained viremic.
13 Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Long-term oral carcinogenicity studies of tenofovir DF in mice and rats were carried out at exposures up to approximately 16 times (mice) and 5 times (rats) those observed in humans at the therapeutic dose for HIV-1 infection. At the high dose in female mice, liver adenomas were increased at exposures 16 times that in humans. In rats, the study was negative for carcinogenic findings at exposures up to 5 times that observed in humans at the therapeutic dose.
Mutagenesis
Tenofovir DF was mutagenic in the in vitro mouse lymphoma assay and negative in an in vitro bacterial mutagenicity test (Ames test). In an in vivo mouse micronucleus assay, tenofovir DF was negative when administered to male mice.
Impairment of Fertility
There were no effects on fertility, mating performance or early embryonic development when tenofovir DF was administered to male rats at a dose equivalent to 10 times the human dose based on body surface area comparisons for 28 days prior to mating and to female rats for 15 days prior to mating through day seven of gestation. There was, however, an alteration of the estrous cycle in female rats.
13.2 Animal Toxicology and/or Pharmacology
Tenofovir and tenofovir DF administered in toxicology studies to rats, dogs, and monkeys at exposures (based on AUCs) greater than or equal to 6 fold those observed in humans caused bone toxicity. In monkeys the bone toxicity was diagnosed as osteomalacia. Osteomalacia observed in monkeys appeared to be reversible upon dose reduction or discontinuation of tenofovir. In rats and dogs, the bone toxicity manifested as reduced bone mineral density. The mechanism(s) underlying bone toxicity is unknown.
Evidence of renal toxicity was noted in 4 animal species. Increases in serum creatinine, BUN, glycosuria, proteinuria, phosphaturia, and/or calciuria and decreases in serum phosphate were observed to varying degrees in these animals. These toxicities were noted at exposures (based on AUCs) 2 to 20 times higher than those observed in humans. The relationship of the renal abnormalities, particularly the phosphaturia, to the bone toxicity is not known.
14 Clinical Studies
14.1 Clinical Efficacy in Adults with HIV-1 Infection
Treatment-Naïve Adult Patients
Study 903
Data through 144 weeks are reported for Study 903, a double-blind, active-controlled multicenter trial comparing tenofovir disoproxil fumarate (300 mg once daily) administered in combination with lamivudine and efavirenz versus stavudine (d4T), lamivudine, and efavirenz in 600 antiretroviral-naïve subjects. Subjects had a mean age of 36 years (range 18 to 64), 74% were male, 64% were Caucasian, and 20% were Black. The mean baseline CD4+ cell count was 279 cells/mm3 (range 3 to 956) and median baseline plasma HIV-1 RNA was 77,600 copies/mL (range 417 to 5,130,000). Subjects were stratified by baseline HIV-1 RNA and CD4+ cell count. Forty-three percent of subjects had baseline viral loads >100,000 copies/mL and 39% had CD4+ cell counts <200 cells/mm3. Treatment outcomes through 48 and 144 weeks are presented in Table 16.
Table 16 Outcomes of Randomized Treatment at Week 48 and 144 (Study 903) a. Subjects achieved and maintained confirmed HIV-1 RNA <400 copies/mL through Week 48 and 144.b. Includes confirmed viral rebound and failure to achieve confirmed <400 copies/mL through Week 48 and 144.c. Includes lost to follow-up, subject’s withdrawal, noncompliance, protocol violation and other reasons. Outcomes At Week 48 At Week 144 Tenofovir disoproxil fumarate +3TC +EFV (N=299) d4T+3TC +EFV (N=301) Tenofovir disoproxil fumarate +3TC +EFV (N=299) d4T+3TC +EFV (N=301) Respondera 79% 82% 68% 62% Virologic failureb 6% 4% 10% 8% Rebound 5% 3% 8% 7% Never suppressed 0% 1% 0% 0% Added an antiretroviral agent 1% 1% 2% 1% Death <1% 1% <1% 2% Discontinued due to adverse event 6% 6% 8% 13% Discontinued for other reasonsc 8% 7% 14% 15%
Achievement of plasma HIV-1 RNA concentrations of less than 400 copies/mL at Week 144 was similar between the two treatment groups for the population stratified at baseline on the basis of HIV-1 RNA concentration (> or ≤100,000 copies/mL) and CD4+ cell count (< or ≥200 cells/mm3). Through 144 weeks of therapy, 62% and 58% of subjects in the tenofovir disoproxil fumarate and stavudine arms, respectively, achieved and maintained confirmed HIV-1 RNA <50 copies/mL. The mean increase from baseline in CD4+ cell count was 263 cells/mm3 for the tenofovir disoproxil fumarate arm and 283 cells/mm3 for the stavudine arm.
