VOTRIENT (pazopanib hydrochloride 200 mg) Dailymed
Generic: pazopanib hydrochloride
Boxed Warning
Warning: Hepatotoxicity

Go PRO for all pill images
Recent Major Changes Section
Warnings and Precautions, Hepatic Toxicity and Hepatic Impairment (5.1 )
04/2015
Warnings and Precautions, Interstitial Lung Disease (ILD)/Pneumonitis (5.9 )
9/2015
Warning: Hepatotoxicity
Severe and fatal hepatotoxicity has been observed in clinical trials. Monitor hepatic function and interrupt, reduce, or discontinue dosing as recommended [see Warnings and Precautions (5.1)].
WARNING: HEPATOTOXICITY
See full prescribing information for complete boxed warning.
Severe and fatal hepatotoxicity has been observed in clinical trials. Monitor hepatic function and interrupt, reduce, or discontinue dosing as recommended [see Warnings and Precautions (5.1 )].
1 Indications And Usage
VOTRIENT¬ģ is indicated for the treatment of patients with advanced renal cell carcinoma (RCC).
VOTRIENT is indicated for the treatment of patients with advanced soft tissue sarcoma (STS) who have received prior chemotherapy.
Limitation of Use: The efficacy of VOTRIENT for the treatment of patients with adipocytic STS or gastrointestinal stromal tumors has not been demonstrated.
VOTRIENT is a kinase inhibitor indicated for the treatment of patients with:
‚ÄĘ advanced renal cell carcinoma. (1 )‚ÄĘ advanced soft tissue sarcoma who have received prior chemotherapy. (1 )
Limitation of Use: The efficacy of VOTRIENT for the treatment of patients with adipocytic soft tissue sarcoma or gastrointestinal stromal tumors has not been demonstrated.
2 Dosage And Administration
‚ÄĘ 800 mg orally once daily without food (at least 1 hour before or 2 hours after a meal). (2.1 )‚ÄĘ Baseline moderate hepatic impairment ‚Äď 200¬†mg orally once daily. Not recommended in patients with severe hepatic impairment. (2.2 )2.1 Recommended Dosing
The recommended starting dose of VOTRIENT is 800 mg orally once daily without food (at least 1 hour before or 2 hours after a meal) [see Clinical Pharmacology (12.3)]. The dose of VOTRIENT should not exceed 800 mg.
Do not crush tablets due to the potential for increased rate of absorption which may affect systemic exposure [see Clinical Pharmacology (12.3)].
If a dose is missed, it should not be taken if it is less than 12 hours until the next dose.
2.2 Dose Modification Guidelines
In RCC, the initial dose reduction should be 400 mg, and additional dose decrease or increase should be in 200-mg steps based on individual tolerability.
In STS, a decrease or increase should be in 200-mg steps based on individual tolerability.
Hepatic Impairment: No dose adjustment is required in patients with mild hepatic impairment. In patients with moderate hepatic impairment, alternatives to VOTRIENT should be considered. If VOTRIENT is used in patients with moderate hepatic impairment, the dose should be reduced to 200 mg per day. VOTRIENT is not recommended in patients with severe hepatic impairment [see Use in Specific Populations (8.6), Clinical Pharmacology (12.3)].
Concomitant Strong CYP3A4 Inhibitors: The concomitant use of strong CYP3A4 inhibitors (e.g., ketoconazole, ritonavir, clarithromycin) increases pazopanib concentrations and should be avoided. Consider an alternate concomitant medication with no or minimal potential to inhibit CYP3A4. If coadministration of a strong CYP3A4 inhibitor is warranted, reduce the dose of VOTRIENT to 400 mg. Further dose reductions may be needed if adverse effects occur during therapy [see Drug Interactions (7.1), Clinical Pharmacology (12.3)].
Concomitant Strong CYP3A4 Inducer: The concomitant use of strong CYP3A4 inducers (e.g., rifampin) may decrease pazopanib concentrations and should be avoided. Consider an alternate concomitant medication with no or minimal enzyme induction potential. VOTRIENT should not be used in patients who cannot avoid chronic use of strong CYP3A4 inducers [see Drug Interactions (7.1)].
3 Dosage Forms And Strengths
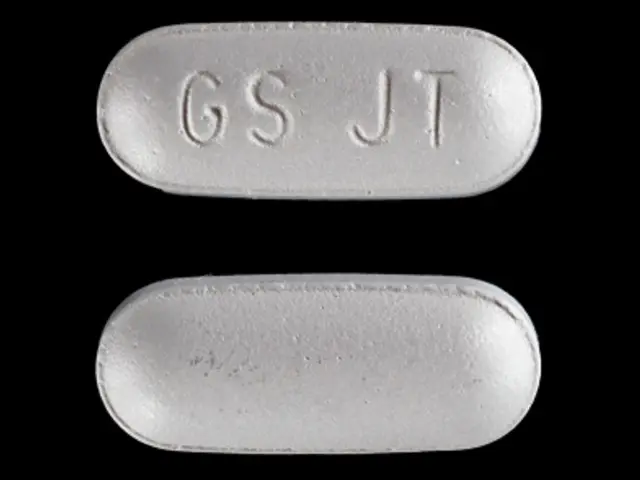
200-mg tablets of VOTRIENT ‚ÄĒ modified capsule-shaped, gray, film-coated with GS¬†JT debossed on one side. Each tablet contains 216.7¬†mg of pazopanib hydrochloride equivalent to 200¬†mg of pazopanib.
200 mg tablets (3 )
4 Contraindications
None.
None (4 )
5 Warnings And Precautions
‚ÄĘ Increases in serum transaminase levels and bilirubin were observed. Severe and fatal hepatotoxicity has occurred. Measure liver chemistries before the initiation of treatment and regularly during treatment. (5.1 )‚ÄĘ Prolonged QT intervals and torsades de pointes have been observed. Use with caution in patients at higher risk of developing QT interval prolongation. Monitoring electrocardiograms and electrolytes should be considered. (5.2 )‚ÄĘ Cardiac dysfunction such as congestive heart failure and decreased left ventricular ejection fraction (LVEF) have occurred. Monitor blood pressure and manage hypertension promptly. Baseline and periodic evaluation of LVEF is recommended in patients at risk of cardiac dysfunction. (5.3 )‚ÄĘ Fatal hemorrhagic events have been reported. VOTRIENT has not been studied in patients who have a history of hemoptysis, cerebral, or clinically significant gastrointestinal hemorrhage in the past 6¬†months and should not be used in those patients. (5.4 )‚ÄĘ Arterial thromboembolic events have been observed and can be fatal. Use with caution in patients who are at increased risk for these events. (5.5 )‚ÄĘ Venous thromboembolic events (VTE) have been observed, including fatal pulmonary emboli (PE). Monitor for signs and symptoms of VTE and PE. (5.6 )‚ÄĘ Thrombotic microangiopathy (TMA), including thrombotic thrombocytopenic purpura (TTP) and hemolytic uremic syndrome (HUS) has been observed. Permanently discontinue VOTRIENT if TMA occurs. (5.7 )‚ÄĘ Gastrointestinal perforation or fistula has occurred. Fatal perforation events have occurred. Use with caution in patients at risk for gastrointestinal perforation or fistula. (5.8 )‚ÄĘ Interstitial lung disease (ILD)/pneumonitis have been observed and can be fatal. Discontinue VOTRIENT in patients developing ILD or pneumonitis. (5.9 )‚ÄĘ Reversible Posterior Leukoencephalopathy Syndrome (RPLS) has been observed and can be fatal. Permanently discontinue VOTRIENT in patients developing RPLS. (5.10 )‚ÄĘ Hypertension including hypertensive crisis has been observed. Blood pressure should be well controlled prior to initiating VOTRIENT. Monitor blood pressure within one week after starting VOTRIENT and frequently thereafter. (5.11 )‚ÄĘ Interruption of therapy with VOTRIENT is recommended in patients undergoing surgical procedures. (5.12 )‚ÄĘ Hypothyroidism may occur. Monitoring of thyroid function tests is recommended. (5.13 )‚ÄĘ Proteinuria: Monitor urine protein. Interrupt treatment for 24-hour urine protein ‚Č•3 grams and discontinue for repeat episodes despite dose reductions. (5.14 )‚ÄĘ Infection: Serious infections (with or without neutropenia), some with fatal outcome, have been reported. Monitor for signs and symptoms and treat active infection promptly. Interrupt or discontinue VOTRIENT. (5.15 )‚ÄĘ Animal studies have demonstrated VOTRIENT can severely affect organ growth and maturation during early post-natal development. The safety and effectiveness in pediatric patients have not been established. (5.17 )‚ÄĘ VOTRIENT can cause fetal harm when administered to a pregnant woman. Advise females of reproductive potential of the potential hazard to the fetus and to use effective contraception. (5.18 ,8.1 )5.1 Hepatic Toxicity and Hepatic Impairment
In clinical trials with VOTRIENT, hepatotoxicity, manifested as increases in serum transaminases (ALT, AST) and bilirubin, was observed. This hepatotoxicity can be severe and fatal.Patients older than 65 years are at greater risk for hepatotoxicity [see Use in Specific Populations (8.5)]. Transaminase elevations occur early in the course of treatment (92.5% of all transaminase elevations of any grade occurred in the first 18 weeks) [see Dosage and Administration (2.2)].
In the randomized RCC trial, ALT >3 X ULN was reported in 18% and 3% of the groups receiving VOTRIENT and placebo, respectively. ALT >10 X ULN was reported in 4% of patients who received VOTRIENT and in <1% of patients who received placebo. Concurrent elevation in ALT >3 X ULN and bilirubin >2 X ULN in the absence of significant alkaline phosphatase >3 X ULN occurred in 2% (5/290) of patients on VOTRIENT and 1% (2/145) on placebo.
In the randomized STS trial, ALT >3 X ULN was reported in 18% and 5% of the groups receiving VOTRIENT and placebo, respectively. ALT >8 X ULN was reported in 5% and 2% of the groups receiving VOTRIENT and placebo, respectively. Concurrent elevation in ALT >3 X ULN and bilirubin >2 X ULN in the absence of significant alkaline phosphatase >3 X ULN occurred in 2% (4/240) of patients on VOTRIENT and <1% (1/123) on placebo.
Two-tenths percent of the patients (2/977) from trials that supported the RCC indication died with disease progression and hepatic failure and 0.4% of patients (1/240) in the randomized STS trial died of hepatic failure.
