Lenvima (lenvatinib 10 mg) Dailymed
Generic: lenvatinib is used for the treatment of Carcinoma, Renal Cell Thyroid Neoplasms


Go PRO for all pill images
1
LENVIMA is a kinase inhibitor that is indicated:
Differentiated Thyroid Cancer (DTC)
- For the treatment of adult patients with locally recurrent or metastatic, progressive, radioactive iodine-refractory differentiated thyroid cancer (DTC). (
1.1 )
Renal Cell Carcinoma (RCC)
- In combination with pembrolizumab, for the first line treatment of adult patients with advanced renal cell carcinoma (RCC). (
1.2 )- In combination with everolimus, for the treatment of adult patients with advanced renal cell carcinoma (RCC) following one prior anti-angiogenic therapy. (
1.2 )
Hepatocellular Carcinoma (HCC)
- For the first-line treatment of patients with unresectable hepatocellular carcinoma (HCC). (
1.3 )
Endometrial Carcinoma (EC)
- In combination with pembrolizumab, for the treatment of patients with advanced endometrial carcinoma (EC) that is mismatch repair proficient (pMMR), as determined by an FDA-approved test, or not microsatellite instability-high (MSI-H), who have disease progression following prior systemic therapy in any setting and are not candidates for curative surgery or radiation. (
1.4 ,2.1 )
LENVIMA is indicated for the treatment of adult patients with locally recurrent or metastatic, progressive, radioactive iodine-refractory differentiated thyroid cancer (DTC).
LENVIMA, in combination with pembrolizumab, is indicated for the first-line treatment of adult patients with advanced renal cell carcinoma (RCC).
LENVIMA, in combination with everolimus, is indicated for the treatment of adult patients with advanced RCC following one prior anti-angiogenic therapy.
LENVIMA is indicated for the first-line treatment of patients with unresectable hepatocellular carcinoma (HCC).
LENVIMA, in combination with pembrolizumab, is indicated for the treatment of patients with advanced endometrial carcinoma (EC) that is mismatch repair proficient (pMMR), as determined by an FDA-approved test, or not microsatellite instability-high (MSI-H), who have disease progression following prior systemic therapy in any setting and are not candidates for curative surgery or radiation [see Dosage and Administration ( 2.1 )].
Dosage & Administration Section
Single Agent Therapy:
- DTC: The recommended dosage is 24 mg orally once daily. (
2.3 )- HCC: The recommended dosage is based on actual body weight: 12 mg orally once daily for patients greater than or equal to 60 kg or 8 mg orally once daily for patients less than 60 kg. (
2.5 )
Combination Therapy:
- EC: The recommended dosage is 20 mg orally once daily in combination with pembrolizumab 200 mg administered as an intravenous infusion over 30 minutes every 3 weeks. (
2.6 )- RCC: The recommended dosage is:
- 20 mg orally once daily with pembrolizumab 200 mg administered as an intravenous infusion over 30 minutes every 3 weeks. (
2.4 )- 18 mg orally once daily with everolimus 5 mg orally once daily. (
2.4 )
Modify the recommended daily dose for certain patients with renal or hepatic impairment. (2.8 ,2.9 )
For the pMMR/not MSI-H advanced endometrial carcinoma indication, select patients for treatment with LENVIMA in combination with pembrolizumab based on MSI or MMR status in tumor specimens [see Clinical Studies ( 14.4 )].
Information on FDA-approved tests for patient selection is available at: http://www.fda.gov/CompanionDiagnostics.
An FDA-approved test for the selection of patients who are not MSI-H is not currently available.
- Reduce the dose for certain patients with renal or hepatic impairment [see Dosage and Administration ( 2.8 , 2.9 )].
- Take LENVIMA once daily, with or without food, at the same time each day [see Clinical Pharmacology ( 12.3 )]. If a dose is missed and cannot be taken within 12 hours, skip that dose and take the next dose at the usual time of administration.
The recommended dosage of LENVIMA is 24 mg orally once daily until disease progression or until unacceptable toxicity.
2.
F irst- L ine Treatment of Patients with Advanced RCC
The recommended dosage of LENVIMA is 20 mg orally once daily in combination with pembrolizumab 200 mg administered as an intravenous infusion over 30 minutes every 3 weeks until disease progression or until unacceptable toxicity or up to 2 years. After completing 2 years of combination therapy, LENVIMA may be administered as a single agent until disease progression or until unacceptable toxicity.
Refer to the pembrolizumab prescribing information for other pembrolizumab dosing information.
Previously Treated RCC
The recommended dosage of LENVIMA is 18 mg in combination with 5 mg everolimus orally once daily until disease progression or until unacceptable toxicity.
Refer to the everolimus prescribing information for recommended everolimus dosing information.
The recommended dosage of LENVIMA is based on actual body weight:
- 12 mg for patients greater than or equal to 60 kg or
- 8 mg for patients less than 60 kg.
Take LENVIMA orally once daily until disease progression or until unacceptable toxicity.
The recommended dosage of LENVIMA is 20 mg orally once daily, in combination with pembrolizumab 200 mg administered as an intravenous infusion over 30 minutes every 3 weeks, until unacceptable toxicity or disease progression.
Refer to the pembrolizumab prescribing information for other pembrolizumab dosing information.
Recommendations for LENVIMA dose interruption, reduction and discontinuation for adverse reactions are uled in Table 1. Table 2 uls the recommended dosage reductions of LENVIMA for adverse reactions.
Table 1 : Recommended Dos ag e Modifications for LENVIMA for Adverse Reactions Adverse Reaction Severity a Dos ag e Modifications for LENVIMA Hypertension [see Warnings and Precautions ( 5.1 )] Grade 3
- Withhold for Grade 3 that persists despite optimal antihypertensive therapy.
- Resume at reduced dose when hypertension is controlled at less than or equal to Grade 2.
Grade 4
- Permanently discontinue.
Cardiac Dysfunction [see Warnings and Precautions ( 5.2 )] Grade 3
- Withhold until improves to Grade 0 to 1 or baseline.
- Resume at a reduced dose or discontinue depending on the severity and persistence of adverse reaction.
Grade 4
- Permanently discontinue.
Arterial Thromboembolic Event [see Warnings and Precautions ( 5.3 )] Any Grade
- Permanently discontinue.
Hepatotoxicity [see Warnings and Precautions ( 5.4 )] Grade 3 or 4
- Withhold until improves to Grade 0 to 1 or baseline.
- Either resume at a reduced dose or discontinue depending on severity and persistence of hepatotoxicity.
- Permanently discontinue for hepatic failure.
Renal Failure or Impairment [see Warnings and Precautions ( 5.5 )] Grade 3 or 4
- Withhold until improves to Grade 0 to 1 or baseline.
- Resume at a reduced dose or discontinue depending on severity and persistence of renal impairment.
Proteinuria [see Warnings and Precautions ( 5.6 )] 2 g or greater proteinuria in 24 hours
- Withhold until less than or equal to 2 grams of proteinuria per 24 hours.
- Resume at a reduced dose.
- Permanently discontinue for nephrotic syndrome.
Gastrointestinal Perforation [see Warnings and Precautions ( 5.8 )] Any Grade
- Permanently discontinue.
Fistula Formation [see Warnings and Precautions ( 5.8 )] Grade 3 or 4
- Permanently discontinue.
QT Prolongation [see Warnings and Precautions ( 5.9 )] Greater than 500 ms or greater than 60 ms increase from baseline
- Withhold until improves to less than or equal to 480 ms or baseline.
- Resume at a reduced dose.
Reversible Posterior Leukoencephalopathy Syndrome [see Warnings and Precautions ( 5.11 )] Any Grade
- Withhold until fully resolved.
- Resume at a reduced dose or discontinue depending on severity and persistence of neurologic symptoms.
Other Adverse Reactions [see Warnings and Precautions ( 5.7 , 5.10 , 5.12 )] Persistent or intolerable Grade 2 or 3 adverse reactionGrade 4 laboratory abnormality
- Withhold until improves to Grade 0 to 1 or baseline.
- Resume at reduced dose.
Grade 4 adverse reaction
- Permanently discontinue.
a. a National Cancer Institute Common Terminology Criteria for Adverse Events, version 4.0.
Table 2: Recommended Dosage Reductions of LENVIMA for Adverse Reactions Indication First Dosage Reduction To Second Dosage Reduction To Third Dosage Reduction To DTC 20 mg once daily 14 mg once daily 10 mgonce daily RCC 14 mg once daily 10 mgonce daily 8 mgonce daily Endometrial Carcinoma 14 mg once daily 10 mgonce daily 8 mgonce daily HCC
- Actual weight 60 kg or greater
8 mg once daily 4 mg once daily 4 mg every other day
- Actual weight less than 60 kg
4 mg once daily 4 mg every other day Discontinue
Recommended Dose Modifications for Adverse Reactions for LENVIMA in Combination with Pembrolizumab
When administering LENVIMA in combination with pembrolizumab, modify the dosage of one or both drugs as appropriate. Withhold, dose reduce, or discontinue LENVIMA as shown in Table 1. Refer to pembrolizumab prescribing information for additional dose modification information.
Recommended Dose Modifications for Adverse Reactions for LENVIMA in Combination with Everolimus
When administering LENVIMA in combination with everolimus, withhold or reduce the LENVIMA dose first and then the everolimus dose for adverse reactions of both LENVIMA and everolimus. Refer to the everolimus prescribing information for additional dose modification information.
The recommended dosage of LENVIMA for patients with DTC, RCC, or endometrial carcinoma and severe renal impairment (creatinine clearance less than 30 mL/min calculated by Cockcroft-Gault equation using actual body weight) is [s ee Warnings and Precautions ( 5.5 ), Use in Specific Populations ( 8.6 )]:
- Differentiated thyroid cancer: 14 mg orally once daily
- Renal cell carcinoma: 10 mg orally once daily
- Endometrial carcinoma: 10 mg orally once daily
The recommended dosage of LENVIMA for patients with DTC, RCC, or endometrial carcinoma and severe hepatic impairment (Child-Pugh C) is [s ee Warnings and Precautions ( 5.4 ), Use in Specific Populations ( 8.7 ) ]:
- Differentiated thyroid cancer: 14 mg taken orally once daily
- Renal cell carcinoma: 10 mg taken orally once daily
- Endometrial carcinoma: 10 mg orally once daily
Administration
Oral: Capsule or Suspension
Capsule
- Swallow LENVIMA capsules whole at the same time each day with or without food [see Clinical Pharmacology ( 12.3 )]. Do not crush or chew the LENVIMA capsules.
Suspension
- Prepare [see Preparation below ] oral suspension with water or apple juice and administer at the same time each day with or without food [see Clinical Pharmacology ( 12.3 )].
Feeding Tube Administration
Suspension
- Prepare [see Preparation below ] suspension for feeding tube administration with water and administer at the same time each day with or without food [see Clinical Pharmacology ( 12.3 )].
Preparation of Suspension
- Place the required number of capsules, up to a maximum of 5, in a small container (approximately 20 mL capacity) or syringe (20 mL). Do not break or crush capsules.
- Add 3 mL of liquid to the container or syringe. Wait 10 minutes for the capsule shell (outer surface) to disintegrate, then stir or shake the mixture for 3 minutes until capsules are fully disintegrated and administer the entire contents.