Through 144 weeks, 11 subjects in the tenofovir disoproxil fumarate group and 9 subjects in the stavudine group experienced a new CDC Class C event.
Study 934
Data through 144 weeks are reported for Study 934, a randomized, open-label, active-controlled multicenter trial comparing emtricitabine + tenofovir disoproxil fumarate administered in combination with efavirenz versus zidovudine/lamivudine fixed-dose combination administered in combination with efavirenz in 511 antiretroviral-naïve subjects. From Weeks 96 to 144 of the trial, subjects received a fixed-dose combination of emtricitabine and tenofovir DF with efavirenz in place of emtricitabine + tenofovir disoproxil fumarate with efavirenz. Subjects had a mean age of 38 years (range 18 to 80), 86% were male, 59% were Caucasian, and 23% were Black. The mean baseline CD4+ cell count was 245 cells/mm3 (range 2 to 1191) and median baseline plasma HIV-1 RNA was 5.01 log10 copies/mL (range 3.56 to 6.54). Subjects were stratified by baseline CD4+ cell count (< or ≥200 cells/mm3); 41% had CD4+ cell counts <200 cells/mm3 and 51% of subjects had baseline viral loads >100,000 copies/mL. Treatment outcomes through 48 and 144 weeks for those subjects who did not have efavirenz resistance at baseline are presented in Table 17.
Table 17 Outcomes of Randomized Treatment at Week 48 and 144 (Study 934) a. Subjects who were responders at Week 48 or Week 96 (HIV-1 RNA <400 copies/mL) but did not consent to continue the trial after Week 48 or Week 96 were excluded from analysis.b. Subjects achieved and maintained confirmed HIV-1 RNA <400 copies/mL through Weeks 48 and 144.c. Includes confirmed viral rebound and failure to achieve confirmed <400 copies/mL through Weeks 48 and 144.d. Includes lost to follow-up, subject withdrawal, noncompliance, protocol violation and other reasons. Outcomes At Week 48 At Week 144 FTC + Tenofovir disoproxil fumarate +EFV (N=244) AZT/3TC +EFV (N=243) FTC + Tenofovir disoproxil fumarate +EFV (N=227)a AZT/3TC +EFV (N=229)a Responderb 84% 73% 71% 58% Virologic failurec 2% 4% 3% 6% Rebound 1% 3% 2% 5% Never suppressed 0% 0% 0% 0% Change in antiretroviral regimen 1% 1% 1% 1% Death <1% 1% 1% 1% Discontinued due to adverse event 4% 9% 5% 12% Discontinued for other reasonsd 10% 14% 20% 22%
Through Week 48, 84% and 73% of subjects in the emtricitabine + tenofovir disoproxil fumarate group and the zidovudine/lamivudine group, respectively, achieved and maintained HIV-1 RNA <400 copies/mL (71% and 58% through Week 144). The difference in the proportion of subjects who achieved and maintained HIV-1 RNA <400 copies/mL through 48 weeks largely results from the higher number of discontinuations due to adverse events and other reasons in the zidovudine/lamivudine group in this open-label trial. In addition, 80% and 70% of subjects in the emtricitabine + tenofovir disoproxil fumarate group and the zidovudine/lamivudine group, respectively, achieved and maintained HIV-1 RNA <50 copies/mL through Week 48 (64% and 56% through Week 144). The mean increase from baseline in CD4+ cell count was 190 cells/mm3 in the EMTRIVA + tenofovir disoproxil fumarate group and 158 cells/mm3 in the zidovudine/lamivudine group at Week 48 (312 and 271 cells/mm3 at Week 144).
Through 48 weeks, 7 subjects in the emtricitabine + tenofovir disoproxil fumarate group and 5 subjects in the zidovudine/lamivudine group experienced a new CDC Class C event (10 and 6 subjects through 144 weeks).
Treatment-Experienced Adult Patients
Study 907
Study 907 was a 24-week, double-blind, placebo-controlled multicenter trial of tenofovir disoproxil fumarate added to a stable background regimen of antiretroviral agents in 550 treatment-experienced subjects. After 24 weeks of blinded trial treatment, all subjects continuing on trial were offered open-label tenofovir disoproxil fumarate for an additional 24 weeks. Subjects had a mean baseline CD4+ cell count of 427 cells/mm3 (range 23 to 1385), median baseline plasma HIV-1 RNA of 2340 (range 50 to 75,000) copies/mL, and mean duration of prior HIV-1 treatment was 5.4 years. Mean age of the subjects was 42 years; 85% were male, 69% were Caucasian, 17% Black, and 12% Hispanic.