‚ÄĘ Monitor serum liver tests before initiation of treatment with VOTRIENT and at Weeks 3, 5, 7, and 9. Thereafter, monitor at Month 3 and at Month 4, and as clinically indicated. Periodic monitoring should then continue after Month¬†4.‚ÄĘ Patients with isolated ALT elevations between 3 X ULN and 8 X ULN may be continued on VOTRIENT with weekly monitoring of liver function until ALT returns to Grade 1 or baseline.‚ÄĘ Patients with isolated ALT elevations of >8 X ULN should have VOTRIENT interrupted until they return to Grade 1 or baseline. If the potential benefit for reinitiating treatment with VOTRIENT is considered to outweigh the risk for hepatotoxicity, then reintroduce VOTRIENT at a reduced dose of no more than 400¬†mg once daily and measure serum liver tests weekly for 8¬†weeks [see Dosage and Administration (2.2)]. Following reintroduction of VOTRIENT, if ALT elevations >3 X ULN recur, then VOTRIENT should be permanently discontinued.‚ÄĘ If ALT elevations >3 X ULN occur concurrently with bilirubin elevations >2 X ULN, VOTRIENT should be permanently discontinued. Patients should be monitored until resolution. VOTRIENT is a uridine diphosphate (UDP)-glucuronosyl transferase 1A1 (UGT1A1) inhibitor. Mild, indirect (unconjugated) hyperbilirubinemia may occur in patients with Gilbert‚Äôs syndrome [see Clinical Pharmacology (12.5)]. Patients with only a mild indirect hyperbilirubinemia, known Gilbert‚Äôs syndrome, and elevation in ALT >3 X ULN should be managed as per the recommendations outlined for isolated ALT elevations.
Concomitant use of VOTRIENT and simvastatin increases the risk of ALT elevations and should be undertaken with caution and close monitoring [see Drug Interactions (7.4)]. Insufficient data are available to assess the risk of concomitant administration of alternative statins and VOTRIENT.
In patients with pre-existing moderate hepatic impairment, the starting dose of VOTRIENT should be reduced or alternatives to VOTRIENT should be considered. Treatment with VOTRIENT is not recommended in patients with pre-existing severe hepatic impairment, defined as total bilirubin >3 X ULN with any level of ALT [see Dosage and Administration (2.2), Use in Specific Populations (8.6), Clinical Pharmacology (12.3)].
5.2 QT Prolongation and Torsades de Pointes
In the RCC trials of VOTRIENT, QT prolongation (‚Č•500¬†msec) was identified on routine electrocardiogram monitoring in 2% (11/558) of patients. Torsades de pointes occurred in <1% (2/977) of patients who received VOTRIENT in the monotherapy trials.
In the randomized RCC and STS trials, 1% (3/290) of patients and 0.4% (1/240) of patients, respectively, who received VOTRIENT had post-baseline values between 500 to 549 msec. Post-baseline QT data were only collected in the STS trial if ECG abnormalities were reported as an adverse reaction. None of the 268 patients who received placebo on the two trials had post-baseline QTc values ‚Č•500 msec.
VOTRIENT should be used with caution in patients with a history of QT interval prolongation, in patients taking antiarrhythmics or other medications that may prolong QT interval, and those with relevant pre-existing cardiac disease. When using VOTRIENT, baseline and periodic monitoring of electrocardiograms and maintenance of electrolytes (e.g., calcium, magnesium, potassium) within the normal range should be performed.
5.3 Cardiac Dysfunction
In clinical trials with VOTRIENT, events of cardiac dysfunction such as decreased left ventricular ejection fraction (LVEF) and congestive heart failure have occurred. In the overall safety population for RCC (N¬†=¬†586), cardiac dysfunction was observed in 0.6% (4/586) of patients without routine on-study LVEF monitoring. In a randomized RCC trial of VOTRIENT compared with sunitinib, myocardial dysfunction was defined as symptoms of cardiac dysfunction or ‚Č•15% absolute decline in LVEF compared with baseline or a decline in LVEF of ‚Č•10% compared with baseline that is also below the lower limit of normal. In patients who had baseline and follow up LVEF measurements, myocardial dysfunction occurred in 13% (47/362) of patients on VOTRIENT compared with 11% (42/369) of patients on sunitinib. Congestive heart failure occurred in 0.5% of patients on each arm.¬†In the randomized STS trial, myocardial dysfunction occurred in 11% (16/142) of patients on VOTRIENT compared with 5% (2/40) of patients on placebo. One percent (3/240) of patients on VOTRIENT in the STS trial had congestive heart failure which did not resolve in one patient.
Fourteen of the 16 patients with myocardial dysfunction treated with VOTRIENT in the STS trial had concurrent hypertension which may have exacerbated cardiac dysfunction in patients at risk (e.g., those with prior anthracycline therapy) possibly by increasing cardiac afterload. Blood pressure should be monitored and managed promptly using a combination of anti-hypertensive therapy and dose modification of VOTRIENT (interruption and re-initiation at a reduced dose based on clinical judgment) [see Warnings and Precautions (5.11)]. Patients should be carefully monitored for clinical signs or symptoms of congestive heart failure. Baseline and periodic evaluation of LVEF is recommended in patients at risk of cardiac dysfunction including previous anthracycline exposure.
5.4 Hemorrhagic Events
Fatal hemorrhage occurred in 0.9% (5/586) in the RCC trials; there were no reports of fatal hemorrhage in the STS trials. In the randomized RCC trial, 13% (37/290) of patients treated with VOTRIENT and 5% (7/145) of patients on placebo experienced at least 1 hemorrhagic event. The most common hemorrhagic events in the patients treated with VOTRIENT were hematuria (4%), epistaxis (2%), hemoptysis (2%), and rectal hemorrhage (1%). Nine of 37 patients treated with VOTRIENT who had hemorrhagic events experienced serious events including pulmonary, gastrointestinal, and genitourinary hemorrhage. One percent (4/290) of patients treated with VOTRIENT died from hemorrhage compared with no (0/145) patients on placebo. In the overall safety population in RCC (N = 586), cerebral/intracranial hemorrhage was observed in <1% (2/586) of patients treated with VOTRIENT.
In the randomized STS trial, 22% (53/240) of patients treated with VOTRIENT compared with 8% (10/123) treated with placebo experienced at least 1 hemorrhagic event. The most common hemorrhagic events were epistaxis (8%), mouth hemorrhage (3%), and anal hemorrhage (2%). Grade 4 hemorrhagic events in the STS population occurred in 1% (3/240) of patients and included intracranial hemorrhage, subarachnoid hemorrhage, and peritoneal hemorrhage.
VOTRIENT has not been studied in patients who have a history of hemoptysis, cerebral hemorrhage, or clinically significant gastrointestinal hemorrhage in the past 6 months and should not be used in those patients.
5.5 Arterial Thromboembolic Events
Fatal arterial thromboembolic events were observed in 0.3% (2/586) of patients in the RCC trials and in no patients in the STS trials. In the randomized RCC trial, 2% (5/290) of patients receiving VOTRIENT experienced myocardial infarction or ischemia, 0.3% (1/290) had a cerebrovascular accident, and 1% (4/290) had an event of transient ischemic attack. In the randomized STS trial, 2% (4/240) of patients receiving VOTRIENT experienced a myocardial infarction or ischemia, 0.4% (1/240) had a cerebrovascular accident, and there were no incidents of transient ischemic attack. No arterial thromboembolic events were reported in patients who received placebo in either trial. VOTRIENT should be used with caution in patients who are at increased risk for these events or who have had a history of these events. VOTRIENT has not been studied in patients who have had an arterial thromboembolic event within the previous 6 months and should not be used in those patients.
5.6 Venous Thromboembolic Events
In RCC and STS trials of VOTRIENT, venous thromboembolic events (VTE) including venous thrombosis and fatal pulmonary embolus (PE) have occurred. In the randomized STS trial, venous thromboembolic events were reported in 5% of patients treated with VOTRIENT compared with 2% with placebo. In the randomized RCC trial, the rate was 1% in both arms. Fatal pulmonary embolus occurred in 1% (2/240) of STS patients receiving VOTRIENT and in no patients receiving placebo. There were no fatal pulmonary emboli in the RCC trial. Monitor for signs and symptoms of VTE and PE.
5.7 Thrombotic Microangiopathy
Thrombotic microangiopathy (TMA), including thrombotic thrombocytopenic purpura (TTP) and hemolytic uremic syndrome (HUS), has been reported in clinical trials of VOTRIENT as monotherapy, in combination with bevacizumab, and in combination with topotecan. VOTRIENT is not indicated for use in combination with other agents. Six of the 7 TMA cases occurred within 90 days of the initiation of VOTRIENT. Improvement of TMA was observed after treatment was discontinued. Monitor for signs and symptoms of TMA. Permanently discontinue VOTRIENT in patients developing TMA. Manage as clinically indicated.
5.8 Gastrointestinal Perforation and Fistula
In the RCC and STS trials, gastrointestinal perforation or fistula occurred in 0.9% (5/586) of patients and 1% (4/382) of patients receiving VOTRIENT, respectively. Fatal perforations occurred in 0.3% (2/586) of these patients in the RCC trials and in 0.3% (1/382) of these patients in the STS trials. Monitor for signs and symptoms of gastrointestinal perforation or fistula.
5.9 Interstitial Lung Disease (ILD)/Pneumonitis
ILD/pneumonitis, which can be fatal, has been reported in association with VOTRIENT. In clinical trials, ILD/pneumonitis occurred in 0.1% of patients treated with VOTRIENT.
Monitor patients for pulmonary symptoms indicative of ILD/pneumonitis and discontinue VOTRIENT in patients developing ILD or pneumonitis.
5.10 Reversible Posterior Leukoencephalopathy Syndrome
Reversible Posterior Leukoencephalopathy Syndrome (RPLS) has been reported in patients receiving VOTRIENT and may be fatal.
RPLS is a neurological disorder which can present with headache, seizure, lethargy, confusion, blindness, and other visual and neurologic disturbances. Mild to severe hypertension may be present. The diagnosis of RPLS is optimally confirmed by magnetic resonance imaging. Permanently discontinue VOTRIENT in patients developing RPLS.
5.11 Hypertension
In clinical trials, hypertension (systolic blood pressure ‚Č•150 or diastolic blood pressure ‚Č•100¬†mm Hg) and hypertensive crisis were observed in patients treated with VOTRIENT. Blood pressure should be well controlled prior to initiating VOTRIENT. Hypertension occurs early in the course of treatment (40% of cases occurred by Day 9 and 90% of cases occurred in the first 18¬†weeks). Blood pressure should be monitored early after starting treatment (no longer than one week) and frequently thereafter to ensure blood pressure control. Approximately 40% of patients who received VOTRIENT experienced hypertension. Grade 3 hypertension was reported in 4% to 7% of patients receiving VOTRIENT [see Adverse Reactions (6.1)].
Increased blood pressure should be treated promptly with standard anti-hypertensive therapy and dose reduction or interruption of VOTRIENT as clinically warranted. VOTRIENT should be discontinued if there is evidence of hypertensive crisis or if hypertension is severe and persistent despite anti-hypertensive therapy and dose reduction. Approximately 1% of patients required permanent discontinuation of VOTRIENT because of hypertension [see Dosage and Administration (2.2)].