- Next, add an additional 2 mL of liquid to the container or syringe using a second syringe or dropper, swirl or shake and administer. Repeat this step at least once and until there is no visible residue to ensure all of the medication is taken.
- If 6 capsules are required for a dose, follow these instructions using 3 capsules at a time.
If LENVIMA suspension is not used at the time of preparation, LENVIMA suspension may be stored in a refrigerator at 36ºF to 46ºF (2ºC to 8ºC) for a maximum of 24 hours in a covered container. If not administered within 24 hours, the suspension should be discarded.
Note: Compatibility has been confirmed for polypropylene syringes and for feeding tubes of at least 5 French diameter (polyvinyl chloride or polyurethane tube) and at least 6 French diameter (silicone tube).
Dosage Forms & Strengths Section
Capsules:
- 4 mg: yellowish-red body and yellowish-red cap, marked in black ink with “Є” on cap and “LENV 4 mg” on body.
- 10 mg: yellow body and yellowish-red cap, marked in black ink with “Є” on cap and “LENV 10 mg” on body.
Capsules: 4 mg and 10 mg. (3 )
Contraindications Section
None.
None. (4 )
Warnings And Precautions Section
- Hypertension: Control blood pressure prior to treatment and monitor during treatment. Withhold for Grade 3 hypertension despite optimal antihypertensive therapy. Discontinue for Grade 4 hypertension. (
2.7 ,5.1 )- Cardiac Dysfunction: Monitor for clinical symptoms or signs of cardiac dysfunction. Withhold or discontinue for Grade 3 cardiac dysfunction. Discontinue for Grade 4 cardiac dysfunction. (
2.7 ,5.2 )- Arterial Thromboembolic Events: Discontinue following an arterial thromboembolic event. (
2.7 ,5.3 )- Hepatotoxicity: Monitor liver function prior to treatment and periodically during treatment. Withhold or discontinue for Grade 3 or 4 hepatotoxicity. Discontinue for hepatic failure. (
2.7 ,5.4 )- Renal Failure or Impairment: Withhold or discontinue for Grade 3 or 4 renal failure or impairment. (
2.7 ,5.5 )- Proteinuria: Monitor for proteinuria prior to treatment and periodically during treatment. Withhold for 2 or more grams of proteinuria per 24 hours. Discontinue for nephrotic syndrome. (
2.7 ,5.6 )- Diarrhea: May be severe and recurrent. Promptly initiate management for severe diarrhea. Withhold or discontinue based on severity. (
2.7 ,5.7 )- Fistula Formation and Gastrointestinal Perforation: Discontinue in patients who develop Grade 3 or 4 fistula or any Grade gastrointestinal perforation. (
2.7 ,5.8 )- QT Interval Prolongation: Monitor and correct electrolyte abnormalities. Withhold for QT interval greater than 500 ms or for 60 ms or greater increase in baseline QT interval. (
2.7 ,5.9 )- Hypocalcemia: Monitor blood calcium levels at least monthly and replace calcium as necessary. Withhold or discontinue based on severity. (
2.7 ,5.10 )- Reversible Posterior Leukoencephalopathy Syndrome (RPLS): Withhold for RPLS until fully resolved or discontinue. (
2.7 ,5.11 )- Hemorrha gic Events: Withhold or discontinue based on severity. (
2.7 ,5.12 )- Impairment of Thyroid Stimulating Hormone Suppression /Thyroid Dysfunction: Monitor thyroid function prior to treatment and monthly during treatment. (
5.13 )- Impaired Wound Healing: Withhold LENVIMA for at least 1 week before elective surgery. Do not administer for at least 2 weeks following major surgery and until adequate wound healing. The safety of resumption of LENVIMA after resolution of wound healing complications has not been established. (
5.14 )- Osteonecrosis of the Jaw: Consider preventive dentistry prior to treatment with LENVIMA. Avoid invasive dental procedures, if possible, particularly in patients at higher risk. (
5.15 )- Embryo -F etal T oxicity: Can cause fetal harm. Advise females of reproductive potential of the potential risk to a fetus and to use effective contraception. (
5.16 ,8.1 ,8.3 )
Hypertension occurred in 73% of patients in SELECT (DTC) receiving LENVIMA 24 mg orally once daily and in 45% of patients in REFLECT (HCC) receiving LENVIMA 8 mg or 12 mg orally once daily. The median time to onset of new or worsening hypertension was 16 days in SELECT and 26 days in REFLECT. Grade 3 hypertension occurred in 44% of patients in SELECT and in 24% in REFLECT. Grade 4 hypertension occurred <1% in SELECT and Grade 4 hypertension was not reported in REFLECT.
In patients receiving LENVIMA 18 mg orally once daily with everolimus in Study 205 (RCC), hypertension was reported in 42% of patients and the median time to onset of new or worsening hypertension was 35 days. Grade 3 hypertension occurred in 13% of patients. Systolic blood pressure ≥160 mmHg occurred in 29% of patients and diastolic blood pressure ≥100 mmHg occurred in 21% [see Adverse Reactions ( 6.1 )].
Serious complications of poorly controlled hypertension have been reported.
Control blood pressure prior to initiating LENVIMA. Monitor blood pressure after 1 week, then every 2 weeks for the first 2 months, and then at least monthly thereafter during treatment. Withhold and resume at a reduced dose when hypertension is controlled or permanently discontinue LENVIMA based on severity [ see Dosage and Administration ( 2.7 ) ].
Serious and fatal cardiac dysfunction can occur with LENVIMA. Across clinical trials in 799 patients with DTC, RCC or HCC, Grade 3 or higher cardiac dysfunction (including cardiomyopathy, left or right ventricular dysfunction, congestive heart failure, cardiac failure, ventricular hypokinesia, or decrease in left or right ventricular ejection fraction of more than 20% from baseline) occurred in 3% of LENVIMA-treated patients.
Monitor patients for clinical symptoms or signs of cardiac dysfunction. Withhold and resume at a reduced dose upon recovery or permanently discontinue LENVIMA based on severity [ see Dosage and Administration ( 2.7 ) ].
Among patients receiving LENVIMA or LENVIMA with everolimus, arterial thromboembolic events of any severity occurred in 2% of patients in Study 205 (RCC), 2% of patients in REFLECT (HCC) and 5% of patients in SELECT (DTC). Grade 3 to 5 arterial thromboembolic events ranged from 2% to 3% across all clinical trials [see Adverse Reactions ( 6.1 )].
Among patients receiving LENVIMA with pembrolizumab, arterial thrombotic events of any severity occurred in 5% of patients in CLEAR, including myocardial infarction (3.4%) and cerebrovascular accident (2.3%).
Permanently discontinue LENVIMA following an arterial thrombotic event [ see Dosage and Administration ( 2.7 ) ]. The safety of resuming LENVIMA after an arterial thromboembolic event has not been established and LENVIMA has not been studied in patients who have had an arterial thromboembolic event within the previous 6 months.
Across clinical studies enrolling 1327 LENVIMA-treated patients with malignancies other than HCC, serious hepatic adverse reactions occurred in 1.4% of patients. Fatal events, including hepatic failure, acute hepatitis and hepatorenal syndrome, occurred in 0.5% of patients.
In REFLECT (HCC), hepatic encephalopathy (including hepatic encephalopathy, encephalopathy, metabolic encephalopathy, and hepatic coma) occurred in 8% of LENVIMA-treated patients and 3% of sorafenib-treated patients. Grade 3 to 5 hepatic encephalopathy occurred in 5% of LENVIMA-treated patients and 2% of sorafenib-treated patients. Grade 3 to 5 hepatic failure occurred in 3% of LENVIMA-treated patients and 3% of sorafenib-treated patients. Two percent of patients discontinued LENVIMA and 0.2% discontinued sorafenib due to hepatic encephalopathy and 1% of patients discontinued LENVIMA or sorafenib due to hepatic failure [see Adverse Reactions ( 6.1 )] .
Monitor liver function prior to initiating LENVIMA, then every 2 weeks for the first 2 months, and at least monthly thereafter during treatment. Monitor patients with HCC closely for signs of hepatic failure, including hepatic encephalopathy. Withhold and resume at a reduced dose upon recovery or permanently discontinue LENVIMA based on severity [ see Dosage and Administration ( 2.7 ) ].
Serious including fatal renal failure or impairment can occur with LENVIMA. Renal impairment occurred in 14% of patients receiving LENVIMA in SELECT (DTC) and in 7% of patients receiving LENVIMA in REFLECT (HCC). Grade 3 to 5 renal failure or impairment occurred in 3% (DTC) and 2% (HCC) of patients, including 1 fatality in each study.
In Study 205 (RCC), renal impairment or renal failure occurred in 18% of patients receiving LENVIMA with everolimus, including Grade 3 in 10% of patients [see Adverse Reactions ( 6.1 )].
Initiate prompt management of diarrhea or dehydration/hypovolemia. Withhold and resume at a reduced dose upon recovery or permanently discontinue LENVIMA for renal failure or impairment based on severity [ see Dosage and Administration ( 2.7 ) ].
Proteinuria occurred in 34% of LENVIMA-treated patients in SELECT (DTC) and in 26% of LENVIMA-treated patients in REFLECT (HCC). Grade 3 proteinuria occurred in 11% and 6% in SELECT and REFLECT, respectively. In Study 205 (RCC), proteinuria occurred in 31% of patients receiving LENVIMA with everolimus and 14% of patients receiving everolimus. Grade 3 proteinuria occurred in 8% of patients receiving LENVIMA with everolimus compared to 2% of patients receiving everolimus [see Adverse Reactions ( 6.1 )].
Monitor for proteinuria prior to initiating LENVIMA and periodically during treatment. If urine dipstick proteinuria greater than or equal to 2+ is detected, obtain a 24-hour urine protein. Withhold and resume at a reduced dose upon recovery or permanently discontinue LENVIMA based on severity [ see Dosage and Administration ( 2.7 ) ].
Of the 737 patients treated with LENVIMA in SELECT (DTC) and REFLECT (HCC), diarrhea occurred in 49% of patients, including Grade 3 in 6%.
In Study 205 (RCC), diarrhea occurred in 81% of patients receiving LENVIMA with everolimus, including Grade 3 in 19%. Diarrhea was the most frequent cause of dose interruption/reduction and diarrhea recurred despite dose reduction [see Adverse Reactions ( 6.1 )].
Promptly initiate management of diarrhea. Withhold and resume at a reduced dose upon recovery or permanently discontinue LENVIMA based on severity [ see Dosage and Administration ( 2.7 ) ].
Of 799 patients treated with LENVIMA or LENVIMA with everolimus in SELECT (DTC), Study 205 (RCC) and REFLECT (HCC), fistula or gastrointestinal perforation occurred in 2%.
Permanently discontinue LENVIMA in patients who develop gastrointestinal perforation of any severity or Grade 3 or 4 fistula [ see Dosage and Administration ( 2.7 ) ].
In SELECT (DTC), QT/QTc interval prolongation occurred in 9% of LENVIMA-treated patients and QT interval prolongation of >500 ms occurred in 2%. In Study 205 (RCC), QTc interval increases of >60 ms occurred in 11% of patients receiving LENVIMA with everolimus and QTc interval >500 ms occurred in 6%. In REFLECT (HCC), QTc interval increases of >60 ms occurred in 8% of LENVIMA-treated patients and QTc interval >500 ms occurred in 2%.