The percent of subjects with HIV-1 RNA <400 copies/mL and outcomes of subjects through 48 weeks are summarized in Table 18.
Table 18 Outcomes of Randomized Treatment (Study 907) a. Subjects with HIV-1 RNA <400 copies/mL and no prior study drug discontinuation at Week 24 and 48, respectively.b. Subjects with HIV-1 RNA ≥400 copies/mL efficacy failure or missing HIV-1 RNA at Week 24 and 48, respectively.c. Includes lost to follow-up, subject withdrawal, noncompliance, protocol violation, and other reasons. Outcomes 0 to 24 weeks 0 to 48 weeks 24 to 48 weeks Tenofovir disoproxil fumarate (N=368) Placebo (N=182) Tenofovir disoproxil fumarate (N=368) Placebo Crossover to Tenofovir disoproxil fumarate (N=170) HIV-1 RNA <400 copies/mL a 40% 11% 28% 30% Virologic failureb 53% 84% 61% 64% Discontinued due to adverse event 3% 3% 5% 5% Discontinued for other reasonsc 3% 3% 5% 1%
At 24 weeks of therapy, there was a higher proportion of subjects in the tenofovir disoproxil fumarate arm compared to the placebo arm with HIV-1 RNA <50 copies/mL (19% and 1%, respectively). Mean change in absolute CD4+ cell counts by Week 24 was +11 cells/mm3 for the tenofovir disoproxil fumarate group and ‒5 cells/mm3 for the placebo group. Mean change in absolute CD4+ cell counts by Week 48 was +4 cells/mm3 for the tenofovir disoproxil fumarate group.
Through Week 24, one subject in the tenofovir disoproxil fumarate group and no subjects in the placebo arm experienced a new CDC Class C event.
14.2 Clinical Efficacy in Adults with Chronic Hepatitis B
HBeAg-Negative Chronic Hepatitis B
Study 0102 was a Phase 3, randomized, double-blind, active-controlled trial of tenofovir disoproxil fumarate 300 mg compared to HEPSERA 10 mg in 375 HBeAg-(anti-HBe+) subjects with compensated liver function, the majority of whom were nucleoside-naïve. The mean age of subjects was 44 years; 77% were male, 25% were Asian, 65% were Caucasian, 17% had previously received alpha-interferon therapy, and 18% were nucleoside-experienced (16% had prior lamivudine experience). At baseline, subjects had a mean Knodell necroinflammatory score of 7.8; mean plasma HBV DNA was 6.9 log10 copies/mL; and mean serum ALT was 140 U/L.
HBeAg-Positive Chronic Hepatitis B
Study 0103 was a Phase 3, randomized, double-blind, active-controlled trial of tenofovir disoproxil fumarate 300 mg compared to HEPSERA 10 mg in 266 HBeAg+ nucleoside-naïve subjects with compensated liver function. The mean age of subjects was 34 years; 69% were male, 36% were Asian, 52% were Caucasian, 16% had previously received alpha-interferon therapy, and <5% were nucleoside experienced. At baseline, subjects had a mean Knodell necroinflammatory score of 8.4; mean plasma HBV DNA was 8.7 log10 copies /mL; and mean serum ALT was 147 U/L.
The primary data analysis was conducted after all subjects reached 48 weeks of treatment and results are summarized below.
The primary efficacy endpoint in both trials was complete response to treatment defined as HBV DNA <400 copies/mL (69 IU/mL) and Knodell necroinflammatory score improvement of at least 2 points, without worsening in Knodell fibrosis at Week 48 (Table 19).