5.12 Wound Healing
No formal trials on the effect of VOTRIENT on wound healing have been conducted. Since vascular endothelial growth factor receptor (VEGFR) inhibitors such as pazopanib may impair wound healing, treatment with VOTRIENT should be stopped at least 7 days prior to scheduled surgery. The decision to resume VOTRIENT after surgery should be based on clinical judgment of adequate wound healing. VOTRIENT should be discontinued in patients with wound dehiscence.
5.13 Hypothyroidism
Hypothyroidism, confirmed based on a simultaneous rise of TSH and decline of T4, was reported in 7% (19/290) of patients treated with VOTRIENT in the randomized RCC trial and in 5% (11/240) of patients treated with VOTRIENT in the randomized STS trial. No patients on the placebo arm of either trial had hypothyroidism. In RCC and STS trials of VOTRIENT, hypothyroidism was reported as an adverse reaction in 4% (26/586) and 5% (20/382) of patients, respectively. Proactive monitoring of thyroid function tests is recommended.
5.14 Proteinuria
In the randomized RCC trial, proteinuria was reported as an adverse reaction in 9% (27/290) of patients receiving VOTRIENT and in no patients receiving placebo. In 2 patients, proteinuria led to discontinuation of treatment with VOTRIENT. In the randomized STS trial, proteinuria was reported as an adverse reaction in 1% (2/240) of patients, and nephrotic syndrome was reported in 1 patient treated with VOTRIENT compared with none in patients receiving placebo. Treatment was withdrawn in the patient with nephrotic syndrome.
Baseline and periodic urinalysis during treatment is recommended with follow up measurement of 24-hour urine protein as clinically indicated. Interrupt VOTRIENT and dose reduce for 24-hour urine protein ‚Č•3 grams; discontinue VOTRIENT for repeat episodes despite dose reductions [see Dosage and Administration (2.2)].
5.15 Infection
Serious infections (with or without neutropenia), including some with fatal outcome, have been reported. Monitor patients for signs and symptoms of infection. Institute appropriate anti-infective therapy promptly and consider interruption or discontinuation of VOTRIENT for serious infections.
5.16 Increased Toxicity with Other Cancer Therapy
VOTRIENT is not indicated for use in combination with other agents. Clinical trials of VOTRIENT in combination with pemetrexed and lapatinib were terminated early due to concerns over increased toxicity and mortality. The fatal toxicities observed included pulmonary hemorrhage, gastrointestinal hemorrhage, and sudden death. A safe and effective combination dose has not been established with these regimens.
5.17 Increased Toxicity in Developing Organs
The safety and effectiveness of VOTRIENT in pediatric patients have not been established. VOTRIENT is not indicated for use in pediatric patients. Based on its mechanism of action, pazopanib may have severe effects on organ growth and maturation during early post-natal development. Administration of pazopanib to juvenile rats less than 21 days old resulted in toxicity to the lungs, liver, heart, and kidney and in death at doses significantly lower than the clinically recommended dose or doses tolerated in older animals. VOTRIENT may potentially cause serious adverse effects on organ development in pediatric patients, particularly in patients younger than 2 years of age [see Use in Specific Populations (8.4)].
5.18 Pregnancy
VOTRIENT can cause fetal harm when administered to a pregnant woman. Based on its mechanism of action, VOTRIENT is expected to result in adverse reproductive effects. In pre-clinical studies in rats and rabbits, pazopanib was teratogenic, embryotoxic, fetotoxic, and abortifacient.
There are no adequate and well-controlled studies of VOTRIENT in pregnant women. If this drug is used during pregnancy, or if the patient becomes pregnant while taking this drug, the patient should be apprised of the potential hazard to the fetus. Women of childbearing potential should be advised to avoid becoming pregnant while taking VOTRIENT [see Use in Specific Populations (8.1)].
6 Adverse Reactions
The most common adverse reactions in patients with advanced renal cell carcinoma (‚Č•20%) are diarrhea, hypertension, hair color changes (depigmentation), nausea, anorexia, and vomiting. (6.1 )
The most common adverse reactions in patients with advanced soft tissue sarcoma (‚Č•20%) are fatigue, diarrhea, nausea, decreased weight, hypertension, decreased appetite, hair color changes, vomiting, tumor pain, dysgeusia, headache, musculoskeletal pain, myalgia, gastrointestinal pain, and dyspnea. (6.1 )
To report SUSPECTED ADVERSE REACTIONS, contact GlaxoSmithKline at 1-888-825-5249 or FDA at 1-800-FDA-1088 orwww.fda.gov/medwatch
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared with rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Potentially serious adverse reactions with VOTRIENT included:
‚ÄĘ Hepatotoxicity [see Warnings and Precautions (5.1)]‚ÄĘ QT prolongation and torsades de pointes [see Warnings and Precautions (5.2)]‚ÄĘ Cardiac dysfunction [see Warnings and Precautions (5.3)]‚ÄĘ Hemorrhagic events [see Warnings and Precautions (5.4)]‚ÄĘ Arterial and venous thromboembolic events [see Warnings and Precautions (5.5 and 5.6)]‚ÄĘ Thrombotic microangiopathy [see Warnings and Precautions (5.7)]‚ÄĘ Gastrointestinal perforation and fistula [see Warnings and Precautions (5.8)]‚ÄĘ Interstitial Lung Disease (ILD)/Pneumonitis [see Warnings and Precautions (5.9)]‚ÄĘ Reversible Posterior Leukoencephalopathy Syndrome (RPLS) [see Warnings and Precautions (5.10)]‚ÄĘ Hypertension [see Warnings and Precautions (5.11)]‚ÄĘ Infection [see Warnings and Precautions (5.15)]‚ÄĘ Increased toxicity with other cancer therapies [see Warnings and Precautions (5.16)]
Renal Cell Carcinoma: The safety of VOTRIENT has been evaluated in 977 patients in the monotherapy trials which included 586 patients with RCC at the time of NDA submission. With a median duration of treatment of 7.4¬†months (range: 0.1 to 27.6), the most commonly observed adverse reactions (‚Č•20%) in the 586 patients were diarrhea, hypertension, hair color change, nausea, fatigue, anorexia, and vomiting.
The data described below reflect the safety profile of VOTRIENT in 290 RCC patients who participated in a randomized, double-blind, placebo-controlled trial [see Clinical Studies (14.1)]. The median duration of treatment was 7.4¬†months (range: 0 to 23) for patients who received VOTRIENT and 3.8¬†months (range: 0 to 22) for the placebo arm. Forty-two percent of patients on VOTRIENT required a dose interruption. Thirty-six percent of patients on VOTRIENT were dose reduced. Table 1 presents the most common adverse reactions occurring in ‚Č•10% of patients who received VOTRIENT.
Table 1. Adverse Reactions Occurring in ‚Č•10% of Patients with RCC Who Received VOTRIENT
VOTRIENT
Placebo
(N = 290)
(N = 145)
All Gradesa
Grade 3
Grade 4
All Gradesa
Grade 3
Grade 4
Adverse Reactions
%
%
%
%
%
%
Diarrhea
52
3
<1
9
<1
0
Hypertension
40
4
0
10
<1
0
Hair color changes
38
<1
0
3
0
0
Nausea
26
<1
0
9
0
0
Anorexia
22
2
0
10
<1
0
Vomiting
21
2
<1
8
2
0
Fatigue
19
2
0
8
1
1
Asthenia
14
3
0
8
0
0
Abdominal pain
11
2
0
1
0
0
Headache
10
0
0
5
0
0
aNational Cancer Institute Common Terminology Criteria for Adverse Events, version 3.
Other adverse reactions observed more commonly in patients treated with VOTRIENT than placebo and that occurred in <10% (any grade) were alopecia (8% versus <1%), chest pain (5% versus 1%), dysgeusia (altered taste) (8% versus <1%), dyspepsia (5% versus <1%), dysphonia (4% versus <1%), facial edema (1% versus 0%), palmar-plantar erythrodysesthesia (hand-foot syndrome) (6% versus <1%), proteinuria (9% versus 0%), rash (8% versus 3%), skin depigmentation (3% versus 0%), and weight decreased (9% versus 3%).
Additional adverse reactions from other clinical trials in RCC patients treated with VOTRIENT are uled below:
Musculoskeletal and Connective Tissue Disorders: Arthralgia, muscle spasms.
Table 2 presents the most common laboratory abnormalities occurring in >10% of patients who received VOTRIENT and more commonly (‚Č•5%) in patients who received VOTRIENT versus placebo.
Table 2. Selected Laboratory Abnormalities Occurring in >10% of Patients with RCC Who Received VOTRIENT and More Commonly (‚Č•5%) in Patients Who Received VOTRIENT versus Placebo
VOTRIENT
Placebo
(N = 290)
(N = 145)
All Grades a
Grade 3
Grade 4
All Grades a
Grade 3
Grade 4
Parameters
%
%
%
%
%
%
Hematologic
   Leukopenia
37
0
0
6
0
0
   Neutropenia
34
1
<1
6
0
0
   Thrombocytopenia
32
<1
<1
5
0
<1
   Lymphocytopenia
31
4
<1
24
1
0
Chemistry
   ALT increased
53
10
2
22
1
0
   AST increased
53
7
<1
19
<1
0
   Glucose increased
41
<1
0
33
1
0
   Total bilirubin increased
36
3
<1
10
1
<1
   Phosphorus decreased
34
4
0
11
0
0
   Sodium decreased
31
4
1
24
4
0
   Magnesium decreased
26
<1
1
14
0
0
   Glucose decreased
17
0
<1
3
0
0
aNational Cancer Institute Common Terminology Criteria for Adverse Events, version 3.
Soft Tissue Sarcoma: The safety of VOTRIENT has been evaluated in 382 patients with advanced soft tissue sarcoma, with a median duration of treatment of 3.6 months (range: 0 to 53). The most commonly observed adverse reactions (‚Č•20%) in the 382 patients were fatigue, diarrhea, nausea, decreased weight, hypertension, decreased appetite, vomiting, tumor pain, hair color changes, musculoskeletal pain, headache, dysgeusia, dyspnea, and skin hypopigmentation.
The data described below reflect the safety profile of VOTRIENT in 240 patients who participated in a randomized, double-blind, placebo-controlled trial [see Clinical Studies (14.2)]. The median duration of treatment was 4.5¬†months (range: 0 to 24) for patients who received VOTRIENT and 1.9¬†months (range: 0 to 24) for the placebo arm. Fifty-eight percent of patients on VOTRIENT required a dose interruption. Thirty-eight percent of patients on VOTRIENT had their dose reduced. Seventeen percent of patients who received VOTRIENT discontinued therapy due to adverse reactions. Table 3 presents the most common adverse reactions occurring in ‚Č•10% of patients who received VOTRIENT.