Monitor and correct electrolyte abnormalities at baseline and periodically during treatment. Monitor electrocardiograms in patients with congenital long QT syndrome, congestive heart failure, bradyarrhythmias, or those who are taking drugs known to prolong the QT interval, including Class Ia and III antiarrhythmics. Withhold and resume at reduced dose of LENVIMA upon recovery based on severity [see Dosage and Administration ( 2.7 )].
In SELECT (DTC), Grade 3 to 4 hypocalcemia occurred in 9% of patients receiving LENVIMA. In 65% of cases, hypocalcemia improved or resolved following calcium supplementation, with or without dose interruption or dose reduction.
In Study 205 (RCC), Grade 3 to 4 hypocalcemia occurred in 6% of patients treated with LENVIMA with everolimus. In REFLECT (HCC), Grade 3 hypocalcemia occurred in 0.8% of LENVIMA-treated patients [see Adverse Reactions ( 6.1 )].
Monitor blood calcium levels at least monthly and replace calcium as necessary during treatment. Withhold and resume at reduced dose upon recovery or permanently discontinue LENVIMA depending on severity [ see Dosage and Administration ( 2.7 ) ].
Across clinical studies of 1823 patients who received LENVIMA as a single agent [see Adverse Reaction s ( 6.1 )], reversible posterior leukoencephalopathy syndrome (RPLS) occurred in 0.3%.
Confirm the diagnosis of RPLS with magnetic resonance imaging. Withhold and resume at a reduced dose upon recovery or permanently discontinue LENVIMA depending on severity and persistence of neurologic symptoms [see Dosage and Administration ( 2.7 )].
Serious including fatal hemorrhagic events can occur with LENVIMA. Across SELECT (DTC), Study 205 (RCC) and REFLECT (HCC), hemorrhagic events of any grade occurred in 29% of the 799 patients treated with LENVIMA as a single agent or in combination with everolimus. The most frequently reported hemorrhagic events (all grades and occurring in at least 5% of patients) were epistaxis and hematuria.
In SELECT, Grade 3 to 5 hemorrhage occurred in 2% of patients receiving LENVIMA, including 1 fatal intracranial hemorrhage among 16 patients who received LENVIMA and had CNS metastases at baseline. In Study 205, Grade 3 to 5 hemorrhage occurred in 8% of patients receiving LENVIMA with everolimus, including 1 fatal cerebral hemorrhage. In REFLECT, Grade 3 to 5 hemorrhage occurred in 5% of patients receiving LENVIMA, including 7 fatal hemorrhagic events [see Adverse Reactions ( 6.1 )].
Serious tumor related bleeds, including fatal hemorrhagic events, occurred in patients treated with LENVIMA in clinical trials and in the post-marketing setting. In post-marketing surveillance, serious and fatal carotid artery hemorrhages were seen more frequently in patients with anaplastic thyroid carcinoma (ATC) than in other tumor types. The safety and effectiveness of LENVIMA in patients with ATC have not been demonstrated in clinical trials.
Consider the risk of severe or fatal hemorrhage associated with tumor invasion or infiltration of major blood vessels (e.g. carotid artery). Withhold and resume at reduced dose upon recovery or permanently discontinue LENVIMA based on the severity [see Dosage and Administration ( 2.7 ) ].
LENVIMA impairs exogenous thyroid suppression. In SELECT (DTC), 88% of all patients had a baseline thyroid stimulating hormone (TSH) level ≤0.5 mU/L. In those patients with a normal TSH at baseline, elevation of TSH level >0.5 mU/L was observed post baseline in 57% of LENVIMA-treated patients.
Grade 1 or 2 hypothyroidism occurred in 24% of patients receiving LENVIMA with everolimus in Study 205 (RCC) and in 21% of patients receiving LENVIMA in REFLECT (HCC). In those patients with a normal or low TSH at baseline, an elevation of TSH was observed post baseline in 70% of patients receiving LENVIMA in REFLECT and 60% of patients receiving LENVIMA with everolimus in Study 205 [see Adverse Reactions ( 6.1 )].
Monitor thyroid function prior to initiating LENVIMA and at least monthly during treatment. Treat hypothyroidism according to standard medical practice.
Impaired wound healing has been reported in patients who received LENVIMA [see Adverse Reactions ( 6.2 )].
Withhold LENVIMA for at least 1 week prior to elective surgery. Do not administer for at least 2 weeks following major surgery and until adequate wound healing. The safety of resumption of LENVIMA after resolution of wound healing complications has not been established.
Osteonecrosis of the Jaw (ONJ) has been reported in patients receiving LENVIMA [see Adverse Reactions ( 6.1 )]. Concomitant exposure to other risk factors, such as bisphosphonates, denosumab, dental disease or invasive dental procedures, may increase the risk of ONJ.
Perform an oral examination prior to treatment with LENVIMA and periodically during LENVIMA treatment. Advise patients regarding good oral hygiene practices. Avoid invasive dental procedures, if possible, while on LENVIMA treatment, particularly in patients at higher risk. Withhold LENVIMA for at least 1 week prior to scheduled dental surgery or invasive dental procedures, if possible. For patients requiring invasive dental procedures, discontinuation of bisphosphonate treatment may reduce the risk of ONJ. Withhold LENVIMA if ONJ develops and restart based on clinical judgement of adequate resolution.
Based on its mechanism of action and data from animal reproduction studies, LENVIMA can cause fetal harm when administered to a pregnant woman. In animal reproduction studies, oral administration of lenvatinib during organogenesis at doses below the recommended clinical doses resulted in embryotoxicity, fetotoxicity, and teratogenicity in rats and rabbits.
Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment with LENVIMA and for 30 days after the last dose [see Use in Specific Populations ( 8.1 , 8.3 ) ].
Adverse Reactions Section
The following adverse reactions are discussed elsewhere in the labeling:
- Hypertension [see Warnings and Precautions ( 5.1 )]
- Cardiac Dysfunction [see Warnings and Precautions ( 5.2 )]
- Arterial Thromboembolic Events [see Warnings and Precautions ( 5.3 )]
- Hepatotoxicity [see Warnings and Precautions ( 5.4 )]
- Renal Failure and Impairment [see Warnings and Precautions ( 5.5 )]
- Proteinuria [see Warnings and Precautions ( 5.6 )]
- Diarrhea [see Warnings and Precautions ( 5.7 )]
- Fistula Formation and Gastrointestinal Perforation [see Warnings and Precautions ( 5.8 )]
- QT Interval Prolongation [see Warnings and Precautions ( 5.9 )]
- Hypocalcemia [see Warnings and Precautions ( 5.10 )]
- Reversible Posterior Leukoencephalopathy Syndrome [see Warnings and Precautions ( 5.11 )]
- Hemorrhagic Events [see Warnings and Precautions ( 5.12 )]
- Impairment of Thyroid Stimulating Hormone Suppression/Thyroid Dysfunction [see Warnings and Precautions ( 5.13 )]
- Impaired Wound Healing [see Warnings and Precautions ( 5.14 )]
- Osteonecrosis of the Jaw (ONJ) [see Warnings and Precautions ( 5.15 )]
- In DTC, the most common adverse reactions (incidence ≥30%) for LENVIMA are hypertension, fatigue, diarrhea, arthralgia/myalgia, decreased appetite, decreased weight, nausea, stomatitis, headache, vomiting, proteinuria, palmar-plantar erythrodysesthesia syndrome, abdominal pain, and dysphonia. (
6.1 )- In RCC:
- The most common adverse reactions (incidence ≥20%) for LENVIMA and pembrolizumab are fatigue, diarrhea, musculoskeletal pain, hypothyroidism, hypertension, stomatitis, decreased appetite, rash, nausea, decreased weight, dysphonia, proteinuria, palmar-plantar erythrodysesthesia syndrome, abdominal pain, hemorrhagic events, vomiting, constipation, hepatotoxicity, headache, and acute kidney injury. (
6.1 )- The most common adverse reactions (incidence ≥30%) for LENVIMA and everolimus are diarrhea, fatigue, arthralgia/myalgia, decreased appetite, vomiting, nausea, stomatitis/oral inflammation, hypertension, peripheral edema, cough, abdominal pain, dyspnea, rash, decreased weight, hemorrhagic events, and proteinuria. (
6.1 )
- In HCC, the most common adverse reactions (incidence ≥20%) for LENVIMA are hypertension, fatigue, diarrhea, decreased appetite, arthralgia/myalgia, decreased weight, abdominal pain, palmar-plantar erythrodysesthesia syndrome, proteinuria, dysphonia, hemorrhagic events, hypothyroidism, and nausea. (
6.1 )- In EC, the most common adverse reactions (incidence ≥20%) for LENVIMA and pembrolizumab are hypothyroidism, hypertension, fatigue, diarrhea, musculoskeletal disorders, nausea, decreased appetite, vomiting, stomatitis, decreased weight, abdominal pain, urinary tract infection, proteinuria, constipation, headache, hemorrhagic events, palmar-plantar erythrodysesthesia, dysphonia, and rash. (
6.1 )
To report SUSPECTED ADVERSE REACTIONS, contact Eisai Inc. at 1-877-873-4724 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
CLINICAL TRIALS EXPERIENCE SECTION
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The data in the Warnings and Precautions reflect exposure to LENVIMA as a single agent in 261 patients with DTC (SELECT) and 476 patients with HCC (REFLECT), LENVIMA with pembrolizumab in 406 patients with endometrial carcinoma (Study 309), LENVIMA with everolimus in 62 patients with RCC (Study 205), and LENVIMA with pembrolizumab in 352 patients with RCC (CLEAR). Safety data obtained in 1823 patients with advanced solid tumors who received LENVIMA as a single agent across multiple clinical studies was used to further characterize the risks of serious adverse reactions. Among the 1823 patients who received LENVIMA as a single agent, the median age was 61 years (20 to 89 years), the dose range was 0.2 mg to 32 mg daily, and the median duration of exposure was 5.6 months.
The data below reflect exposure to LENVIMA in 1557 patients enrolled in randomized, active-controlled trials (REFLECT; Study 205; CLEAR; Study 309), and a randomized, placebo-controlled trial (SELECT). The median duration of exposure to LENVIMA across these five studies ranged from 6 to 16 months. The demographic and exposure data for each clinical trial population are described in the subsections below.
Differentiated Thyroid Cancer
The safety of LENVIMA was evaluated in SELECT, in which patients with radioactive iodine-refractory differentiated thyroid cancer were randomized (2:1) to LENVIMA (n=261) or placebo (n=131) [ see Clinical Studies ( 14.1 ) ]. The median treatment duration was 16.1 months for LENVIMA. Among 261 patients who received LENVIMA, median age was 64 years, 52% were females, 80% were White, 18% were Asian, and 2% were Black; and 4% were Hispanic/Latino.
The most common adverse reactions observed in LENVIMA-treated patients (≥30%) were, in order of decreasing frequency, hypertension, fatigue, diarrhea, arthralgia/myalgia, decreased appetite, decreased weight, nausea, stomatitis, headache, vomiting, proteinuria, palmar-plantar erythrodysesthesia (PPE) syndrome, abdominal pain, and dysphonia. The most common serious adverse reactions (at least 2%) were pneumonia (4%), hypertension (3%), and dehydration (3%).