Table 19 Histological, Virological, Biochemical, and Serological Response at Week 48 a. Knodell necroinflammatory score improvement of at least 2 points without worsening in Knodell fibrosis.b. The population used for analysis of ALT normalization included only subjects with ALT above ULN at baseline.c. NA = Not Applicable! 0102 (HBeAg-) 0103 (HBeAg+) Tenofovir disoproxil fumarate (N=250) HEPSERA (N=125) Tenofovir disoproxil fumarate (N=176) HEPSERA (N=90) Complete Response 71% 49% 67% 12% Histology Histological Responsea 72% 69% 74% 68% HBV DNA <400 copies/mL (<69 IU/mL) 93% 63% 76% 13% ALT Normalized ALTb 76% 77% 68% 54% Serology HBeAg Loss/ Seroconversion NAc NAc 20% / 19% 16% / 16% HBsAg Loss/ Seroconversion 0/0 0/0 3% / 1% 0/0
Treatment Beyond 48 Weeks
In Studies 0102 (HBeAg-negative) and 0103 (HBeAg-positive), subjects who completed double-blind treatment (389 and 196 subjects who were originally randomized to tenofovir disoproxil fumarate and HEPSERA, respectively) were eligible to roll over to open-label tenofovir disoproxil fumarate with no interruption in treatment.
In Study 0102, 266 of 347 subjects who entered the open-label period (77%) continued in the study through Week 384. Among subjects randomized to tenofovir disoproxil fumarate followed by open-label treatment with tenofovir disoproxil fumarate, 73% had HBV DNA <400 copies/ml (69 IU/ml), and 63% had ALT normalization at Week 384. Among subjects randomized to HEPSERA followed by open-label treatment with tenofovir disoproxil fumarate, 80% had HBV DNA <400 copies/mL (69 IU/mL) and 70% had ALT normalization through Week 384. At Week 384, both HBsAg loss and seroconversion were approximately 1% in both treatment groups.
In Study 0103, 146 of 238 subjects who entered the open-label period (61%) continued in the study through Week 384. Among subjects randomized to tenofovir disoproxil fumarate, 49% had HBV DNA <400 copies/mL (69 IU/mL), 42% had ALT normalization, and 20% had HBeAg loss (13% seroconversion to anti-HBe antibody) through Week 384. Among subjects randomized to HEPSERA followed by open-label treatment with tenofovir disoproxil fumarate, 56% had HBV DNA <400 copies/mL (69 IU/mL), 50% had ALT normalization, and 28% had HBeAg loss (19% seroconversion to anti-HBe antibody) through Week 384. At Week 384, HBsAg loss and seroconversion were 11% and 8%, respectively, in subjects initially randomized to tenofovir disoproxil fumarate and 12% and 10%, respectively, in subjects initially randomized to HEPSERA.
Of the originally randomized and treated 641 subjects in the two studies, liver biopsy data from 328 subjects who received continuing open-label treatment with tenofovir disoproxil fumarate monotherapy were available for analysis at baseline, Week 48, and Week 240. There were no apparent differences between the subset of subjects who had liver biopsy data at Week 240 and those subjects remaining on open-label tenofovir disoproxil fumarate without biopsy data that would be expected to affect histological outcomes at Week 240. Among the 328 subjects evaluated, the observed histological response rates were 80% and 88% at Week 48 and Week 240, respectively. In the subjects without cirrhosis at baseline (Ishak fibrosis score 0 to 4), 92% (216/235) and 95% (223/235) had either improvement or no change in Ishak fibrosis score at Week 48 and Week 240, respectively. In subjects with cirrhosis at baseline (Ishak fibrosis score 5 to 6), 97% (90/93) and 99% (92/93) had either improvement or no change in Ishak fibrosis score at Week 48 and Week 240, respectively. Twenty-nine percent (27/93) and 72% (67/93) of subjects with cirrhosis at baseline experienced regression of cirrhosis by Week 48 and Week 240, respectively, with a reduction in Ishak fibrosis score of at least 2 points. No definitive conclusions can be established about the remaining study population who were not part of this subset analysis.
Patients with Lamivudine-Resistant Chronic Hepatitis B
Study 121 was a randomized, double-blind, active-controlled trial evaluating the safety and efficacy of tenofovir disoproxil fumarate compared to an unapproved antiviral regimen in subjects with chronic hepatitis B, persistent viremia (HBV DNA ≥1,000 IU/mL), and genotypic evidence of lamivudine resistance (rtM204I/V +/-rtL180M). One hundred forty-one adult subjects were randomized to the tenofovir disoproxil fumarate treatment arm. The mean age of subjects randomized to tenofovir disoproxil fumarate was 47 years (range 18 to 73), 74% were male, 59% were Caucasian, and 37% were Asian. At baseline, 54% of subjects were HBeAg-negative, 46% were HBeAg-positive, and 56% had abnormal ALT. Subjects had a mean HBV DNA of 6.4 log10 copies/mL and mean serum ALT of 71 U/L at baseline.