Table 3. Adverse Reactions Occurring in ‚Č•10% of Patients with STS Who Received VOTRIENT
VOTRIENT
Placebo
(N = 240)
(N = 123)
All Gradesa
Grade 3
Grade 4
All Gradesa
Grade 3
Grade 4
Adverse Reactions
%
%
%
%
%
%
Fatigue
65
13
1
48
4
1
Diarrhea
59
5
0
15
1
0
Nausea
56
3
0
22
2
0
Weight decreased
48
4
0
15
0
0
Hypertension
42
7
0
6
0
0
Appetite decreased
40
6
0
19
0
0
Hair color changes
39
0
0
2
0
0
Vomiting
33
3
0
11
1
0
Tumor pain
29
8
0
21
7
2
Dysgeusia
28
0
0
3
0
0
Headache
23
1
0
8
0
0
Musculoskeletal pain
23
2
0
20
2
0
Myalgia
23
2
0
9
0
0
Gastrointestinal pain
23
3
0
9
4
0
Dyspnea
20
5
<1
17
5
1
Exfoliative rash
18
<1
0
9
0
0
Cough
17
<1
0
12
<1
0
Peripheral edema
14
2
0
9
2
0
Mucositis
12
2
0
2
0
0
Alopecia
12
0
0
1
0
0
Dizziness
11
1
0
4
0
0
Skin disorderb
11
2
0
1
0
0
Skin hypopigmentation
11
0
0
0
0
0
Stomatitis
11
<1
0
3
0
0
Chest pain
10
2
0
6
0
0
aNational Cancer Institute Common Terminology Criteria for Adverse Events, version 3.
b27 of the 28 cases of skin disorder were palmar-plantar erythrodysesthesia.
Other adverse reactions observed more commonly in patients treated with VOTRIENT that occurred in ‚Č•5% of patients and at an incidence of more than 2% difference from placebo included insomnia (9% versus 6%), hypothyroidism (8% versus 0%), dysphonia (8% versus 2%), epistaxis (8% versus 2%), left ventricular dysfunction (8% versus 4%), dyspepsia (7% versus 2%), dry skin (6% versus <1%), chills (5% versus 1%), vision blurred (5% versus 2%), and nail disorder (5% versus 0%).
Table 4 presents the most common laboratory abnormalities occurring in >10% of patients who received VOTRIENT and more commonly (‚Č•5%) in patients who received VOTRIENT versus placebo.
Table 4. Selected Laboratory Abnormalities Occurring in >10% of Patients with STS Who Received VOTRIENT and More Commonly (‚Č•5%) in Patients Who Received VOTRIENT versus Placebo
VOTRIENT
Placebo
(N = 240)
(N = 123)
All Grades a
Grade 3
Grade 4
All Grades a
Grade 3
Grade 4
Parameters
%
%
%
%
%
%
Hematologic
   Leukopenia
44
1
0
15
0
0
   Lymphocytopenia
43
10
0
36
9
2
   Thrombocytopenia
36
3
1
6
0
0
   Neutropenia
33
4
0
7
0
0
Chemistry
   AST increased
51
5
3
22
2
0
   ALT increased
46
8
2
18
2
1
   Glucose increased
45
<1
0
35
2
0
   Albumin decreased
34
1
0
21
0
0
   Alkaline phosphatase increased
32
3
0
23
1
0
   Sodium decreased
31
4
0
20
3
0
   Total bilirubin increased
29
1
0
7
2
0
   Potassium increased
16
1
0
11
0
0
aNational Cancer Institute Common Terminology Criteria for Adverse Events, version 3.
Diarrhea: Diarrhea occurred frequently and was predominantly mild to moderate in severity in both the RCC and STS clinical trials. Patients should be advised how to manage mild diarrhea and to notify their healthcare provider if moderate to severe diarrhea occurs so appropriate management can be implemented to minimize its impact.
Lipase Elevations: In a single-arm RCC trial, increases in lipase values were observed for 27% (48/181) of patients. Elevations in lipase as an adverse reaction were reported for 4% (10/225) of patients and were Grade 3 for 6 patients and Grade 4 for 1 patient. In the RCC trials of VOTRIENT, clinical pancreatitis was observed in <1% (4/586) of patients.
Pneumothorax: Two of 290 patients treated with VOTRIENT and no patient on the placebo arm in the randomized RCC trial developed a pneumothorax. In the randomized trial of VOTRIENT for the treatment of STS, pneumothorax occurred in 3% (8/240) of patients treated with VOTRIENT and in no patients on the placebo arm.
Bradycardia: In the randomized trial of VOTRIENT for the treatment of RCC, bradycardia based on vital signs (<60 beats per minute) was observed in 19% (52/280) of patients treated with VOTRIENT and in 11% (16/144) of patients on the placebo arm. Bradycardia was reported as an adverse reaction in 2% (7/290) of patients treated with VOTRIENT compared with <1% (1/145) of patients treated with placebo. In the randomized trial of VOTRIENT for the treatment of STS, bradycardia based on vital signs (<60 beats per minute) was observed in 19% (45/238) of patients treated with VOTRIENT and in 4% (5/121) of patients on the placebo arm. Bradycardia was reported as an adverse reaction in 2% (4/240) of patients treated with VOTRIENT compared with <1% (1/123) of patients treated with placebo.
6.2 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of VOTRIENT. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate the frequency or establish a causal relationship to drug exposure.
Eye Disorders: Retinal detachment/tear.
Gastrointestinal Disorders: Pancreatitis.
7 Drug Interactions
‚ÄĘ CYP3A4 Inhibitors: Avoid use of strong CYP3A4 inhibitors. If coadministration is warranted, reduce the dose of VOTRIENT to 400¬†mg. (7.1 )‚ÄĘ CYP3A4 Inducers: Consider an alternate concomitant medication with no or minimal enzyme induction potential or avoid VOTRIENT. (7.1 )‚ÄĘ CYP Substrates: Concomitant use of VOTRIENT with agents with narrow therapeutic windows that are metabolized by CYP3A4, CYP2D6, or CYP2C8 is not recommended. (7.3 )‚ÄĘ Concomitant use of VOTRIENT and simvastatin increases the risk of ALT elevations and should be undertaken with caution and close monitoring. (7.4 )‚ÄĘ Drugs that Raise Gastric pH: Avoid concomitant use of VOTRIENT with drugs that raise gastric pH. Consider short-acting antacids in place of proton pump inhibitors (PPIs) and H2 receptor antagonists. Separate antacid and pazopanib dosing by several hours. (7.5 )7.1 Drugs that Inhibit or Induce Cytochrome P450 3A4 Enzymes
In vitro studies suggested that the oxidative metabolism of pazopanib in human liver microsomes is mediated primarily by CYP3A4, with minor contributions from CYP1A2 and CYP2C8. Therefore, inhibitors and inducers of CYP3A4 may alter the metabolism of pazopanib.
CYP3A4 Inhibitors: Coadministration of pazopanib with strong inhibitors of CYP3A4 (e.g., ketoconazole, ritonavir, clarithromycin) increases pazopanib concentrations and should be avoided. Consider an alternate concomitant medication with no or minimal potential to inhibit CYP3A4 [see Clinical Pharmacology (12.3)]. If coadministration of a strong CYP3A4 inhibitor is warranted, reduce the dose of VOTRIENT to 400 mg [see Dosage and Administration (2.2)]. Grapefruit or grapefruit juice should be avoided as it inhibits CYP3A4 activity and may also increase plasma concentrations of pazopanib.
CYP3A4 Inducers: CYP3A4 inducers such as rifampin may decrease plasma pazopanib concentrations. Consider an alternate concomitant medication with no or minimal enzyme induction potential. VOTRIENT should not be used if chronic use of strong CYP3A4 inducers cannot be avoided [see Dosage and Administration (2.2)].
7.2 Drugs that Inhibit Transporters
In vitro studies suggested that pazopanib is a substrate of P-glycoprotein (P-gp) and breast cancer resistance protein (BCRP). Therefore, absorption and subsequent elimination of pazopanib may be influenced by products that affect P-gp and BCRP.
Concomitant treatment with strong inhibitors of P-gp or BCRP should be avoided due to risk of increased exposure to pazopanib. Selection of alternative concomitant medicinal products with no or minimal potential to inhibit P-gp or BCRP should be considered.
7.3 Effects of Pazopanib on CYP Substrates
Results from drug-drug interaction trials conducted in cancer patients suggest that pazopanib is a weak inhibitor of CYP3A4, CYP2C8, and CYP2D6 in vivo, but had no effect on CYP1A2, CYP2C9, or CYP2C19 [see Clinical Pharmacology (12.3)].
Concomitant use of VOTRIENT with agents with narrow therapeutic windows that are metabolized by CYP3A4, CYP2D6, or CYP2C8 is not recommended. Coadministration may result in inhibition of the metabolism of these products and create the potential for serious adverse events [see Clinical Pharmacology (12.3)].
7.4 Effect of Concomitant Use of VOTRIENT and Simvastatin
Concomitant use of VOTRIENT and simvastatin increases the incidence of ALT elevations. Across monotherapy trials with VOTRIENT, ALT >3 X ULN was reported in 126/895 (14%) of patients who did not use statins, compared with 11/41 (27%) of patients who had concomitant use of simvastatin. If a patient receiving concomitant simvastatin develops ALT elevations, follow dosing guidelines for VOTRIENT or consider alternatives to VOTRIENT [see Warnings and Precautions (5.1)]. Alternatively, consider discontinuing simvastatin [see Warnings and Precautions (5.1)]. Insufficient data are available to assess the risk of concomitant administration of alternative statins and VOTRIENT.
7.5 Drugs that Raise Gastric pH
In a drug interaction trial in patients with solid tumors, concomitant administration of pazopanib with esomeprazole, a proton pump inhibitor (PPI), decreased the exposure of pazopanib by approximately 40% (AUC and Cmax). Therefore, concomitant use of VOTRIENT with drugs that raise gastric pH should be avoided. If such drugs are needed, short-acting antacids should be considered in place of PPIs and H2 receptor antagonists. Separate antacid and pazopanib dosing by several hours to avoid a reduction in pazopanib exposure [see Clinical Pharmacology (12.3)].
8 Use In Specific Populations
8.1 Pregnancy
Pregnancy Category D [see Warnings and Precautions (5.18)].
VOTRIENT can cause fetal harm when administered to a pregnant woman. There are no adequate and well-controlled studies of VOTRIENT in pregnant women.
In developmental toxicity studies in rats and rabbits, pazopanib was teratogenic, embryotoxic, fetotoxic, and abortifacient. Administration of pazopanib to pregnant rats during organogenesis at a dose level of ‚Č•3¬†mg/kg/day (approximately 0.1 times the human clinical exposure based on AUC) resulted in teratogenic effects including cardiovascular malformations (retroesophageal subclavian artery, missing innominate artery, changes in the aortic arch) and incomplete or absent ossification. In addition, there was reduced fetal body weight, and pre- and post-implantation embryolethality in rats administered pazopanib at doses ‚Č•3¬†mg/kg/day. In rabbits, maternal toxicity (reduced food consumption, increased post-implantation loss, and abortion) was observed at doses ‚Č•30¬†mg/kg/day (approximately 0.007 times the human clinical exposure). In addition, severe maternal body weight loss and 100% litter loss were observed at doses ‚Č•100¬†mg/kg/day (0.02 times the human clinical exposure), while fetal weight was reduced at doses ‚Č•3¬†mg/kg/day (AUC not calculated).