Adverse reactions led to dose reductions in 68% of patients receiving LENVIMA; 18% of patients discontinued LENVIMA for adverse reactions. The most common adverse reactions (at least 10%) resulting in dose reductions of LENVIMA were hypertension (13%), proteinuria (11%), decreased appetite (10%), and diarrhea (10%); the most common adverse reactions (at least 1%) resulting in discontinuation of LENVIMA were hypertension (1%) and asthenia (1%).
Table 3 presents adverse reactions occurring at a higher rate in LENVIMA-treated patients than patients receiving placebo in the double-blind phase of the study.
Table 3 : Adverse Reactions Occurring in Patients with a Between-Group Difference of ≥5% in All Grades or ≥2% in Grades 3 and 4 in SELECT (DTC) Adverse Reaction LENVIMA 24 mg N=261 Placebo N=131 All Grades (%) Grades 3-4 (%) All Grades (%) Grades 3- 4 (%) Vascular Hypertensiona 73 44 16 4 Hypotension 9 2 2 0 Gastrointestinal Diarrhea 67 9 17 0 Nausea 47 2 25 1 Stomatitisb 41 5 8 0 Vomiting 36 2 15 0 Abdominal painc 31 2 11 1 Constipation 29 0.4 15 1 Oral paind 25 1 2 0 Dry mouth 17 0.4 8 0 Dyspepsia 13 0.4 4 0 General Fatiguee 67 11 35 4 Edema peripheral 21 0.4 8 0 Musculoskeletal and Connective Tissue Arthralgia/Myalgiaf 62 5 28 3 Metabolism and Nutrition Decreased appetite 54 7 18 1 Decreased weight 51 13 15 1 Dehydration 9 2 2 1 Nervous System Headache 38 3 11 1 Dysgeusia 18 0 3 0 Dizziness 15 0.4 9 0 Renal and Urinary Proteinuria 34 11 3 0 Skin and Subcutaneous Tissue Palmar-plantar erythrodysesthesia 32 3 1 0 Rashg 21 0.4 3 0 Alopecia 12 0 5 0 Hyperkeratosis 7 0 2 0 Respiratory, Thoracic and Mediastinal Dysphonia 31 1 5 0 Cough 24 0 18 0 Epistaxis 12 0 1 0 Psychiatric Insomnia 12 0 3 0 Infections Urinary tract infection 11 1 5 0 Dental and oral infectionsh 10 1 1 0 Cardiac Electrocardiogram QT prolonged 9 2 2 0 a. Includes hypertension, hypertensive crisis, increased blood pressure diastolic, and increased blood pressure b. Includes aphthous stomatitis, stomatitis, glossitis, mouth ulceration, and mucosal inflammationc. Includes abdominal discomfort, abdominal pain, lower abdominal pain, upper abdominal pain, abdominal tenderness, epigastric discomfort, and gastrointestinal pain d. Includes oral pain, glossodynia, and oropharyngeal paine. Includes asthenia, fatigue, and malaisef. Includes musculoskeletal pain, back pain, pain in extremity, arthralgia, and myalgia g. Includes macular rash, maculo-papular rash, generalized rash, and rash h. Includes gingivitis, oral infection, parotitis, pericoronitis, periodontitis, sialoadenitis, tooth abscess, and tooth infection
Clinically important adverse reactions occurring more frequently in LENVIMA-treated patients than patients receiving placebo, but with an incidence of <5% were pulmonary embolism (3%, including fatal reports vs 2%, respectively) and osteonecrosis of the jaw (0.4% vs 0%, respectively).
Laboratory abnormalities with a difference of ≥2% in Grade 3 – 4 events and at a higher incidence in the LENVIMA arm are presented in Table 4.
Table 4: Laboratory Abnormalities with a Difference of ≥2% in Grade 3 - 4 Events and at a Higher Incidence in the LENVIMA Arm a, b in SELECT (DTC) Laboratory A bnormality LENVIMA 24 mg Placebo Grades 3-4 (%) Grades 3-4 (%) Chemistry Hypocalcemia 9 2 Hypokalemia 6 1 Increased aspartate aminotransferase (AST) 5 0 Increased alanine aminotransferase (ALT) 4 0 Increased lipase 4 1 Increased creatinine 3 0 Hematology Thrombocytopenia 2 0 a. With at least 1 grade increase from baseline b. Laboratory Abnormality percentage is based on the number of patients who had both baseline and at least one post baseline laboratory measurement for each parameter. LENVIMA (n = 253 to 258), Placebo (n = 129 to 131)
The following laboratory abnormalities (all Grades) occurred in >5% of LENVIMA-treated patients and at a rate that was two-fold or higher than in patients who received placebo: hypoalbuminemia, increased alkaline phosphatase, hypomagnesemia, hypoglycemia, hyperbilirubinemia, hypercalcemia, hypercholesterolemia, increased serum amylase, and hyperkalemia.
First-Line Treatment of Renal Cell Carcinoma in Combination withP embrolizumab (CLEAR)
The safety of LENVIMA in combination with pembrolizumab was investigated in CLEAR [see Clinical Studies ( 14.2 )]. Patients received LENVIMA 20 mg orally once daily in combination with pembrolizumab 200 mg intravenously every 3 weeks (n=352), or LENVIMA 18 mg orally once daily in combination with everolimus 5 mg orally once daily (n=355), or sunitinib 50 mg orally once daily for 4 weeks then off treatment for 2 weeks (n=340). The median duration of exposure to the combination therapy of LENVIMA and pembrolizumab was 17 months (range: 0.1 to 39).
Fatal adverse reactions occurred in 4.3% of patients receiving LENVIMA in combination with pembrolizumab, including cardio-respiratory arrest (0.9%), sepsis (0.9%), and one case (0.3%) each of arrhythmia, autoimmune hepatitis, dyspnea, hypertensive crisis, increased blood creatinine, multiple organ dysfunction syndrome, myasthenic syndrome, myocarditis, nephritis, pneumonitis, ruptured aneurysm and subarachnoid hemorrhage.
Serious adverse reactions occurred in 51% of patients receiving LENVIMA and pembrolizumab. Serious adverse reactions in ≥2% of patients were hemorrhagic events (5%), diarrhea (4%), hypertension (3%), myocardial infarction (3%), pneumonitis (3%), vomiting (3%), acute kidney injury (2%), adrenal insufficiency (2%), dyspnea (2%), and pneumonia (2%).
Permanent discontinuation of LENVIMA, pembrolizumab, or both due to an adverse reaction occurred in 37% of patients; 26% LENVIMA only, 29% pembrolizumab only, and 13% both drugs. The most common adverse reactions (≥2%) leading to permanent discontinuation of LENVIMA, pembrolizumab, or both were pneumonitis (3%), myocardial infarction (3%), hepatotoxicity (3%), acute kidney injury (3%), rash (3%), and diarrhea (2%).
Dose interruptions of LENVIMA, pembrolizumab, or both due to an adverse reaction occurred in 78% of patients receiving LENVIMA in combination with pembrolizumab. LENVIMA was interrupted in 73% of patients and both drugs were interrupted in 39% of patients. LENVIMA was dose reduced in 69% of patients. The most common adverse reactions (≥5%) resulting in dose reduction or interruption of LENVIMA were diarrhea (26%), fatigue (18%), hypertension (17%), proteinuria (13%), decreased appetite (12%), palmar-plantar erythrodysesthesia (11%), nausea (9%), stomatitis (9%), musculoskeletal pain (8%), rash (8%), increased lipase (7%), abdominal pain (6%), and vomiting (6%), increased ALT (5%), and increased amylase (5%).
Tables 5 and 6 summarize the adverse reactions and laboratory abnormalities, respectively, that occurred in ≥20% of patients treated with LENVIMA and pembrolizumab in CLEAR.
Table 5 : Adverse Reactions in ≥20% of Patients on LENVIMA plus Pembrolizumab in CLEAR (RCC) LENVIMA 20 mg in combination with Pembrolizumab 200mg N=352 Sunitinib 50 mg N=340 Adverse Reactions All Grades (%) Grade 3-4 (%) All Grades (%) Grade 3-4 (%) General Fatiguea 63 9 56 8 Gastrointestinal Diarrheab 62 10 50 6 Stomatitisc 43 2 43 2 Nausea 36 3 33 1 Abdominal paind 27 2 18 1 Vomiting 26 3 20 1 Constipation 25 1 19 0 Musculoskeletal and connective tissue Musculoskeletal paine 58 4 41 3 Endocrine Hypothyroidismf 57 1 32 0 Vascular Hypertensiong 56 29 43 20 Hemorrhagic eventsh 27 5 26 4 Metabolism Decreased appetitei 41 4 31 1 Skin and subcutaneous tissue Rashj 37 5 17 1 Palmar-plantar erythrodysaesthesia syndromek 29 4 38 4 Respiratory, thoracic, and mediastinal Dysphonia 30 0 4 0 Renal and urinary Proteinurial 30 8 13 3 Acute kidney injurym 21 5 16 2 Investigations Weight decreased 30 8 9 0 Hepatobiliary Hepatotoxicityn 25 9 21 5 Nervous system Headache 23 1 16 1 a. Includes asthenia, fatigue, lethargy and malaise b. Includes diarrhea and gastroenteritis c. Includes aphthous ulcer, gingival pain, glossitis, glossodynia, mouth ulceration, mucosal inflammation, oral discomfort, oral mucosal bulering, oral pain, oropharyngeal pain, pharyngeal inflammation, and stomatitis d. Includes abdominal discomfort, abdominal pain, abdominal rigidity, abdominal tenderness, epigastric discomfort, lower abdominal pain, and upper abdominal pain e. Includes arthralgia, arthritis, back pain, bone pain, breast pain, musculoskeletal chest pain, musculoskeletal discomfort, musculoskeletal pain, musculoskeletal stiffness, myalgia, neck pain, non-cardiac chest pain, pain in extremity, and pain in jaw f. Includes hypothyroidism, increased blood thyroid stimulating hormone and secondary hypothyroidism g. Includes essential hypertension, increased blood pressure, increased diastolic blood pressure, hypertension, hypertensive crisis, hypertensive retinopathy, and labile blood pressure h. Includes all hemorrhage terms. Hemorrhage terms that occurred in 1 or more subjects in either treatment group include: Anal hemorrhage, aneurysm ruptured, blood buler, blood loss anemia, blood urine present, catheter site hematoma, cerebral microhemorrhage, conjunctival hemorrhage, contusion, diarrhea hemorrhagic, disseminated intravascular coagulation, ecchymosis, epistaxis, eye hemorrhage, gastric hemorrhage, gastritis hemorrhagic, gingival bleeding, hemorrhage urinary tract, hemothorax, hematemesis, hematoma, hematochezia, hematuria, hemoptysis, hemorrhoidal hemorrhage, increased tendency to bruise, injection site hematoma, injection site hemorrhage, intra-abdominal hemorrhage, lower gastrointestinal hemorrhage, Mallory-Weiss syndrome, melaena, petechiae, rectal hemorrhage, renal hemorrhage, retroperitoneal hemorrhage, small intestinal hemorrhage, splinter hemorrhages, subcutaneous hematoma, subdural hematoma, subarachnoid hemorrhage, thrombotic thrombocytopenic purpura, tumor hemorrhage, traumatic hematoma, and upper gastrointestinal hemorrhage i. Includes decreased appetite and early satiety j. Includes genital rash, infusion site rash, penile rash, perineal rash, rash, rash erythematous, rash macular, rash maculo-papular, rash papular, rash pruritic, and rash pustular k. Includes palmar erythema, palmar-plantar erythrodysesthesia syndrome and plantar erythema l. Includes hemoglobinuria, nephrotic syndrome, and proteinuria m. Includes acute kidney injury, azotaemia, blood creatinine increased, creatinine renal clearance decreased, hypercreatininaemia, renal failure, renal impairment, oliguria, glomerular filtration rate decreased, and nephropathy toxic n. Includes alanine aminotransferase increased, aspartate aminotransferase increased, blood bilirubin increased, drug-induced liver injury, hepatic enzyme increased, hepatic failure, hepatic function abnormal, hepatocellular injury, hepatotoxicity, hyperbilirubinemia, hypertransaminasemia, immune-mediated hepatitis, liver function test increased, liver injury, transaminases increased, and gamma-glutamyltransferase increased
Clinically relevant adverse reactions (<20%) that occurred in patients receiving LENVIMA/pembrolizumab were myocardial infarction (3%) and angina pectoris (1%).