After 96 weeks of treatment, 126 of 141 subjects (89%) randomized to tenofovir disoproxil fumarate had HBV DNA <400 copies/mL (69 IU/mL), and 49 of 79 subjects (62%) with abnormal ALT at baseline had ALT normalization. Among the HBeAg-positive subjects randomized to tenofovir disoproxil fumarate, 10 of 65 subjects (15%) experienced HBeAg loss and 7 of 65 subjects (11%) experienced anti-HBe seroconversion through Week 96. The proportion of subjects with HBV DNA concentrations below 400 copies/mL (69 IU/mL) at Week 96 was similar between the tenofovir disoproxil fumarate monotherapy and the comparator arms.
Across the combined chronic hepatitis B treatment trials, the number of subjects with adefovir-resistance associated substitutions at baseline was too small to establish efficacy in this subgroup.
Patients with Chronic Hepatitis B and Decompensated Liver Disease
Tenofovir disoproxil fumarate was studied in a small randomized, double-blind, active-controlled trial evaluating the safety of tenofovir disoproxil fumarate compared to other antiviral drugs in subjects with chronic hepatitis B and decompensated liver disease through 48 weeks (Study 0108).
Forty-five adult subjects (37 males and 8 females) were randomized to the tenofovir disoproxil fumarate treatment arm. At baseline, 69% subjects were HBeAg-negative and 31% were HBeAg-positive. Subjects had a mean Child-Pugh score of 7, a mean MELD score of 12, mean HBV DNA of 5.8 log10 copies/mL, and mean serum ALT of 61 U/L at baseline. Trial endpoints were discontinuation due to an adverse event and confirmed increase in serum creatinine ≥0.5 mg/dL or confirmed serum phosphorus of <2 mg/dL [See Adverse Reactions (6.1)].
At 48 weeks, 31/44 (70%) and 12/26 (46%) tenofovir disoproxil fumarate-treated subjects achieved an HBV DNA <400 copies/mL (69 IU/mL), and normalized ALT, respectively. The trial was not designed to evaluate treatment impact on clinical endpoints such as progression of liver disease, need for liver transplantation, or death.
16 How Supplied/storage And Handling
Tenofovir disoproxil fumarate tablets 300 mg are supplied as blue colored, oval shaped, film-coated, and debossed with “LA16” on one side and plain on the other, containing 300 mg of tenofovir DF, which is equivalent to 245 mg of tenofovir disoproxil. These are packaged as follows: Bottle of 30 NDC 42385-901-03 Bottle of 500 NDC 42385-901-50 Store Tenofovir disoproxil fumarate tablets at 20° to 25°C (68° to 77°F); excursions permitted between 15° to 30°C (59° to 86°F) [see USP Controlled Room Temperature]. Keep the bottle tightly closed.
17 Patient Counseling Information
Advise the patient to read the FDA-approved patient labeling (Patient Information). Inform patients that tenofovir disoproxil fumarate tablets is not a cure for HIV-1 infection and patients may continue to experience illnesses associated with HIV-1 infection, including opportunistic infections. Patients should remain under the care of a physician when using tenofovir disoproxil fumarate tablets. Advise Patients to avoid doing things that can spread HIV or HBV to others. • Do not share needles or other injection equipment. • Do not share personal lis that can have blood or body fluids on them, like toothbrushes and razor blades. • Do not have any kind of sex without protection. Always practice safer sex by using a latex or polyurethane condom to lower the chance of sexual contact with semen, vaginal secretions, or blood. • Do not breastfeed. Tenofovir is excreted in breast milk and it is not known whether it can harm the baby. Mothers with HIV-1 should not breastfeed because HIV-1 can be passed to the baby in the breast milk. Inform patients that: • The long-term effects of tenofovir disoproxil fumarate tablets are unknown. • Tenofovir disoproxil fumarate tablets are for oral ingestion only. • Tenofovir disoproxil fumarate tablets should not be discontinued without first informing their physician. • If you have HIV-1 infection, with or without HBV coinfection, it is important to take tenofovir disoproxil fumarate tablets with combination therapy. • It is important to take tenofovir disoproxil fumarate tablets on a regular dosing schedule and to avoid missing doses. • Severe acute exacerbations of hepatitis have been reported in patients who are infected with HBV or coinfected with HBV and HIV-1 and have discontinued tenofovir disoproxil fumarate tablets [See Warnings and Precautions (5.1)]. • Renal impairment, including cases of acute renal failure and Fanconi syndrome, has been reported. Tenofovir disoproxil fumarate tablets should be avoided with concurrent or recent use of a nephrotoxic agent (e.g., high-dose or multiple NSAIDs) [See Warnings and Precautions (5.2)]. • Lactic acidosis and severe hepatomegaly with steatosis, including fatal cases, have been reported. Treatment with tenofovir disoproxil fumarate tablets should be suspended in any patient