If this drug is used during pregnancy, or if the patient becomes pregnant while taking this drug, the patient should be apprised of the potential hazard to the fetus. Advise females of reproductive potential to use effective contraception during treatment and for at least 2 weeks after the last dose of VOTRIENT.
8.3 Nursing Mothers
It is not known whether this drug is excreted in human milk. Because many drugs are excreted in human milk and because of the potential for serious adverse reactions in nursing infants from VOTRIENT, a decision should be made whether to discontinue nursing or to discontinue the drug, taking into account the importance of the drug to the mother.
8.4 Pediatric Use
The safety and effectiveness of VOTRIENT in pediatric patients have not been established.
In rats, weaning occurs at Day 21 postpartum which approximately equates to a human pediatric age of 2 years. In a juvenile animal toxicology study performed in rats, when animals were dosed from Day 9 through Day 14 postpartum (pre-weaning), pazopanib caused abnormal organ growth/maturation in the kidney, lung, liver, and heart at approximately 0.1 times the clinical exposure, based on AUC in adult patients receiving VOTRIENT. At approximately 0.4 times the clinical exposure (based on the AUC in adult patients), pazopanib administration resulted in mortality.
In repeat-dose toxicology studies in rats including 4-week, 13-week, and 26-week administration, toxicities in bone, teeth, and nail beds were observed at doses ‚Č•3¬†mg/kg/day (approximately 0.07 times the human clinical exposure based on AUC). Doses of 300¬†mg/kg/day (approximately 0.8 times the human clinical exposure based on AUC) were not tolerated in 13- and 26-week studies and animals required dose reductions due to body weight loss and morbidity. Hypertrophy of epiphyseal growth plates, nail abnormalities (including broken, overgrown, or absent nails) and tooth abnormalities in growing incisor teeth (including excessively long, brittle, broken, and missing teeth, and dentine and enamel degeneration and thinning) were observed in rats at doses ‚Č•30¬†mg/kg/day (approximately 0.35 times the human clinical exposure based on AUC) at 26¬†weeks, with the onset of tooth and nail bed alterations noted clinically after 4 to 6¬†weeks. Similar findings were noted in repeat-dose studies in juvenile rats dosed with pazopanib beginning Day 21 postpartum (post-weaning). In the post-weaning animals, the occurrence of changes in teeth and bones occurred earlier and with greater severity than in older animals. There was evidence of tooth degeneration and decreased bone growth at doses ‚Č•30¬†mg/kg (approximately 0.1 to 0.2 times the AUC in human adults at the clinically recommended dose). Pazopanib exposure in juvenile rats was lower than that seen at the same dose levels in adult animals, based on comparative AUC values. At pazopanib doses approximately 0.5 to 0.7 times the exposure in adult patients at the clinically recommended dose, decreased bone growth in juvenile rats persisted even after the end of the dosing period. Finally, despite lower pazopanib exposures than those reported in adult animals or adult humans, juvenile animals administered 300¬†mg/kg/dose pazopanib required dose reduction within 4¬†weeks of dosing initiation due to significant toxicity, although adult animals could tolerate this same dose for at least 3 times as long [see Warnings and Precautions (5.17)].
8.5 Geriatric Use
In pooled clinical trials with VOTRIENT, 30% (618/2,080) of patients were aged >65¬†years. Patients aged >65¬†years had an increase in ALT elevations of >3 X ULN compared to patients aged <65¬†years (23% versus 18%) [see Warnings and Precautions (5.1)]. In clinical trials with VOTRIENT for the treatment of RCC, 33% (196/582) of patients were aged ‚Č•65¬†years. No overall differences in safety or effectiveness of VOTRIENT were observed between these patients and younger patients. In the STS trials, 24% (93/382) of patients were aged ‚Č•65¬†years. Patients aged ‚Č•65 years had increased Grade 3 or 4 fatigue (19% versus 12% for <65), hypertension (10% versus 6%), decreased appetite (11% versus 2%), and ALT (3% versus 2%) or AST elevations (4% versus 1%). Other reported clinical experience has not identified differences in responses between elderly and younger patients, but greater sensitivity of some older individuals cannot be ruled out.
8.6 Hepatic Impairment
In clinical trials for VOTRIENT, patients with total bilirubin ‚ȧ1.5 X ULN and AST and ALT ‚ȧ2 X ULN were included [see Warnings and Precautions (5.1)].
An analysis of data from a pharmacokinetic trial of pazopanib in patients with varying degrees of hepatic dysfunction suggested that no dose adjustment is required in patients with mild hepatic impairment (either total bilirubin within normal limit [WNL] with ALT >ULN or bilirubin >1 X to 1.5 X ULN regardless of the ALT value). The maximum tolerated dose in patients with moderate hepatic impairment (total bilirubin >1.5 X to 3 X ULN regardless of the ALT value) was 200 mg per day (N = 11). The median steady-state Cmax and AUC(0-24) achieved at this dose was approximately 40% and 29%, respectively, of that seen in patients with normal hepatic function at the recommended daily dose of 800 mg. The maximum dose explored in patients with severe hepatic impairment (total bilirubin >3 X ULN regardless of the ALT value) was 200 mg per day (N = 14). This dose was not well tolerated. Median exposures achieved at this dose were approximately 18% and 15% of those seen in patients with normal liver function at the recommended daily dose of 800 mg. Therefore, VOTRIENT is not recommended in these patients [see Clinical Pharmacology (12.3)].
8.7 Renal Impairment
Patients with renal cell cancer and mild/moderate renal impairment (creatinine clearance ‚Č•30¬†mL/min) were included in clinical trials for VOTRIENT.
There are no clinical or pharmacokinetic data in patients with severe renal impairment or in patients undergoing peritoneal dialysis or hemodialysis. However, renal impairment is unlikely to significantly affect the pharmacokinetics of pazopanib since <4% of a radiolabeled oral dose was recovered in the urine. In a population pharmacokinetic analysis using 408 patients with various cancers, creatinine clearance (30 to 150 mL/min) did not influence clearance of pazopanib. Therefore, renal impairment is not expected to influence pazopanib exposure, and dose adjustment is not necessary.
10 Overdosage
Pazopanib doses up to 2,000 mg have been evaluated in clinical trials. Dose-limiting toxicity (Grade 3 fatigue) and Grade 3 hypertension were each observed in 1 of 3 patients dosed at 2,000 mg daily and 1,000 mg daily, respectively.
Treatment of overdose with VOTRIENT should consist of general supportive measures. There is no specific antidote for overdosage of VOTRIENT.
Hemodialysis is not expected to enhance the elimination of VOTRIENT because pazopanib is not significantly renally excreted and is highly bound to plasma proteins.
11 Description
VOTRIENT (pazopanib) is a tyrosine kinase inhibitor (TKI). Pazopanib is presented as the hydrochloride salt, with the chemical name 5-[[4-[(2,3-dimethyl-2H-indazol-6-yl)methylamino]-2-pyrimidinyl]amino]-2-methylbenzenesulfonamide monohydrochloride. It has the molecular formula C21H23N7O2S‚ÄĘHCl and a molecular weight of 473.99. Pazopanib hydrochloride has the following chemical structure:
![]()
Pazopanib hydrochloride is a white to slightly yellow solid. It is very slightly soluble at pH 1 and practically insoluble above pH 4 in aqueous media.
Tablets of VOTRIENT are for oral administration. Each 200-mg tablet of VOTRIENT contains 216.7 mg of pazopanib hydrochloride, equivalent to 200 mg of pazopanib free base.
The inactive ingredients of VOTRIENT are: Tablet Core: Magnesium stearate, microcrystalline cellulose, povidone, sodium starch glycolate. Coating: Gray film-coat: Hypromellose, iron oxide black, macrogol/polyethylene glycol 400 (PEG 400), polysorbate 80, titanium dioxide.
12 Clinical Pharmacology
12.1 Mechanism of Action
Pazopanib is a multi-tyrosine kinase inhibitor of vascular endothelial growth factor receptor (VEGFR)-1, VEGFR-2, VEGFR-3, platelet-derived growth factor receptor (PDGFR)-őĪ and -ő≤, fibroblast growth factor receptor (FGFR)-1 and -3, cytokine receptor (Kit), interleukin-2 receptor-inducible T-cell kinase (Itk), leukocyte-specific protein tyrosine kinase (Lck), and transmembrane glycoprotein receptor tyrosine kinase (c-Fms). In vitro, pazopanib inhibited ligand-induced autophosphorylation of VEGFR-2, Kit, and PDGFR-ő≤ receptors. In vivo, pazopanib inhibited VEGF-induced VEGFR-2 phosphorylation in mouse lungs, angiogenesis in a mouse model, and the growth of some human tumor xenografts in mice.
12.2 Pharmacodynamics
Increases in blood pressure have been observed and are related to steady-state trough plasma pazopanib concentrations.
The QT prolongation potential of pazopanib was assessed in a randomized, blinded, parallel trial (N = 96) using moxifloxacin as a positive control. Pazopanib 800 mg was dosed under fasting conditions on Days 2 to 8 and 1,600 mg was dosed on Day 9 after a meal in order to increase exposure to pazopanib and its metabolites. No large changes (i.e., >20 msec) in QTc interval following the treatment of pazopanib were detected in this QT trial. The trial was not able to exclude small changes (<10 msec) in QTc interval, because assay sensitivity below this threshold (<10 msec) was not established in this trial [see Warnings and Precautions (5.2)].
12.3 Pharmacokinetics
Absorption: Pazopanib is absorbed orally with median time to achieve peak concentrations of 2 to 4¬†hours after the dose. Daily dosing at 800¬†mg results in geometric mean AUC and Cmax of 1,037¬†mcg‚ÄĘh/mL and 58.1¬†mcg/mL (equivalent to 132¬†őľM), respectively. There was no consistent increase in AUC or Cmax at pazopanib doses above 800¬†mg.
Administration of a single pazopanib 400-mg crushed tablet increased AUC(0-72) by 46% and Cmax by approximately 2 fold and decreased Tmax by approximately 2 hours compared with administration of the whole tablet. These results indicate that the bioavailability and the rate of pazopanib oral absorption are increased after administration of the crushed tablet relative to administration of the whole tablet. Therefore, due to this potential for increased exposure, tablets of VOTRIENT should not be crushed.
Systemic exposure to pazopanib is increased when administered with food. Administration of pazopanib with a high-fat or low-fat meal results in an approximately 2-fold increase in AUC and Cmax. Therefore, pazopanib should be administered at least 1 hour before or 2 hours after a meal [see Dosage and Administration (2.1)].