Table 6: Laboratory Abnormalities in ≥20% (All Grades) of Patients on LENVIMA plus Pembrolizumab in CLEAR (RCC) LENVIMA 20 mg in combination with Pembrolizumab 200 mg Sunitinib 50 mg Laboratory Abnormality a All Grades % b Grades 3-4 % b All Grades % b Grade 3-4 % b Chemistry Hypertriglyceridemia 80 15 71 15 Hypercholesterolemia 64 5 43 1 Increased lipase 61 34 59 28 Increased creatinine 61 5 61 2 Increased amylase 59 17 41 9 Increased aspartate aminotransferase (AST) 58 7 57 3 Hyperglycemia 55 7 48 3 Increased alanine aminotransferase (ALT) 52 7 49 4 Hyperkalemia 44 9 28 6 Hypoglycemia 44 2 27 1 Hyponatremia 41 12 28 9 Decreased albumin 34 0.3 22 0 Increased alkaline phosphatase 32 4 32 1 Hypocalcemia 30 2 22 1 Hypophosphatemia 29 7 50 8 Hypomagnesemia 25 2 15 3 Increased creatine phosphokinase 24 6 36 5 Hypermagnesemia 23 2 22 3 Hypercalcemia 21 1 11 1 Hematology Lymphopenia 54 9 66 15 Thrombocytopenia 39 2 73 13 Anemia 38 3 66 8 Leukopenia 34 1 77 8 Neutropenia 31 4 72 16 a. With at least 1 grade increase from baseline b. Laboratory abnormality percentage is based on the number of patients who had both baseline and at least one post baseline laboratory measurement for each parameter. LENVIMA/pembrolizumab (n= 343 to 349) and sunitinib (n= 329 to 335).
Grade 3 and 4 increased ALT or AST was seen in 9% of patients. Grade ≥2 increased ALT or AST was reported in 64 (18%) patients, of whom 20 (31%) received ≥40 mg daily oral prednisone equivalent. Recurrence of Grade ≥2 increased ALT or AST was observed in 3 patients on rechallenge in patients receiving LENVIMA and 10 patients receiving both LENVIMA and pembrolizumab.
Previously T reated Renal Cell Carcinoma in Combination with Everolimus (Study 205)
The safety of LENVIMA was evaluated in Study 205, in which patients with unresectable advanced or metastatic renal cell carcinoma (RCC) were randomized (1:1:1) to LENVIMA 18 mg orally once daily with everolimus 5 mg orally once daily (n=51), LENVIMA 24 mg orally once daily (n=52), or everolimus 10 mg orally once daily (n=50) [see Clinical Studies ( 14.2 ) ]. This data also includes patients on the dose escalation portion of the study who received LENVIMA with everolimus (n=11). The median treatment duration was 8.1 months for LENVIMA with everolimus. Among 62 patients who received LENVIMA with everolimus, the median age was 61 years, 71% were men, and 98% were White.
The most common adverse reactions observed in the LENVIMA with everolimus-treated group (≥30%) were, in order of decreasing frequency, diarrhea, fatigue, arthralgia/myalgia, decreased appetite, vomiting, nausea, stomatitis/oral inflammation, hypertension, peripheral edema, cough, abdominal pain, dyspnea, rash, decreased weight, hemorrhagic events, and proteinuria. The most common serious adverse reactions (≥5%) were renal failure (11%), dehydration (10%), anemia (6%), thrombocytopenia (5%), diarrhea (5%), vomiting (5%), and dyspnea (5%).
Adverse reactions led to dose reductions or interruption in 89% of patients receiving LENVIMA with everolimus. The most common adverse reactions (≥5%) resulting in dose reductions in the LENVIMA with everolimus-treated group were diarrhea (21%), fatigue (8%), thrombocytopenia (6%), vomiting (6%), nausea (5%), and proteinuria (5%).
Treatment discontinuation due to an adverse reaction occurred in 29% of patients in the LENVIMA with everolimus-treated group.
Table 7 presents the adverse reactions in >15% of patients in the LENVIMA with everolimus arm. Study 205 was not designed to demonstrate a statistically significant difference in adverse reaction rates for LENVIMA in combination with everolimus, as compared to everolimus for any specific adverse reaction uled in Table 7.
Table 7 : Adverse Reactions Occurring in >15% of Patients in the LENVIMA with Everolimus Arm in Study 205 (RCC) LENVIMA 18 mg with Everolimus 5 mg N=62 Everolimus 10 mg N=50 Adverse Reactions Grade 1-4 (%) Grade 3-4 (%) Grade 1-4 (%) Grade 3-4 (%) Endocrine Hypothyroidism 24 0 2 0 Gastrointestinal Diarrhea 81 19 34 2 Vomiting 48 7 12 0 Nausea 45 5 16 0 Stomatitis/Oral inflammationa 44 2 50 4 Abdominal painb 37 3 8 0 Oral painc 23 2 4 0 Dyspepsia/Gastro-esophageal reflux 21 0 12 0 Constipation 16 0 18 0 General Fatigued 73 18 40 2 Peripheral edema 42 2 20 0 Pyrexia/Increased body temperature 21 2 10 2 Metabolism and Nutrition Decreased appetite 53 5 18 0 Decreased weight 34 3 8 0 Musculoskeletal and Connective Tissue Arthralgia/Myalgiae 55 5 32 0 Musculoskeletal chest pain 18 2 4 0 Nervous System Headache 19 2 10 2 Psychiatric Insomnia 16 2 2 0 Renal and Urinary Proteinuria/Urine protein present 31 8 14 2 Renal failure eventf 18 10 12 2 Respiratory, Thoracic and Mediastinal Cough 37 0 30 0 Dyspnea/Exertional dyspnea 35 5 28 8 Dysphonia 18 0 4 0 Skin and Subcutaneous Tissue Rashg 35 0 40 0 Vascular Hypertension/Increased blood pressure 42 13 10 2 Hemorrhagic eventsh 32 6 26 2 a Includes aphthous stomatitis, gingival inflammation, glossitis, and mouth ulcerationb Includes abdominal discomfort, gastrointestinal pain, lower abdominal pain, and upper abdominal pain c Includes gingival pain, glossodynia, and oropharyngeal paind Includes asthenia, fatigue, lethargy and malaise e Includes arthralgia, back pain, extremity pain, musculoskeletal pain, and myalgiaf Includes blood creatinine increased, blood urea increased, creatinine renal clearance decreased, nephropathy toxic, renal failure, renal failure acute, and renal impairment g Includes erythema, erythematous rash, genital rash, macular rash, maculo-papular rash, papular rash, pruritic rash, pustular rash, and septic rash h Includes hemorrhagic diarrhea, epistaxis, gastric hemorrhage, hemarthrosis, hematoma, hematuria, hemoptysis, lip hemorrhage, renal hematoma, and scrotal hematocele
In Table 8, Grade 3-4 laboratory abnormalities occurring in ≥3% of patients in the LENVIMA with everolimus arm are presented.
Table 8 : Grade 3-4 Laboratory Abnormalities Occurring in ≥3% of Patients in the LENVIMA with Everolimus Arm a , b in Study 205 (RCC) Laboratory Abnormality LENVIMA 18 mg with Everolimus 5 mg Everolimus 10 mg Grades 3-4 (%) Grades 3-4 (%) Chemistry Hypertriglyceridemia 18 18 Increased lipase 13 12 Hypercholesterolemia 11 0 Hyponatremia 11 6 Hypophosphatemia 11 6 Hyperkalemia 6 2 Hypocalcemia 6 2 Hypokalemia 6 2 Increased aspartate aminotransferase (AST) 3 0 Increased alanine aminotransferase (ALT) 3 2 Increased alkaline phosphatase 3 0 Hyperglycemia 3 16 Increased creatine kinase 3 4 Hematology Lymphopenia 10 20 Anemia 8 16 Thrombocytopenia 5 0 a. With at least 1 grade increase from baseline b. Laboratory Abnormality percentage is based on the number of patients who had both baseline and at least one post baseline laboratory measurement for each parameter. LENVIMA with Everolimus (n = 62), Everolimus (n = 50).
Hepatocellular Carcinoma
The safety of LENVIMA was evaluated in REFLECT, which randomized (1:1) patients with unresectable hepatocellular carcinoma (HCC) to LENVIMA (n=476) or sorafenib (n=475) [ see Clinical Studies ( 14.3 ) ]. The dose of LENVIMA was 12 mg orally once daily for patients with a baseline body weight of ≥60 kg and 8 mg orally once daily for patients with a baseline body weight of <60 kg. The dose of sorafenib was 400 mg orally twice daily. Duration of treatment was ≥6 months in 49% and 32% of patients in the LENVIMA and sorafenib groups, respectively. Among the 476 patients who received LENVIMA in REFLECT, the median age was 63 years, 85% were men, 28% were White and 70% were Asian.
The most common adverse reactions observed in the LENVIMA-treated patients (≥20%) were, in order of decreasing frequency, hypertension, fatigue, diarrhea, decreased appetite, arthralgia/myalgia, decreased weight, abdominal pain, palmar-plantar erythrodysesthesia syndrome, proteinuria, dysphonia, hemorrhagic events, hypothyroidism, and nausea.
The most common serious adverse reactions (≥2%) in LENVIMA-treated patients were hepatic encephalopathy (5%), hepatic failure (3%), ascites (3%), and decreased appetite (2%).
Adverse reactions led to dose reduction or interruption in 62% of patients receiving LENVIMA. The most common adverse reactions (≥5%) resulting in dose reduction or interruption of LENVIMA were fatigue (9%), decreased appetite (8%), diarrhea (8%), proteinuria (7%), hypertension (6%), and palmar-plantar erythrodysesthesia syndrome (5%).
Treatment discontinuation due to adverse reactions occurred in 20% of patients in the LENVIMA-treated group. The most common adverse reactions (≥1%) resulting in discontinuation of LENVIMA were fatigue (1%), hepatic encephalopathy (2%), hyperbilirubinemia (1%), and hepatic failure (1%).