who develops clinical symptoms suggestive of lactic acidosis or pronounced hepatotoxicity [See Warnings and Precautions (5.3)]. • Tenofovir disoproxil fumarate tablets should not be coadministered with ATRIPLA, COMPLERA, DESCOVY, GENVOYA, ODEFSEY, STRIBILD, TRUVADA, or VEMLIDY [See Warnings and Precautions (5.4)]. • Tenofovir disoproxil fumarate tablets should not be administered in combination with HEPSERA [See Warnings and Precautions (5.4)]. • Decreases in bone mineral density have been observed with the use of tenofovir disoproxil fumarate tablets. Bone mineral density monitoring should be considered in patients who have a history of pathologic bone fracture or at risk for osteopenia [See Warnings and Precautions (5.6)]. • In some patients treated with combination antiretroviral therapy, including tenofovir disoproxil fumarate tablets, signs and symptoms of inflammation from previous infections may occur soon after anti-HIV treatment is started. It is believed that these symptoms are due to an improvement in the body’s immune response, enabling the body to fight infections that may have been present with no obvious symptoms. Advise patients to inform their healthcare provider immediately of any symptoms of infection [See Warnings and Precautions (5.7)]. • In the treatment of chronic hepatitis B, the optimal duration of treatment is unknown. The relationship between response and long-term prevention of outcomes such as hepatocellular carcinoma is not known.
Patient Information
Tenofovir disoproxil fumarate (ten of’ oh vir dye’ soe prox’ il fue’ ma rate) Tablets Read this Patient Information before you start taking tenofovir disoproxil fumarate tablets and each time you get a refill. There may be new information. This information does not take the place of talking with your healthcare provider about your medical condition or your treatment. What is the most important information I should know about Tenofovir disoproxil fumarate tablets? Tenofovir disoproxil fumarate tablets can cause serious side effects, including: Worsening of your Hepatitis B infection. Your hepatitis B Virus (HBV) infection may become worse (flare-up) if you take tenofovir disoproxil fumarate tablets and then stop it. A “flare-up” is when your HBV infection suddenly returns in a worse way than before. • Do not let your tenofovir disoproxil fumarate tablets run out. Refill your prescription or talk to your healthcare provider before your tenofovir disoproxil fumarate tablets are all gone. • Do not stop taking tenofovir disoproxil fumarate tablets without first talking to your healthcare provider. • If you stop taking tenofovir disoproxil fumarate tablets, your healthcare provider will need to check your health often and do blood tests regularly to check your HBV infection. Tell your healthcare provider about any new or unusual symptoms you may have after you stop taking tenofovir disoproxil fumarate tablets. Talk to your doctor about taking an HIV test before starting treatment with tenofovir disoproxil fumarate tablets for chronic hepatitis B. You should also get a test for HBV if you are taking tenofovir disoproxil fumarate tablets for treatment of HIV. What is Tenofovir disoproxil fumarate tablets? Tenofovir disoproxil fumarate tablets are a prescription medicine used: 1. with other antiviral medicines to treat Human Immunodeficiency Virus-1 (HIV-1) in people 2 years of age and older. HIV is the virus that causes AIDS (Acquired Immune Deficiency Syndrome). • When used with other HIV medicines, tenofovir disoproxil fumarate tablets may reduce the amount of HIV in your blood (called “viral load”). Tenofovir disoproxil fumarate tablets may also help to increase the number of CD4 (T) cells in your blood which help fight off other infections. Reducing the amount of HIV and increasing the CD4 (T) cell count may improve your immune system. This may reduce your risk of death or infections that can happen when your immune system is weak (opportunistic infections). • Tenofovir disoproxil fumarate tablets does not cure HIV infection or AIDS. People taking tenofovir disoproxil fumarate tablets may still develop infections or other conditions associated with HIV infection. • You must stay on continuous HIV therapy to control infection and decrease HIV-related illnesses. • It is very important that you stay under the care of your healthcare provider. • It is not known if tenofovir disoproxil fumarate tablets are safe and effective for the treatment of HIV-1 infection in children under the age of 2 years. 