Distribution: Binding of pazopanib to human plasma protein in vivo was greater than 99% with no concentration dependence over the range of 10 to 100 mcg/mL. In vitro studies suggest that pazopanib is a substrate for P-gp and BCRP.
Metabolism: In vitro studies demonstrated that pazopanib is metabolized by CYP3A4 with a minor contribution from CYP1A2 and CYP2C8.
Elimination: Pazopanib has a mean half-life of 30.9 hours after administration of the recommended dose of 800 mg. Elimination is primarily via feces with renal elimination accounting for <4% of the administered dose.
Hepatic Impairment: Mild hepatic impairment was defined as either total bilirubin WNL with ALT >ULN or bilirubin >1 X to 1.5 X ULN regardless of the ALT value. The median steady-state pazopanib Cmax and AUC(0-24) after a once-daily dose of 800¬†mg/day in patients (N¬†=¬†12) with mild impairment were 34¬†mcg/mL (range: 11 to 104) and 774¬†mcg‚ÄĘh/mL (range: 215 to 2,034), respectively. These were in a similar range as the median steady-state pazopanib Cmax and AUC(0-24) in patients (N¬†=¬†18) with no hepatic impairment (52¬†mcg/mL, range: 17 to 86 and 888¬†mcg‚ÄĘh/mL, range: 346 to 1,482, respectively) [see Dosage and Administration (2.2)].
Moderate hepatic impairment was defined as total bilirubin >1.5 X to 3 X ULN regardless of the ALT value. The maximum tolerated pazopanib dose in patients with moderate impairment was 200¬†mg once daily. The median (N¬†=¬†11) steady-state Cmax with that regimen was 22¬†mcg/mL (range: 4.2 to 33), and the median AUC(0-24) was 257¬†mcg‚ÄĘh/mL (range: 66 to 488). These values were approximately 43% and 29% of the corresponding median values after administration of 800 mg once daily in patients with normal hepatic function (N¬†=¬†18) [see Dosage and Administration (2.2)].
Severe hepatic impairment was defined as total bilirubin >3 X ULN regardless of the ALT value. Median exposures in patients with severe hepatic impairment receiving 200 mg once daily (N¬†=¬†14) were unexpectedly lower than those observed in patients with moderate hepatic impairment receiving 200 mg once daily. The median steady-state Cmax was 9.4¬†mcg/mL (range: 2.4 to 24), and the median AUC(0-24) was 131 mcg‚ÄĘh/mL (range: 47 to 473). These values were approximately 18% and 15% of the corresponding median values after administration of 800¬†mg once daily in patients with normal hepatic function. Despite the observed concentrations, the dose of 200¬†mg was not well tolerated in patients with severe hepatic impairment. Use of VOTRIENT is not recommended in patients with severe hepatic impairment [see Use in Specific Populations (8.6)].
Drug Interactions: Coadministration of multiple doses of oral pazopanib 400 mg with multiple doses of oral ketoconazole 400 mg (strong CYP3A4/P-gp inhibitor) resulted in a 1.7-fold increase in the AUC(0-24) and a 1.5-fold increase in the Cmax of pazopanib compared with pazopanib administered alone. Concurrent administration of a single dose of pazopanib eye drops with ketoconazole in healthy volunteers resulted in a 2-fold and 1.5-fold increase in mean AUC(0-t) and Cmax values, respectively [see Dosage and Administration (2.2), Drug Interactions (7.1)].
Administration of 1,500 mg lapatinib, a substrate and weak inhibitor of CYP3A4, P-gp, and BCRP, with 800 mg pazopanib resulted in an approximately 50% to 60% increase in mean pazopanib AUC(0-24) and Cmax compared with administration of 800 mg pazopanib alone.
In vitro studies with human liver microsomes showed that pazopanib inhibited the activities of CYP enzymes 1A2, 3A4, 2B6, 2C8, 2C9, 2C19, 2D6, and 2E1. Potential induction of human CYP3A4 was demonstrated in an in vitro human pregnane X receptor (PXR) assay. Clinical pharmacology studies, using pazopanib 800 mg once daily, have demonstrated that pazopanib does not have a clinically relevant effect on the pharmacokinetics of caffeine (CYP1A2 probe substrate), warfarin (CYP2C9 probe substrate), or omeprazole (CYP2C19 probe substrate) in cancer patients. Pazopanib resulted in an increase of approximately 30% in the mean AUC and Cmax of midazolam (CYP3A4 probe substrate) and increases of 33% to 64% in the ratio of dextromethorphan to dextrorphan concentrations in the urine after oral administration of dextromethorphan (CYP2D6 probe substrate). Coadministration of pazopanib 800 mg once daily and paclitaxel 80 mg/m2 (CYP3A4 and CYP2C8 substrate) once weekly resulted in a mean increase of 26% and 31% in paclitaxel AUC and Cmax, respectively [see Drug Interactions (7.3)].
Pazopanib exhibits pH-dependent solubility. In a drug interaction trial in patients with solid tumors, concomitant administration of pazopanib with esomeprazole, a PPI, decreased the exposure of pazopanib by approximately 40% (AUC and Cmax).
In vitro studies also showed that pazopanib inhibits UGT1A1 and organic anion-transporting polypeptide (OATP1B1) with IC50s of 1.2 and 0.79 őľM, respectively. Pazopanib may increase concentrations of drugs eliminated by UGT1A1 and OATP1B1.
12.5 Pharmacogenomics
Pazopanib can increase serum total bilirubin levels [see Warnings and Precautions (5.1)]. In vitro studies showed that pazopanib inhibits UGT1A1, which glucuronidates bilirubin for elimination. A pooled pharmacogenetic analysis of 236 Caucasian patients evaluated the TA-repeat polymorphism of UGT1A1 and its potential association with hyperbilirubinemia during pazopanib treatment. In this analysis, the (TA)7/(TA)7 genotype (UGT1A1*28/*28) (underlying genetic susceptibility to Gilbert’s syndrome) was associated with a statistically significant increase in the incidence of hyperbilirubinemia relative to the (TA)6/(TA)6 and (TA)6/(TA)7 genotypes.
In a pooled pharmacogenetic analysis of data from 31 clinical studies of pazopanib administered as either monotherapy or in combination with other agents, ALT > 3 X ULN (NCI CTC Grade 2) occurred in 32% (42/133) of HLA-B*57:01 allele carriers and in 19% (397/2101) of non-carriers and ALT > 5 X ULN (NCI CTC Grade 3) occurred in 19% (25/133) of HLA-B*57:01 allele carriers and in 10% (213/2101) of non-carriers. In this dataset, 6% (133/2234) of the patients carried the HLA-B*57:01 allele. Liver function should be monitored in all subjects receiving pazopanib, regardless of genotype [see Warnings and Precautions (5.1)].
13 Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
The carcinogenic potential of pazopanib was evaluated in CD-1 mice and Sprague-Dawley rats. Administration of pazopanib to mice for 2 years did not result in increased incidence of neoplasms at doses up to 100 mg/kg/day (approximately 1.4 times human clinical exposure based on AUC). Administration of pazopanib to rats for 2 years resulted in findings of duodenal adenocarcinoma in males at 30 mg/kg/day (approximately 0.3 times human clinical exposure based on AUC) and in females at ‚Č• 10 mg/kg/day (approximately 0.3 times the human clinical exposure based on AUC). The human relevance of these neoplastic findings is unclear.
Pazopanib did not induce mutations in the microbial mutagenesis (Ames) assay and was not clastogenic in both the in vitro cytogenetic assay using primary human lymphocytes and in the in vivo rat micronucleus assay.
Pazopanib may impair fertility in humans. In female rats, reduced fertility including increased pre-implantation loss and early resorptions were noted at dosages ‚Č•30¬†mg/kg/day (approximately 0.4 times the human clinical exposure based on AUC). Total litter resorption was seen at 300 mg/kg/day (approximately 0.8 times the human clinical exposure based on AUC). Post-implantation loss, embryolethality, and decreased fetal body weight were noted in females administered doses ‚Č•10¬†mg/kg/day (approximately 0.3 times the human clinical exposure based on AUC). Decreased corpora lutea and increased cysts were noted in mice given ‚Č•100¬†mg/kg/day for 13¬†weeks and ovarian atrophy was noted in rats given ‚Č•300¬†mg/kg/day for 26¬†weeks (approximately 1.3 and 0.85 times the human clinical exposure based on AUC, respectively). Decreased corpora lutea was also noted in monkeys given 500¬†mg/kg/day for up to 34¬†weeks (approximately 0.4 times the human clinical exposure based on AUC).
Pazopanib did not affect mating or fertility in male rats. However, there were reductions in sperm production rates and testicular sperm concentrations at doses ‚Č•3 mg/kg/day, epididymal sperm concentrations at doses ‚Č•30¬†mg/kg/day, and sperm motility at ‚Č•100¬†mg/kg/day following 15¬†weeks of dosing. Following 15 and 26¬†weeks of dosing, there were decreased testicular and epididymal weights at doses of ‚Č•30¬†mg/kg/day (approximately 0.35 times the human clinical exposure based on AUC); atrophy and degeneration of the testes with aspermia, hypospermia and cribiform change in the epididymis was also observed at this dose in the 6-month toxicity studies in male rats.
14 Clinical Studies
14.1 Renal Cell Carcinoma
The safety and efficacy of VOTRIENT in renal cell carcinoma (RCC) were evaluated in a randomized, double-blind, placebo-controlled, multicenter, Phase 3 trial. Patients (N = 435) with locally advanced and/or metastatic RCC who had received either no prior therapy or one prior cytokine-based systemic therapy were randomized (2:1) to receive VOTRIENT 800 mg once daily or placebo once daily. The primary objective of the trial was to evaluate and compare the 2 treatment arms for progression-free survival (PFS); the secondary endpoints included overall survival (OS), overall response rate (RR), and duration of response.
Of the total of 435 patients enrolled in this trial, 233 patients had no prior systemic therapy (treatment-na√Įve subgroup) and 202 patients received one prior IL-2 or INFőĪ-based therapy (cytokine-pretreated subgroup). The baseline demographic and disease characteristics were balanced between the arms receiving VOTRIENT and placebo. The majority of patients were male (71%) with a median age of 59¬†years. Eighty-six percent of patients were Caucasian, 14% were Asian, and less than 1% were other. Forty-two percent were ECOG performance status 0 and 58% were ECOG performance status 1. All patients had clear cell histology (90%) or predominantly clear cell histology (10%). Approximately 50% of all patients had 3 or more organs involved with metastatic disease. The most common metastatic sites at baseline were lung (74%), lymph nodes (56%), bone (27%), and liver (25%).
A similar proportion of patients in each arm were treatment-na√Įve and cytokine-pretreated (see Table 5). In the cytokine-pretreated subgroup, the majority (75%) had received interferon-based treatment. Similar proportions of patients in each arm had prior nephrectomy (89% and 88% for VOTRIENT and placebo, respectively).