Table 9 summarizes the adverse reactions that occurred in ≥10% of patients receiving LENVIMA in REFLECT. REFLECT was not designed to demonstrate a statistically significant reduction in adverse reaction rates for LENVIMA, as compared to sorafenib, for any specified adverse reaction uled in Table 9.
Table 9 : Adverse Reactions Occurring in ≥10% of Patients in the LENVIMA Arm in REFLECT (HCC) Adverse Reaction LENVIMA 8 mg/12 mg N=476 Sorafenib 800 mg N=475 Grade 1-4 (%) Grade 3-4 (%) Grade 1-4 (%) Grade 3-4 (%) Endocrine Hypothyroidisma 21 0 3 0 Gastrointestinal Diarrhea 39 4 46 4 Abdominal painb 30 3 28 4 Nausea 20 1 14 1 Vomiting 16 1 8 1 Constipation 16 1 11 0 Ascitesc 15 4 11 3 Stomatitisd 11 0.4 14 1 General Fatiguee 44 7 36 6 Pyrexiaf 15 0 14 0.2 Peripheral edema 14 1 7 0.2 Metabolism and Nutrition Decreased appetite 34 5 27 1 Decreased weight 31 8 22 3 Musculoskeletal and Connective Tissue Arthralgia/Myalgiag 31 1 20 2 Nervous System Headache 10 1 8 0 Renal and Urinary Proteinuriah 26 6 12 2 Respiratory, Thoracic and Mediastinal Dysphonia 24 0.2 12 0 Skin and Subcutaneous Tissue Palmar-plantar erythrodysesthesia syndrome 27 3 52 11 Rashi 14 0 24 2 Vascular Hypertensionj 45 24 31 15 Hemorrhagic eventsk 23 4 15 4 a. Includes hypothyroidism, blood thyroid stimulating hormone increased. b. Includes abdominal discomfort, abdominal pain, abdominal tenderness, epigastric discomfort, gastrointestinal pain, lower abdominal pain, and upper abdominal pain c. Includes ascites and malignant ascites d. Includes aphthous ulcer, gingival erosion, gingival ulceration, glossitis, mouth ulceration, oral mucosal bulering, and stomatitis e. Includes asthenia, fatigue, lethargy and malaise f. Includes increased body temperature, pyrexia g. Includes arthralgia, back pain, extremity pain, musculoskeletal chest pain, musculoskeletal discomfort, musculoskeletal pain, and myalgia h. Includes proteinuria, increased urine protein, protein urine present i. Includes erythema, erythematous rash, exfoliative rash, genital rash, macular rash, maculo-papular rash, papular rash, pruritic rash, pustular rash and rash j. Includes increased diastolic blood pressure, increased blood pressure, hypertension and orthostatic hypertension k. Includes all hemorrhage terms. Hemorrhage terms that occurred in 5 or more subjects in either treatment group include: epistaxis, hematuria, gingival bleeding, hemoptysis, esophageal varices hemorrhage, hemorrhoidal hemorrhage, mouth hemorrhage, rectal hemorrhage and upper gastrointestinal hemorrhage
In Table 10, Grade 3-4 laboratory abnormalities occurring in ≥2% of patients in the LENVIMA arm in REFLECT (HCC) are presented.
Table 10 : Grade 3-4 Laboratory Abnormalities Occurring in ≥2% of Patients in the LENVIMA Arm a,b in REFLECT (HCC) Laboratory Abnormality LENVIMA (%) Sorafenib (%) Chemistry Increased GGT 17 20 Hyponatremia 15 9 Hyperbilirubinemia 13 10 Increased aspartate aminotransferase (AST) 12 18 Increased alanine aminotransferase (ALT) 8 9 Increased alkaline phosphatase 7 5 Increased lipase 6 17 Hypokalemia 3 4 Hyperkalemia 3 2 Decreased albumin 3 1 Increased creatinine 2 2 Hematology Thrombocytopenia 10 8 Lymphopenia 8 9 Neutropenia 7 3 Anemia 4 5 a. With at least 1 grade increase from baseline b. Laboratory Abnormality percentage is based on the number of patients who had both baseline and at least one post baseline laboratory measurement for each parameter. LENVIMA (n=278 to 470) and sorafenib (n=260 to 473)
Endometrial Carcinoma
The safety of LENVIMA in combination with pembrolizumab was investigated in Study 309, a multicenter, open-label, randomized (1:1), active-controlled trial in patients with advanced endometrial carcinoma previously treated with at least one prior platinum-based chemotherapy regimen in any setting, including in the neoadjuvant and adjuvant settings [see Clinical Studies ( 14.4 )]. Patients with endometrial carcinoma that are pMMR or not MSI-H received LENVIMA 20 mg orally once daily with pembrolizumab 200 mg intravenously every 3 weeks (n=342); or received doxorubicin or paclitaxel (n= 325).
For patients with pMMR or not MSI-H status, the median duration of study treatment was 7.2 months (range 1 day to 26.8 months) and the median duration of exposure to LENVIMA was 6.7 months (range: 1 day to 26.8 months).
Fatal adverse reactions among these patients occurred in 4.7% of those treated with LENVIMA and pembrolizumab, including 2 cases of pneumonia, and 1 case of the following: acute kidney injury, acute myocardial infarction, colitis, decreased appetite, intestinal perforation, lower gastrointestinal hemorrhage, malignant gastrointestinal obstruction, multiple organ dysfunction syndrome, myelodysplastic syndrome, pulmonary embolism, and right ventricular dysfunction.
Serious adverse reactions occurred in 50% of these patients receiving LENVIMA and pembrolizumab. Serious adverse reactions with frequency ≥3% were hypertension (4.4%), and urinary tract infection (3.2%).
Discontinuation of LENVIMA due to an adverse reaction occurred in 26% of these patients. The most common (≥1 %) adverse reactions leading to discontinuation of LENVIMA were hypertension (2%), asthenia (1.8%), diarrhea (1.2%), decreased appetite (1.2%), proteinuria (1.2%), and vomiting (1.2%).
Dose reductions of LENVIMA due to adverse reactions occurred in 67% of patients. The most common (≥5%) adverse reactions resulting in dose reduction of LENVIMA were hypertension (18%), diarrhea (11%), palmar-plantar erythrodysesthesia syndrome (9%), proteinuria (7%), fatigue (7%), decreased appetite (6%), asthenia (5%), and weight decreased (5%).
Dose interruptions of LENVIMA due to an adverse reaction occurred in 58% of these patients. The most common (≥2%) adverse reactions leading to interruption of LENVIMA were hypertension (11%), diarrhea (11%), proteinuria (6%), decreased appetite (5%), vomiting (5%), increased alanine aminotransferase (3.5%), fatigue (3.5%), nausea (3.5%), abdominal pain (2.9%), weight decreased (2.6%), urinary tract infection (2.6%), increased aspartate aminotransferase (2.3%), asthenia (2.3%), and palmar-plantar erythrodysesthesia (2%).
Tables 11 and 12 summarize adverse reactions and laboratory abnormalities, respectively, in patients receiving LENVIMA in Study 309.
Table 11 : Adverse Reactions in ≥20% of Patients Receiving LENVIMA plus Pembrolizumab in Study 309 (EC) Endometrial Carcinoma ( pMMR or not MSI-H) LENVIMA 20 mg in combination with Pembrolizumab 200 mg N=342 Doxorubicin or Paclitaxel N=325 Adverse Reaction All Grades a (%) Grades 3-4 (%) All Grades a (%) Grades 3-4 (%) Endocrine Hypothyroidismb 67 0.9 0.9 0 Vascular Hypertensionc 67 39 6 2.5 Hemorrhagic eventsd 25 2.6 15 0.9 General Fatiguee 58 11 54 6 Gastrointestinal Diarrheaf 55 8 20 2.8 Nausea 49 2.9 47 1.5 Vomiting 37 2.3 21 2.2 Stomatitisg 35 2.6 26 1.2 Abdominal painh 34 2.6 21 1.2 Constipation 27 0 25 0.6 Musculoskeletal and Connective Tissue Musculoskeletal disordersi 53 5 27 0.6 Metabolism Decreased appetitej 44 7 21 0 Investigations Decreased weight 34 10 6 0.3 Renal and Urinary Proteinuriak 29 6 3.4 0.3 Infections Urinary tract infectionl 31 5 13 1.2 Nervous System Headache 26 0.6 9 0.3 Respiratory, Thoracic and Mediastinal Dysphonia 22 0 0.6 0 Skin and Subcutaneous Tissue Palmar-plantar erythrodysesthesiam 23 2.9 0.9 0 Rashn 20 2.3 4.9 0 a. Graded per NCI CTCAE v4.03 b. Includes hypothyroidism, blood thyroid stimulating hormone increased, thyroiditis, primary hypothyroidism, and secondary hypothyroidism c. Includes hypertension, blood pressure increased, hypertensive crisis, secondary hypertension, blood pressure abnormal, hypertensive encephalopathy, and blood pressure fluctuation d. Includes epistaxis, vaginal hemorrhage, hematuria, gingival bleeding, metrorrhagia, rectal hemorrhage, contusion, hematochezia, cerebral hemorrhage, conjunctival hemorrhage, gastrointestinal hemorrhage, hemoptysis, hemorrhage urinary tract, lower gastrointestinal hemorrhage, mouth hemorrhage, petechiae, uterine hemorrhage, anal hemorrhage, blood buler, eye hemorrhage, hematoma, hemorrhage intracranial, hemorrhagic stroke, injection site hemorrhage, melena, purpura, stoma site hemorrhage, upper gastrointestinal hemorrhage, wound hemorrhage, blood urine present, coital bleeding, ecchymosis, hematemesis, hemorrhage subcutaneous, hepatic hematoma, injection site bruising, intestinal hemorrhage, laryngeal hemorrhage, pulmonary hemorrhage, subdural hematoma, umbilical hemorrhage, and vessel puncture site bruise e. Includes fatigue, asthenia, malaise, and lethargy f. Includes diarrhea and gastroenteritis g. Includes stomatitis, mucosal inflammation, oropharyngeal pain, aphthous ulcer, mouth ulceration, cheilitis, oral mucosal erythema, and tongue ulceration h. Includes abdominal pain, abdominal pain upper, abdominal pain lower, abdominal discomfort, gastrointestinal pain, abdominal tenderness, and epigastric discomfort i. Includes arthralgia, myalgia, back pain, pain in extremity, bone pain, neck pain, musculoskeletal pain, arthritis, musculoskeletal chest pain, musculoskeletal stiffness, non-cardiac chest pain, pain in jaw j. Includes decreased appetite and early satiety k. Includes proteinuria, protein urine present, hemoglobinuria l. Includes urinary tract infection, cystitis, and pyelonephritis m. Includes palmar-plantar erythrodysesthesia syndrome, palmar erythema, plantar erythema, and skin reaction n. Includes rash, rash maculo-papular, rash pruritic, rash erythematous, rash macular, rash pustular, rash papular, rash vesicular, and application site rash
Table 12 : Laboratory Abnormalities Worsened from Baseline a Occurring in ≥20% (All Grades) or ≥3% (Grades 3-4) of Patients Receiving LENVIMA plus Pembrolizumab in Study 309 (EC) Endometrial Carcinoma ( pMMR or not MSI-H) LENVIMA 20 mg in combination with Pembrolizumab 200 mg N=342 Doxorubicin or Paclitaxel N=325 Laboratory Test b All Grades c (%) Grades 3-4 (%) All Grades c (%) Grades 3-4 (%) Chemistry Hypertriglyceridemia 70 6 45 1.7 Hypoalbuminemia 60 2.7 42 1.6 Increased aspartate aminotransferase 58 9 23 1.6 Hyperglycemia 58 8 45 4.4 Hypomagnesemia 53 6 32 3.8 Increased alanine aminotransferase 55 9 21 1.2 Hypercholesteremia 53 3.2 23 0.7 Hyponatremia 46 15 28 7 Increased alkaline phosphatase 43 4.7 18 0.9 Hypocalcemia 40 4.7 21 1.7 Increased lipase 36 14 13 3.9 Increased creatinine 35 4.7 18 1.9 Hypokalemia 34 10 24 5 Hypophosphatemia 26 8 17 3.2 Increased amylase 25 7 8 1 Hyperkalemia 23 2.4 12 1.2 Increased creatine kinase 19 3.7 7 0 Increased bilirubin 18 3.6 6 1.6 Hematology Lymphopenia 50 16 65 20 Thrombocytopenia 50 8 30 4.7 Anemia 49 8 84 14 Leukopenia 43 3.5 83 43 Neutropenia 31 6 76 58 a. With at least 1 grade increase from baseline b. Laboratory abnormality percentage is based on the number of patients who had both baseline and at least one post-baseline laboratory measurement for each parameter: LENVIMA/pembrolizumab (range: 263 to 340 patients) and doxorubicin or paclitaxel (range: 240 to 322 patients). c. Graded per NCI CTCAE v4.03 6.2
pancreatitis, increased amylase
The following adverse reactions have been identified during post-approval use of LENVIMA. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Gastrointestinal :
General: impaired wound healing
Hepatobiliary: cholecystitis
Renal and Urinary: nephrotic syndrome
Vascular: arterial (including aortic) aneurysms, dissections, and rupture
Drug Interactions Section
LENVIMA has been reported to prolong the QT/QTc interval. Avoid coadministration of LENVIMA with medicinal products with a known potential to prolong the QT/QTc interval [ see Warnings and Precaution s ( 5.9 ) ].