2. to treat chronic (long-lasting) hepatitis B virus (HBV) in people 12 years of age and older. • Tenofovir disoproxil fumarate tablets will not cure HBV. • Tenofovir disoproxil fumarate tablets may lower the amount of HBV in your body. • Tenofovir disoproxil fumarate tablets may improve the condition of your liver • The long-term effects of taking tenofovir disoproxil fumarate tablets for treatment of chronic hepatitis B infection are not known. • It is not known if tenofovir disoproxil fumarate tablets are safe and effective for treatment of chronic hepatitis B in children under the age of 12 years. What should I tell my healthcare provider before taking tenofovir disoproxil fumarate tablets? Before you take tenofovir disoproxil fumarate tablets, tell your healthcare provider if you: • have liver problems, including hepatitis B (HBV) infection. • have kidney problems. • have bone problems. • have any other medical conditions, including HIV infection. • are pregnant or plan to become pregnant. It is not known if tenofovir disoproxil fumarate tablets will harm your unborn baby. Pregnancy Registry. There is a pregnancy registry for women who take antiviral medicines during pregnancy. Its purpose is to collect information about the health of you and your baby. Talk to your healthcare provider about how you can take part in this registry. • are breastfeeding or plan to breastfeed. Do not breastfeed if you are taking tenofovir disoproxil fumarate tablets. Tenofovir passes into your breast milk. You should not breastfeed because of the risk of passing HIV to your baby. Talk to your healthcare provider about the best way to feed your baby. Tell your healthcare provider about all the medicines you take, including prescription and non-prescription medicines, vitamins and herbal supplements. Tenofovir disoproxil fumarate tablets may affect the way other medicines work, and other medicines may affect how tenofovir disoproxil fumarate tablets work. Do not take tenofovir disoproxil fumarate tablets if you also take: • other medicines that contain tenofovir (ATRIPLA®, COMPLERA®, DESCOVY®, GENVOYA®, ODEFSEY®, STRIBILD®, TRUVADA®, VEMLIDY®) • adefovir (HEPSERA®) Especially tell your healthcare provider if you take the following medications • didanosine (Videx, Videx EC) • atazanavir (Reyataz) • darunavir (Prezista) • lopinavir with ritonavir (Kaletra) • ledipasvir with sofosbuvir (HARVONI®) • sofosbuvir with velpatasvir (EPCLUSA®) Know the medicines you take. Keep a ul of them to show your healthcare provider or pharmacist when you get a new medicine. How should I take tenofovir disoproxil fumarate tablets? • See “What is the most important information I should know about tenofovir disoproxil fumarate tablets?” • Take tenofovir disoproxil fumarate tablets exactly as your healthcare provider tells you to take it. • Take tenofovir disoproxil fumarate tablets at the same time every day. • For adults and children 12 years of age and older, the usual dose of tenofovir disoproxil fumarate is one 300 mg tablet each day. • If you are an adult with kidney problems, your healthcare provider may tell you to take tenofovir disoproxil fumarate tablets less often. • Take tenofovir disoproxil fumarate tablets by mouth, with or without food. • Do not miss a dose of tenofovir disoproxil fumarate tablets. If you miss a dose of tenofovir disoproxil fumarate tablets, take the missed dose as soon as you remember. If it is almost time for your next dose of tenofovir disoproxil fumarate tablets, do not take the missed dose. Take the next dose of tenofovir disoproxil fumarate tablets at your regular time. • If you take too much tenofovir disoproxil fumarate tablets, call your local poison control center or go right away to the nearest hospital emergency room. What are the possible side effects of tenofovir disoproxil fumarate tablets? Tenofovir disoproxil fumarate tablets may cause serious side effects, including: • See “What is the most important information I should know about tenofovir disoproxil fumarate tablets?” • New or worse kidney problems, including kidney failure, can happen in some people who take tenofovir disoproxil fumarate tablets. Your healthcare provider should do blood tests to check your kidneys before you start treatment with tenofovir disoproxil fumarate tablets. If you have had kidney problems in the past or need to take another medicine that can cause kidney problems, your healthcare provider may need to do blood tests to check your kidneys during your treatment with tenofovir disoproxil fumarate tablets. • Too much lactic acid in your blood (lactic acidosis). Too much lactic acid is a serious but rare medical emergency that can lead to death. Tell your healthcare provider right away if you get these symptoms: weakness or being more tired than usual, unusual muscle pain, being short of breath or fast breathing, stomach pain with nausea and vomiting, cold or blue hands and feet, feel dizzy or lightheaded, or a fast or abnormal heartbeat. • Severe liver problems. In rare cases, severe liver problems can happen that can lead to death. Tell your healthcare provider right away if you get these symptoms: skin or the white part of your eyes turns yellow, dark “tea-colored” urine, light-colored stools, loss of appetite for several days