The analysis of the primary endpoint PFS was based on disease assessment by independent radiological review in the entire trial population. Efficacy results are presented in Table 5 and Figure 1.
Table 5. Efficacy Results in RCC Patients by Independent Assessment
Endpoint/Trial Population
VOTRIENT
Placebo
HR
(95% CI)
PFS
Overall ITT
N = 290
N = 145
   Median (months)
9.2
4.2
0.46a
(0.34, 0.62)
Treatment-na√Įve subgroup
N = 155 (53%)
N = 78 (54%)
   Median (months)
11.1
2.8
0.40
(0.27, 0.60)
Cytokine pre-treated subgroup
N = 135 (47%)
N = 67 (46%)
   Median (months)
7.4
4.2
0.54
(0.35, 0.84)
Response Rate (CR + PR)
N = 290
N = 145
   % (95% CI)
30 (25.1, 35.6)
3 (0.5, 6.4)
‚Äď
Duration of response
   Median (weeks) (95% CI)
58.7 (52.1, 68.1)
‚Äďb
HR = Hazard Ratio; ITT = Intent to Treat; PFS = Progression-free Survival; CR = Complete Response; PR = Partial Response.
a P value <0.001.
bThere were only 5 objective responses.
Figure 1. Kaplan-Meier Curve for Progression-free Survival in RCC by Independent Assessment for the Overall Population (Treatment-na√Įve and Cytokine Pre-treated Populations)
![]()
At the protocol-specified final analysis of OS, the median OS was 22.9 months for patients randomized to VOTRIENT and 20.5 months for the placebo arm [HR = 0.91 (95% CI: 0.71, 1.16)]. The median OS for the placebo arm includes 79 patients (54%) who discontinued placebo treatment because of disease progression and crossed over to treatment with VOTRIENT. In the placebo arm, 95 (66%) patients received at least one systemic anti-cancer treatment after progression compared with 88 (30%) patients randomized to VOTRIENT.
14.2 Soft Tissue Sarcoma
The safety and efficacy of VOTRIENT in patients with STS were evaluated in a randomized, double-blind, placebo-controlled, multicenter trial. Patients (N = 369) with metastatic STS who had received prior chemotherapy, including anthracycline treatment, or were unsuited for such therapy, were randomized (2:1) to receive VOTRIENT 800 mg once daily or placebo. Patients with gastrointestinal stromal tumors (GIST) or adipocytic sarcoma were excluded from the trial. Randomization was stratified by the factors of WHO performance status (WHO PS) 0 or 1 at baseline and the number of lines of prior systemic therapy for advanced disease (0 or 1 versus 2+). Progression-free survival (PFS) was assessed by independent radiological review. Other efficacy endpoints included overall survival (OS), overall response rate, and duration of response.
The majority of patients were female (59%) with a median age of 55 years. Seventy-two percent of patients were Caucasian, 22% were Asian, and 6% were other. Forty-three percent of patients had leiomyosarcoma, 10% had synovial sarcoma, and 47% had other soft tissue sarcomas. Fifty-six percent of patients had received 2 or more lines of prior systemic therapy and 44% had received 0 or 1 lines of prior systemic therapy. The median duration of treatment was 4.5 months for patients on the pazopanib arm and 1.9 months for patients on the placebo arm.
Efficacy results are presented in Table 6 and Figure 2.
Table 6. Efficacy Results in STS Patients by Independent Assessment
Endpoint/Trial Population
VOTRIENT
Placebo
HR
(95% CI)
PFS
Overall ITT
N = 246
N = 123
0.35a
   Median (months)
4.6
1.6
(0.26, 0.48)
Leiomyosarcoma subgroup
N = 109
N = 49
0.37
   Median (months)
4.6
1.9
(0.23, 0.60)
Synovial sarcoma subgroup
N = 25
N = 13
0.43
   Median (months)
4.1
0.9
(0.19, 0.98)
‚ÄėOther soft tissue sarcoma‚Äô subgroup
N = 112
N = 61
0.39
   Median (months)
4.6
1.0
(0.25, 0.60)
Response Rate (CR + PR)
   % (95% CI)
4 (2.3, 7.9)b
0 (0.0, 3.0)
‚Äď
Duration of response
   Median (months) (95% CI)
9.0 (3.9, 9.2)
HR = Hazard Ratio; ITT = Intent to Treat; PFS = Progression-free Survival; CR = Complete Response; PR = Partial Response.
a P value <0.001.
bThere were 11 partial responses and 0 complete responses.
Figure 2. Kaplan-Meier Curve for Progression-free Survival in STS by Independent Assessment for the Overall Population
![]()
At the protocol-specified final analysis of OS, the median OS was 12.6 months for patients randomized to VOTRIENT and 10.7 months for the placebo arm [HR = 0.87 (95% CI: 0.67, 1.12)].
16 How Supplied/storage And Handling
The 200-mg tablets of VOTRIENT are modified capsule-shaped, gray, film-coated with GS JT debossed on one side and are available in:
Bottles of 120 tablets: NDC 0173-0804-09
Store at room temperature between 20¬įC and 25¬įC (68¬įF to 77¬įF); excursions permitted to 15¬įC to 30¬įC (59¬įF to 86¬įF) [see USP Controlled Room Temperature].
17 Patient Counseling Information
Advise the patient to read the FDA-approved patient labeling (Medication Guide). The Medication Guide is contained in a separate leaflet that accompanies the product.
However, inform patients of the following:
‚ÄĘ Therapy with VOTRIENT may result in hepatobiliary laboratory abnormalities. Monitor serum liver tests (ALT, AST, and bilirubin) prior to initiation of VOTRIENT and at Weeks 3, 5, 7, and 9. Thereafter, monitor at Month 3 and at Month 4, and as clinically indicated. Inform patients that they should report signs and symptoms of liver dysfunction to their healthcare provider right away.‚ÄĘ Prolonged QT intervals and torsades de pointes have been observed. Patients should be advised that ECG monitoring may be performed. Patients should be advised to inform their physicians of concomitant medications.‚ÄĘ ILD has been reported in association with VOTRIENT. Patients should be advised to report pulmonary signs or symptoms indicative of ILD or pneumonitis.‚ÄĘ Cardiac dysfunction (such as CHF and LVEF decrease) has been observed in patients at risk (e.g., prior anthracycline therapy) particularly in association with development or worsening of hypertension. Patients should be advised to report hypertension or signs and symptoms of congestive heart failure.‚ÄĘ Serious hemorrhagic events have been reported. Patients should be advised to report unusual bleeding.‚ÄĘ Arterial thrombotic events have been reported. Patients should be advised to report signs or symptoms of an arterial thrombosis.‚ÄĘ Reports of pneumothorax and venous thromboembolic events, including pulmonary embolus, have been reported. Patients should be advised to report if new onset of dyspnea, chest pain, or localized limb edema occurs.‚ÄĘ Advise patients to inform their doctor if they have worsening of neurological function consistent with RPLS (headache, seizure, lethargy, confusion, blindness, and other visual and neurologic disturbances).‚ÄĘ Hypertension and hypertensive crisis have been reported. Patients should be advised to monitor blood pressure early in the course of therapy and frequently thereafter and report increases of blood pressure or symptoms such as blurred vision, confusion, severe headache, or nausea and vomiting.‚ÄĘ GI perforation or fistula has occurred. Advise patients to report signs and symptoms of a GI perforation or fistula.‚ÄĘ VEGFR-inhibitors such as VOTRIENT may impair wound healing. Advise patients to stop VOTRIENT at least 7 days prior to a scheduled surgery.‚ÄĘ Hypothyroidism and proteinuria have been reported. Advise patients that thyroid function testing and urinalysis will be performed during treatment.‚ÄĘ Serious infections, including some with fatal outcomes, have been reported. Advise patients to promptly report any signs or symptoms of infection.‚ÄĘ Advise females of reproductive potential of the potential hazard to the fetus and to use effective contraception during treatment and for at least 2 weeks after the last dose of VOTRIENT.‚ÄĘ Gastrointestinal adverse reactions such as diarrhea, nausea, and vomiting have been reported with VOTRIENT. Patients should be advised how to manage diarrhea and to notify their healthcare provider if moderate to severe diarrhea occurs.‚ÄĘ Patients should be advised to inform their healthcare providers of all concomitant medications, vitamins, or dietary and herbal supplements.‚ÄĘ Patients should be advised that depigmentation of the hair or skin may occur during treatment with VOTRIENT.‚ÄĘ Patients should be advised to take VOTRIENT without food (at least 1¬†hour before or 2¬†hours after a meal).
VOTRIENT is a registered trademark of the GSK group of companies.
GlaxoSmithKline
Research Triangle Park, NC 27709
©2016 the GSK group of companies. All rights reserved.
VTR:16PI
Spl Medguide Section
MEDICATION GUIDE VOTRIENT¬ģ (VO-tree-ent) (pazopanib) tablets
What is the most important information I should know about VOTRIENT?
‚ÄĘ VOTRIENT can cause serious liver problems including death. Your healthcare provider will do blood tests to check your liver before you start and while you take VOTRIENT.
Tell your healthcare provider right away if you get any of these signs of liver problems during treatment with VOTRIENT:
¬†¬†¬†¬†¬†¬†¬†¬†¬†‚Äʬ†¬†yellowing of your skin or the whites of your eyes¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†‚Äʬ†¬†loss of appetite
¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†(jaundice)¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†‚Äʬ†¬†pain on the right side of your stomach area
¬†¬†¬†¬†¬†¬†¬†¬†¬†‚Äʬ†¬†dark urine¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†(abdomen)
¬†¬†¬†¬†¬†¬†¬†¬†¬†‚Äʬ†¬†tiredness¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†‚Äʬ†¬†bruise easily
¬†¬†¬†¬†¬†¬†¬†¬†¬†‚Äʬ†¬†nausea or vomiting
  Your healthcare provider may need to prescribe a lower dose of VOTRIENT for you or tell you to stop taking VOTRIENT if you develop liver problems during treatment.
What is VOTRIENT?
  VOTRIENT is a prescription medicine used to treat people with:
‚ÄĘ advanced renal cell cancer (RCC)‚ÄĘ advanced soft tissue sarcoma (STS) who have received chemotherapy in the past
It is not known if VOTRIENT is effective in treating certain soft tissue sarcomas or certain gastrointestinal tumors.
It is not known if VOTRIENT is safe and effective in children under 18 years of age.
What should I tell my healthcare provider before taking VOTRIENT?