Use In Specific Populations Section
- Lactation: Advise not to breastfeed. (
8.2 )PREGNANCY SECTION
Risk Summary
Based on findings from animal studies and its mechanism of action, LENVIMA can cause fetal harm when administered to a pregnant woman [see Clinical Pharmacology ( 12.1 )]. In animal reproduction studies, oral administration of lenvatinib during organogenesis at doses below the recommended human doses resulted in embryotoxicity, fetotoxicity, and teratogenicity in rats and rabbits ( see Data ). There are no available human data informing the drug-associated risk. Advise pregnant women of the potential risk to a fetus.
In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Data
Animal Data
In an embryofetal development study, daily oral administration of lenvatinib mesylate at doses ≥0.3 mg/kg [approximately 0.14 times the recommended clinical dose of 24 mg based on body surface area (BSA)] to pregnant rats during organogenesis resulted in dose-related decreases in mean fetal body weight, delayed fetal ossifications, and dose-related increases in fetal external (parietal edema and tail abnormalities), visceral, and skeletal anomalies. Greater than 80% post-implantation loss was observed at 1.0 mg/kg/day (approximately 0.5 times the recommended clinical dose of 24 mg based on BSA).
Daily oral administration of lenvatinib mesylate to pregnant rabbits during organogenesis resulted in fetal external (short tail), visceral (retroesophageal subclavian artery), and skeletal anomalies at doses greater than or equal to 0.03 mg/kg (approximately 0.03 times the recommended clinical dose of 24 mg based on BSA). At the 0.03 mg/kg dose, increased post-implantation loss, including 1 fetal death, was also observed. Lenvatinib was abortifacient in rabbits, resulting in late abortions in approximately one-third of the rabbits treated at a dose level of 0.5 mg/kg/day (approximately 0.5 times the recommended clinical dose of 24 mg based on BSA).
LACTATION SECTION
Risk Summary
It is not known whether LENVIMA is present in human milk; however, lenvatinib and its metabolites are excreted in rat milk at concentrations higher than those in maternal plasma ( see Data ). Because of the potential for serious adverse reactions in breastfed children, advise women to discontinue breastfeeding during treatment with LENVIMA and for 1 week after the last dose.
Data
Animal Data
Following administration of radiolabeled lenvatinib to lactating Sprague Dawley rats, lenvatinib-related radioactivity was approximately 2 times higher [based on area under the curve (AUC)] in milk compared to maternal plasma.
FEMALES & MALES OF REPRODUCTIVE POTENTIAL SECTION
Based on animal data and its mechanism of action, LENVIMA can cause fetal harm when administered to a pregnant woman [see Use in Specific Populations ( 8.1 )].
Pregnancy Testing
Verify the pregnancy status of females of reproductive potential prior to initiating LENVIMA [see Use in Specific Populations ( 8.1 )].
Contraception
Females
Advise females of reproductive potential of the potential risk to a fetus and to use effective contraception during treatment with LENVIMA and for 30 days after the last dose.
Infertility
LENVIMA may impair fertility in males and females of reproductive potential [ see Nonclinical Toxicology ( 13.1 )].
PEDIATRIC USE SECTION
The safety and effectiveness of LENVIMA in pediatric patients have not been established.
The safety and efficacy of LENVIMA alone and in combination were investigated but not established in four open label studies (NCT02432274, NCT04154189, NCT04447755, NCT03245151) in 232 patients aged 2 to <17 years with relapsed or refractory solid tumors, including osteosarcoma, Ewing sarcoma, rhabdomyosarcoma, and high-grade glioma. Hypothyroidism and pneumothorax were observed at a higher rate in pediatric patients compared to that of adult patients. The pharmacokinetics (PK) of lenvatinib in pediatric patients were within range of values previously observed in adults at the approved recommended dose of 24 mg.
Juvenile Animal Data
Daily oral administration of lenvatinib mesylate to juvenile rats for 8 weeks starting on postnatal day 21 (approximately equal to a human pediatric age of 2 years) resulted in growth retardation (decreased body weight gain, decreased food consumption, and decreases in the width and/or length of the femur and tibia) and secondary delays in physical development and reproductive organ immaturity at doses greater than or equal to 2 mg/kg (approximately 1.2 to 5 times the human exposure based on AUC at the recommended clinical dose of 24 mg). Decreased length of the femur and tibia persisted following 4 weeks of recovery. In general, the toxicologic profile of lenvatinib was similar between juvenile and adult rats, though toxicities including broken teeth at all dose levels and mortality at the 10 mg/kg/day dose level (attributed to primary duodenal lesions) occurred at earlier treatment time-points in juvenile rats.
GERIATRIC USE SECTION
Of the 261 patients with differentiated thyroid cancer (DTC) who received LENVIMA in SELECT, 45% were ≥65 years of age and 11% were ≥75 years of age. No overall differences in safety or effectiveness were observed between these subjects and younger subjects.
Of the 352 patients with renal cell carcinoma (RCC) who received LENVIMA with pembrolizumab in CLEAR, 45% were ≥65 years of age and 13% were ≥75 years of age. No overall differences in safety or effectiveness were observed between these elderly patients and younger patients.
Of the 62 patients with RCC who received LENVIMA with everolimus in Study 205, 36% were ≥65 years of age. Conclusions are limited due to the small sample size, but there appeared to be no overall differences in safety or effectiveness between these subjects and younger subjects.
Of the 476 patients with hepatocellular carcinoma (HCC) who received LENVIMA in REFLECT, 44% were ≥65 years of age and 12% were ≥75 years of age. No overall differences in safety or effectiveness were observed between patients ≥65 and younger subjects. Patients ≥75 years of age showed reduced tolerability to LENVIMA.
Of 406 adult patients with endometrial carcinoma (EC) who were treated with LENVIMA in combination with pembrolizumab in Study 309, 201 (50%) were 65 years and over. No overall differences in safety or effectiveness were observed between elderly patients and younger patients.
RENAL IMPAIRMENT SUBSECTION
No dose adjustment is recommended for patients with mild (CLcr 60-89 mL/min) or moderate (CLcr 30-59 mL/min) renal impairment. Lenvatinib concentrations may increase in patients with DTC, RCC, or endometrial carcinoma and severe (CLcr 15-29 mL/min) renal impairment. Reduce the dose of LENVIMA for patients with RCC, DTC, or endometrial carcinoma and severe renal impairment [see Dosage and Administration ( 2.7 )]. There is no recommended dose of LENVIMA for patients with HCC and severe renal impairment. LENVIMA has not been studied in patients with end stage renal disease [ see Warnings and Precautions ( 5.5 ) , Clinical Pharmacology ( 12.3 ) ].
HEPATIC IMPAIRMENT SUBSECTION
No dose adjustment is recommended for patients with HCC and mild hepatic impairment (Child-Pugh A). There is no recommended dose for patients with HCC with moderate or severe hepatic impairment.
No dose adjustment is recommended for patients with DTC, RCC, or endometrial carcinoma and mild or moderate hepatic impairment (Child-Pugh A or B). Lenvatinib concentrations may increase in patients with DTC, RCC, or endometrial carcinoma and severe hepatic impairment (Child-Pugh C). Reduce the dose of lenvatinib for patients with DTC, RCC, or endometrial carcinoma and severe hepatic impairment [see Dosage and Administration ( 2.7 ) , Clinical Pharmacology ( 12.3 ) ].
Overdosage Section
Due to the high plasma protein binding, lenvatinib is not expected to be dialyzable [see Clinical Pharmacology ( 12.3 )]. Death due to multiorgan dysfunction occurred in a patient who received a single dose of LENVIMA 120 mg orally.
Description Section
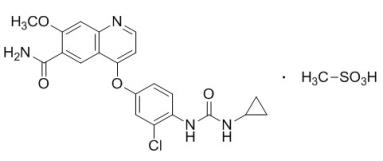
LENVIMA, a kinase inhibitor, is the mesylate salt of lenvatinib. Its chemical name is 4-[3-chloro-4-(N’-cyclopropylureido)phenoxy]-7-methoxyquinoline-6-carboxamide methanesulfonate. The molecular formula is C21H19ClN4O4 • CH4O3S, and the molecular weight of the mesylate salt is 522.96. The chemical structure of lenvatinib mesylate is:
Lenvatinib mesylate is a white to pale reddish yellow powder. It is slightly soluble in water and practically insoluble in ethanol (dehydrated). The dissociation constant (pKa value) of lenvatinib mesylate is 5.05 at 25°C. The partition coefficient (log P value) is 3.3.
LENVIMA capsules for oral administration contain 4 mg or 10 mg of lenvatinib, equivalent to 4.90 mg or 12.25 mg of lenvatinib mesylate, respectively. The inactive ingredients are: calcium carbonate, hydroxypropyl cellulose, low-substituted hydroxypropyl cellulose, mannitol, microcrystalline cellulose, and talc.