or longer, nausea, or stomach-area pain. • Bone problems can happen in some people who take tenofovir disoproxil fumarate tablets. Bone problems include bone pain, softening or thinning (which may lead to fractures). Your healthcare provider may need to do additional tests to check your bones. • Changes in your immune system (Immune Reconstitution Syndrome) can happen when you start taking HIV medicines. Your immune system may get stronger and begin to fight infections that have been hidden in your body for a long time. Tell your healthcare provider if you start having new symptoms after starting your HIV medicine. The most common side effects in all people who take tenofovir disoproxil fumarate tablets are: • nausea • rash • diarrhea • headache • pain • depression • weakness In some people with advanced HBV-infection, other common side effects may include: • sleeping problems • itching • vomiting • dizziness • fever Tell your healthcare provider if you have any side effect that bothers you or that does not go away. These are not all the possible side effects of tenofovir disoproxil fumarate tablets. For more information, ask your healthcare provider or pharmacist. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. How should I store tenofovir disoproxil fumarate tablets? • Store tenofovir disoproxil fumarate tablets at room temperature between 68° to 77°F (20° to 25°C). • Keep the bottle tightly closed. Keep tenofovir disoproxil fumarate tablets and all medicines out of the reach of children General information about tenofovir disoproxil fumarate tablets: Medicines are sometimes prescribed for purposes other than those uled in a Patient Information leaflet. Do not use tenofovir disoproxil fumarate tablets for a condition for which it was not prescribed. Do not give tenofovir disoproxil fumarate tablets to other people, even if they have the same condition you have. It may harm them. Avoid doing things that can spread HIV-1 or HBV infection to others. Do not share or re-use needles or other injection equipment. Do not share personal lis that can have blood or body fluids on them, like toothbrushes and razor blades. Do not have any kind of sex without protection. Always practice safe sex by using a latex or polyurethane condom to lower the chance of sexual contact with semen, vaginal secretions, or blood. A vaccine is available to protect people at risk for becoming infected with HBV. You can ask your healthcare provider for information about this vaccine. This leaflet summarizes the most important information about tenofovir disoproxil fumarate tablets. If you would like more information, talk with your healthcare provider. You can ask your pharmacist or healthcare provider for information about tenofovir disoproxil fumarate tablets that is written for health professionals. For more information call Laurus Generics Inc. at 1-833-3-LAURUS (1-833-352-8787). What are the ingredients in tenofovir disoproxil fumarate tablets? Active Ingredient: Tenofovir disoproxil fumarate Inactive Ingredients: Tenofovir disoproxil fumarate tablets: lactose monohydrate, microcrystalline cellulose, pregelatinized starch, croscarmellose sodium and magnesium stearate. Tablet Coating: Tenofovir disoproxil fumarate tablets 300mg: Opadry II Blue 32K505023, which contains FD&C Blue 2/Indigo Carmine Aluminium Lake, Hypromellose 2910, Lactose monohydrate, Titanium dioxide and Triacetin. This Patient Information has been approved by the U.S. Food and Drug Administration. Trademarks are the property of their respective owners. Rx Only Manufactured for: Laurus Generics Inc. 400 Connell DriveSuite 5200Berkeley Heights, NJ 07922 Manufactured by: Laurus Labs Limited Visakhapatnam-531011India Revised: 12/2017
Principal Display Panel - Container Label (30's Count)
NDC 42385-901-03 Tenofovir Disoproxil Fumarate Tablets 300 mg 30 Tablets Rx Only LAURUS Labs
Principal Display Panel - Container Label (500's Count)
NDC 42385-901-50 Tenofovir Disoproxil Fumarate Tablets300 mg 500 Tablets Rx Only LAURUS Labs
DISCLAIMER:
"This tool does not provide medical advice, and is for informational and educational purposes only, and is not a substitute for professional medical advice, treatment or diagnosis. Call your doctor to receive medical advice. If you think you may have a medical emergency, please dial 911."
"Do not rely on openFDA to make decisions regarding medical care. While we make every effort to ensure that data is accurate, you should assume all results are unvalidated. We may limit or otherwise restrict your access to the API in line with our Terms of Service."
"This product uses publicly available data from the U.S. National Library of Medicine (NLM), National Institutes of Health, Department of Health and Human Services; NLM is not responsible for the product and does not endorse or recommend this or any other product."
PillSync may earn a commission via links on our site