  Before you take VOTRIENT, tell your healthcare provider if you:
‚ÄĘ have or had liver problems. You may need a lower dose of VOTRIENT or your healthcare provider may prescribe a different medicine to treat your advanced renal cell cancer or advanced soft tissue sarcoma.‚ÄĘ have high blood pressure‚ÄĘ have heart problems or an irregular heartbeat including QT prolongation‚ÄĘ have a history of a stroke‚ÄĘ have headaches, seizures, or vision problems‚ÄĘ have coughed up blood in the last 6 months‚ÄĘ had bleeding of your stomach or intestines in the last 6 months‚ÄĘ have a history of a tear (perforation) in your stomach or intestine, or an abnormal connection between two parts of your gastrointestinal tract (fistula)‚ÄĘ have had blood clots in a vein or in the lung‚ÄĘ have thyroid problems‚ÄĘ had recent surgery (within the last 7 days) or are going to have surgery‚ÄĘ have any other medical conditions‚ÄĘ are pregnant or plan to become pregnant. VOTRIENT can harm your unborn baby. You should not become pregnant while you are taking VOTRIENT. You should use effective birth control during treatment with VOTRIENT and for 2 weeks after your last dose of VOTRIENT. Talk to your healthcare provider about types of birth control that may be right for you during this time.‚ÄĘ are breastfeeding or plan to breastfeed. It is not known if VOTRIENT passes into your breast milk. You and your healthcare provider should decide if you will take VOTRIENT or breastfeed. You should not do both.¬† Tell your healthcare provider about all the medicines you take including prescription and over-the-counter medicines, vitamins, and herbal supplements. VOTRIENT may affect the way other medicines work and other medicines may affect how VOTRIENT works.¬† Especially, tell your healthcare provider if you:‚ÄĘ take medicines that can affect how your liver enzymes work such as:
¬†¬†¬†¬†¬†¬†¬†¬†¬†‚Äʬ†¬†certain antibiotics (used to treat infections)¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†‚Äʬ†¬†certain medicines used to treat depression
¬†¬†¬†¬†¬†¬†¬†¬†¬†‚Äʬ†¬†certain medicines used to treat HIV¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†‚Äʬ†¬†medicines used to treat irregular heart beats
‚ÄĘ take a medicine that contains simvastatin to treat high cholesterol levels‚ÄĘ take medicines that reduce stomach acid (e.g., esomeprazole)‚ÄĘ drink grapefruit juice
Ask your healthcare provider if you are not sure if your medicine is one that is uled above.
Know the medicines you take. Keep a ul of them and show it to your healthcare provider and pharmacist when you get a new medicine.
How should I take VOTRIENT?
‚ÄĘ Take VOTRIENT exactly as your healthcare provider tells you. Your healthcare provider will tell you how much VOTRIENT to take.‚ÄĘ Your healthcare provider may change your dose.‚ÄĘ Take VOTRIENT on an empty stomach, at least 1 hour before or 2 hours after food.‚ÄĘ Do not crush VOTRIENT tablets.‚ÄĘ Do not eat grapefruit or drink grapefruit juice during treatment with VOTRIENT. Grapefruit products may increase the amount of VOTRIENT in your body.‚ÄĘ If you miss a dose, take it as soon as you remember. Do not take it if it is close (within 12¬†hours) to your next dose. Just take the next dose at your regular time. Do not take more than 1 dose of VOTRIENT at a time.‚ÄĘ Your healthcare provider will test your urine, blood, and heart before you start and while you take VOTRIENT.‚ÄĘ Tell your healthcare provider if you plan to have surgery while taking VOTRIENT. You will need to stop taking VOTRIENT at least 7 days before surgery because VOTRIENT may affect healing after surgery.
What are the possible side effects of VOTRIENT?
VOTRIENT may cause serious side effects including:
‚ÄĘ See ‚ÄúWhat is the most important information I should know about VOTRIENT?‚ÄĚ‚ÄĘ irregular or fast heartbeat or fainting‚ÄĘ heart failure. This is a condition where your heart does not pump as well as it should and may cause you to have shortness of breath.‚ÄĘ heart attack or stroke. Heart attack and stroke can happen with VOTRIENT and may cause death. Symptoms may include: chest pain or pressure, pain in your arms, back, neck or jaw, shortness of breath, numbness or weakness on one side of your body, trouble talking, headache, or dizziness.‚ÄĘ blood clots. Blood clots may form in a vein, especially in your legs (deep vein thrombosis or DVT). Pieces of a blood clot may travel to your lungs (pulmonary embolism). This may be life-threatening and cause death. Symptoms may include: new chest pain, trouble breathing or shortness of breath that starts suddenly, leg pain, and swelling of the arms and hands, or legs and feet, a cool or pale arm or leg.‚ÄĘ thrombotic microangiopathy (TMA) including thrombotic thrombocytopenia purpura (TTP) and hemolytic uremic syndrome (HUS). TMA is a condition involving blood clots that can happen while taking VOTRIENT. TMA is accompanied by a decrease in red blood cells and cells that are involved in clotting. TMA may harm organs such as the brain and kidneys.‚ÄĘ bleeding problems. These bleeding problems may be severe and cause death. Symptoms may include: unusual bleeding, bruising, or wounds that do not heal.‚ÄĘ tear in your stomach or intestinal wall (perforation) or an abnormal connection between two parts of your gastrointestinal tract (fistula). Symptoms may include: pain, swelling in your stomach area, vomiting blood, and black sticky stools.‚ÄĘ lung problems. VOTRIENT may cause lung problems that may lead to death. Tell your healthcare provider right away if you get a cough that will not go away or shortness of breath.‚ÄĘ Reversible Posterior Leukoencephalopathy Syndrome (RPLS). RPLS is a condition that can happen while taking VOTRIENT that may cause death. Symptoms may include: headaches, seizures, lack of energy, confusion, high blood pressure, loss of speech, blindness or changes in vision, and problems thinking.‚ÄĘ high blood pressure. High blood pressure can happen with VOTRIENT, including a sudden and severe rise in blood pressure which may be life-threatening. These blood pressure increases usually happen in the first several months of treatment. Your blood pressure should be well controlled before you start taking VOTRIENT. Your healthcare provider should begin checking your blood pressure within 1 week of you starting VOTRIENT and often during treatment to make sure that your blood pressure is well controlled. Have someone call your healthcare provider or get medical help right away for you, if you get symptoms of a severe increase in blood pressure, including: severe chest pain, severe headache, blurred vision, confusion, nausea and vomiting, severe anxiety, shortness of breath, seizures, or you pass out (become unconscious).‚ÄĘ thyroid problems. Your healthcare provider should check you for this during treatment with VOTRIENT.‚ÄĘ protein in your urine. Your healthcare provider will check you for this problem. If there is too much protein in your urine, your healthcare provider may tell you to stop taking VOTRIENT.‚ÄĘ serious infections.Serious infections can happen with VOTRIENT and can cause death. Symptoms of an infection may include: fever, cold symptoms, such as runny nose or sore throat that do not go away, flu symptoms, such as cough, tiredness, and body aches; pain when urinating, cuts, scrapes, or wounds that are red, warm, swollen or painful.‚ÄĘ collapsed lung (pneumothorax). A collapsed lung can happen with VOTRIENT. Air may get trapped in the space between your lung and chest wall. This may cause you to have shortness of breath.¬† Call your healthcare provider right away if you have any of the symptoms uled above.
The most common side effects in people who take VOTRIENT include:
¬†¬†¬†¬†¬†¬†¬†¬†¬†‚Äʬ†¬†diarrhea¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†‚Äʬ†¬†nausea or vomiting
¬†¬†¬†¬†¬†¬†¬†¬†¬†‚Äʬ†¬†change in hair color¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†‚Äʬ†¬†loss of appetite
Other common side effects in people with advanced soft tissue sarcoma who take VOTRIENT include:
¬†¬†¬†¬†¬†¬†¬†¬†¬†‚Äʬ†¬†feeling tired¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†‚Äʬ†¬†headache
¬†¬†¬†¬†¬†¬†¬†¬†¬†‚Äʬ†¬†decreased weight¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†‚Äʬ†¬†taste changes
¬†¬†¬†¬†¬†¬†¬†¬†¬†‚Äʬ†¬†tumor pain¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†‚Äʬ†¬†trouble breathing
¬†¬†¬†¬†¬†¬†¬†¬†¬†‚Äʬ†¬†muscle or bone pain ¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†‚Äʬ†¬†change in skin color
Tell your healthcare provider if you have any side effect that bothers you or that does not go away.
These are not all the possible side effects of VOTRIENT. For more information, ask your healthcare provider or pharmacist. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store VOTRIENT tablets?
Store VOTRIENT at room temperature between 68¬įF and 77¬įF (20¬įC to 25¬įC).
Keep VOTRIENT and all medicines out of the reach of children.
General information about the safe and effective use of VOTRIENT.
Medicines are sometimes prescribed for purposes other than those uled in a Medication Guide. Do not use VOTRIENT for a condition for which it was not prescribed. Do not give VOTRIENT to other people even if they have the same symptoms that you have. It may harm them.
If you would like more information, talk with your healthcare provider. You can ask your pharmacist or healthcare provider for information about VOTRIENT that is written for healthcare professionals. For more information, go to www.VOTRIENT.com or call 1-888-825-5249.
What are the ingredients in VOTRIENT?
Active ingredient: pazopanib.
Inactive ingredients:Tablet core: Magnesium stearate, microcrystalline cellulose, povidone, sodium starch glycolate. Coating: Gray film-coat: Hypromellose, iron oxide black, macrogol/polyethylene glycol 400 (PEG 400), polysorbate 80, titanium dioxide.
GlaxoSmithKline
Research Triangle Park, NC 27709
VOTRIENT is a registered trademark of the GSK group of companies.
©2016 the GSK group of companies. All rights reserved.
VTR:11MG
This Medication Guide has been approved by the U.S. Food and Drug Administration Revised: May 2016
Package Label.principal Display Panel
PRINCIPAL DISPLAY PANEL
NDC 0173-0804-09
Votrient¬ģ
(pazopanib)
Tablets
200 mg
120 Tablets
Rx only
Each tablet contains 217 mg of pazopanib hydrochloride, equivalent to 200 mg of pazopanib free base.
Dispense the Medication Guide, attached or provided separately, to each patient pursuant to Federal Law.
Store at room temperature between 20¬įC and 25¬įC (68¬įF to 77¬įF); excursions permitted to 15¬į to 30¬įC (59¬į to 86¬įF) [See USP Controlled Room Temperature].
Do not use if printed safety seal under cap is broken or missing.
Dosage: See accompanying prescribing information.
GlaxoSmithKline
RTP, NC 27709
Made in Singapore
Rev. 11/14
10000000128445
![]()
DISCLAIMER:
"This tool does not provide medical advice, and is for informational and educational purposes only, and is not a substitute for professional medical advice, treatment or diagnosis. Call your doctor to receive medical advice. If you think you may have a medical emergency, please dial 911."
"Do not rely on openFDA to make decisions regarding medical care. While we make every effort to ensure that data is accurate, you should assume all results are unvalidated. We may limit or otherwise restrict your access to the API in line with our Terms of Service."
"This product uses publicly available data from the U.S. National Library of Medicine (NLM), National Institutes of Health, Department of Health and Human Services; NLM is not responsible for the product and does not endorse or recommend this or any other product."
PillSync may earn a commission via links on our site