In addition, the capsule shell contains ferric oxide red, ferric oxide yellow, hypromellose, and titanium dioxide. The printing ink contains black iron oxide, potassium hydroxide, propylene glycol, and shellac.
Clinical Pharmacology Section
MECHANISM OF ACTION SECTION
Lenvatinib is a kinase inhibitor that inhibits the kinase activities of vascular endothelial growth factor (VEGF) receptors VEGFR1 (FLT1), VEGFR2 (KDR), and VEGFR3 (FLT4). Lenvatinib inhibits other kinases that have been implicated in pathogenic angiogenesis, tumor growth, and cancer progression in addition to their normal cellular functions, including fibroblast growth factor (FGF) receptors FGFR1, 2, 3, and 4; platelet derived growth factor receptor alpha (PDGFRA), KIT, and RET. Lenvatinib also exhibited antiproliferative activity in hepatocellular carcinoma cell lines dependent on activated FGFR signaling with a concurrent inhibition of FGF-receptor substrate 2 alpha (FRS2) phosphorylation.
In syngeneic mouse tumor models, lenvatinib decreased tumor-associated macrophages, increased activated cytotoxic T cells, and demonstrated greater antitumor activity in combination with an anti-PD-1 monoclonal antibody compared to either treatment alone.
The combination of lenvatinib and everolimus showed increased antiangiogenic and antitumor activity as demonstrated by decreases in human endothelial cell proliferation, tube formation, and VEGF signaling in vitro, and by decreases in tumor volume in mouse xenograft models of human renal cell cancer that were greater than those with either drug alone.
PHARMACODYNAMICS SECTION
Exposure-Response Relationships
In a multicenter randomized (1:1) double blind trial of 152 patients with radioactive iodine (RAI)-refractory DTC, a dose-response relationship for overall response rate (ORR) was observed over the dose range of 18 mg (0.75 times the recommended dose of 24 mg) and 24 mg. A higher ORR was observed at the recommended lenvatinib dose.
No dose-response relationships for adverse reactions, serious adverse reactions, adverse reactions leading to study drug discontinuation, and adverse reactions leading to study drug interruption were observed over the same dose range.
PHARMACOKINETICS SECTION
In patients with solid tumors administered single and multiple doses of LENVIMA once daily, the maximum lenvatinib plasma concentration (Cmax) and the area under the concentration-time curve (AUC) increased proportionally over the dose range of 3.2 mg (0.1 times the recommended clinical dose of 24 mg) to 32 mg (1.33 times the recommended clinical dose of 24 mg) with a median accumulation index of 0.96 (20 mg) to 1.54 (6.4 mg).
Geometric mean Cmax and AUC values at steady state for RCC, DTC and HCC are summarized in the Table 13.
Table 13: Lenvatinib C max and AUC in Patients with Solid Tumors a Tumor Type Dose Parameter N Geometric Mean %CV RCC 18 mg Cmax (ng/mL) 350 267 36.7 AUC (ng∙h/mL) 350 3148 42.5 20 mg Cmax (ng/mL) 346 275 32.6 AUC (ng∙h/mL) 346 3135 41.3 DTC 24 mg Cmax (ng/mL) 251 323 33.3 AUC (ng∙h/mL) 251 3483 34.7 HCC (body weight < 60 kg) 8 mg Cmax (ng/mL) 150 154 25.4 AUC (ng∙h/mL) 150 1835 34.0 HCC (body weight ≥ 60 kg) 12 mg Cmax (ng/mL) 318 172 23.1 AUC (ng∙h/mL) 318 2013 29.3
a Model-predicted steady-state pharmacokinetic parameters are presented.
Absorption
The time to peak plasma concentration (Tmax) typically occurred from 1 to 4 hours post-dose.
Food Effect
Administration with a high fat meal (approximately 900 calories of which approximately 55% were from fat, 15% from protein, and 30% from carbohydrates) did not affect the extent of absorption, but decreased the rate of absorption and delayed the median Tmax from 2 hours to 4 hours.
Distribution
Model-predicted geometric mean steady state volume of distribution is 97 L (%CV, 30.2%). Protein binding of lenvatinib is 97% to 99%, which is independent of concentration, and is not impacted by hepatic or renal function. The blood-to-plasma concentration ratio ranged from 0.59 to 0.61 at concentrations of 0.1 to 10 μg/mL in vitro.
Elimination
The terminal elimination half-life of lenvatinib was approximately 28 hours.
Metabolism
The main metabolic pathways for lenvatinib in humans were identified as enzymatic (CYP3A and aldehyde oxidase) and non-enzymatic processes.
Excretion
Ten days after a single administration of radiolabeled lenvatinib, approximately 64% and 25% of the radiolabel were eliminated in the feces and urine, respectively.
Specific Populations
Age (18 to 92 years), sex, race/ethnicity (White, Black and Asian), tumor types (DTC, RCC, HCC and other tumor types) and renal impairment (creatinine clearance: 15-89 mL/min) did not have a significant effect on apparent oral clearance (CL/F). Subjects with end stage renal disease were not studied.
Patients with Hepatic Impairment
The pharmacokinetics of lenvatinib following a single 10 mg dose were evaluated in subjects with mild (Child-Pugh A) or moderate (Child-Pugh B) hepatic impairment. The pharmacokinetics of a single 5 mg dose were evaluated in subjects with severe (Child-Pugh C) hepatic impairment. Compared to subjects with normal hepatic function, the dose-adjusted AUC0-inf of lenvatinib for subjects with mild, moderate, and severe hepatic impairment were 119%, 107%, and 180%, respectively.
Apparent oral clearance of lenvatinib in patients with HCC and mild hepatic impairment was similar to patients with HCC and moderate hepatic impairment.
Body Weight
Lenvatinib exposures in patients with HCC in REFLECT were comparable between those weighing <60 kg who received a starting dose of 8 mg and those ≥60 kg who received a starting dose of 12 mg.
Drug Interaction Studies
Effect of Other D rugs on L envatinib
CYP3A, P-gp, and BCRP Inhibitor s: Ketoconazole (400 mg daily for 18 days) increased lenvatinib (administered as a single 5 mg dose on Day 5) AUC by 15% and Cmax by 19%.
P-gp Inhibitor: Rifampicin (600 mg as a single dose) increased lenvatinib (24 mg as a single dose) AUC by 31% and Cmax by 33%.
CYP3A and P-gp Inducers: Rifampicin (600 mg daily for 21 days) decreased lenvatinib (24 mg as a single dose on Day 15) AUC by 18%. The Cmax was unchanged.
Population pharmacokinetic analysis demonstrated that neither everolimus nor pembrolizumab meaningfully affect the pharmacokinetics of lenvatinib.
In Vitro Studies with Transporters: Lenvatinib is a substrate of P-gp and BCRP but not a substrate for organic anion transporter (OAT) 1, OAT3, organic anion transporting polypeptide (OATP) 1B1, OATP1B3, organic cation transporter (OCT) 1, OCT2, multidrug and toxin extrusion (MATE) 1, MATE2-K, or the bile salt export pump (BSEP).
Effect of L envatinib on O ther D rugs
CYP2C8 Substrate : There is no projected significant drug-drug interaction risk between lenvatinib and repaglinide.
CYP3A4 Substrate : Co-administration of lenvatinib with midazolam had no effect on the pharmacokinetics of midazolam.
Population pharmacokinetic analysis demonstrated that lenvatinib does not meaningfully affect the pharmacokinetics of either everolimus or pembrolizumab.
In Vitro Studies with Substrates of CYP or UDP-glucuronosyltransferase (UGT) : Lenvatinib inhibits CYP2C8, CYP1A2, CYP2B6, CYP2C9, CYP2C19, CYP2D6, and CYP3A. Lenvatinib does not inhibit CYP2A6 and CYP2E1. Lenvatinib induces CYP3A, but it does not induce CYP1A1, CYP1A2, CYP2B6, and CYP2C9.
Lenvatinib inhibits UGT1A1, UGT1A4, and UGT1A9 in vitro, but likely only inhibits UGT1A1 in vivo in the gastrointestinal tract based on the expression of the enzyme in tissues. Lenvatinib does not inhibit UGT1A6, UGT2B7 or aldehyde oxidase. Lenvatinib does not induce UGT1A1, UGT1A4, UGT1A6, UGT1A9, or UGT2B7.
In Vitro Studies with Substrates of Transporter s :
Lenvatinib does not have the potential to inhibit MATE1, MATE2-K, OCT1, OCT2, OAT1, OAT3, BSEP, OATP1B1, or OATP1B3 in vivo.
Nonclinical Toxicology Section
CARCINOGENESIS & MUTAGENESIS & IMPAIRMENT OF FERTILITY SECTION
Carcinogenicity studies have not been conducted with lenvatinib. Lenvatinib mesylate was not mutagenic in the in vitro bacterial reverse mutation (Ames) assay. Lenvatinib was not clastogenic in the in vitro mouse lymphoma thymidine kinase assay or the in vivo rat micronucleus assay.
No specific studies with lenvatinib have been conducted in animals to evaluate the effect on fertility; however, results from general toxicology studies in rats, monkeys, and dogs suggest there is a potential for lenvatinib to impair fertility. Male dogs exhibited testicular hypocellularity of the seminiferous epithelium and desquamated seminiferous epithelial cells in the epididymides at lenvatinib exposures approximately 0.02 to 0.09 times the AUC at the recommended clinical dose of 24 mg once daily. Follicular atresia of the ovaries was observed in monkeys and rats at exposures 0.2 to 0.8 times and 10 to 44 times the AUC at the recommended clinical dose of 24 mg once daily, respectively. In addition, in monkeys, a decreased incidence of menstruation was reported at lenvatinib exposures lower than those observed in humans at the recommended clinical dose of 24 mg once daily.
Clinical Studies Section
A multicenter, randomized (2:1), double-blind, placebo-controlled study (SELECT; NCT01321554) was conducted in 392 patients with locally recurrent or metastatic radioactive iodine-refractory differentiated thyroid cancer and radiographic evidence of disease progression within 12 months prior to randomization, confirmed by independent radiologic review. Radioactive iodine (RAI)-refractory was defined as 1 or more measurable lesions with no iodine uptake on RAI scan, iodine uptake with progression within 12 months of RAI therapy, or having received cumulative RAI activity >600 mCi (22 GBq) with the last dose administered at least 6 months prior to study entry. Patients were randomized to receive LENVIMA 24 mg once daily (n=261) or placebo (n=131) until disease progression. Randomization was stratified by geographic region, prior VEGF/VEGFR-targeted therapy, and age. The major efficacy outcome measure was progression-free survival (PFS) as determined by blinded independent radiologic review using Response Evaluation Criteria in Solid Tumors (RECIST) 1.1. Independent review confirmation of disease progression was required prior to discontinuing patients from the randomization phase of the study. Other efficacy outcome measures included objective response rate (ORR) and overall survival (OS). Patients in the placebo arm could receive LENVIMA following independent review confirmation of disease progression.
Of the 392 patients randomized, 51% were male, the median age was 63 years, 40% were 65 years or older, 79% were White, 54% had an Eastern Cooperative Oncology Group (ECOG) performance status (PS) of 0, and 24% had received 1 prior VEGF/VEGFR-targeted therapy. Metastases were present in 99% of the patients: lungs in 89%, lymph nodes in 52%, bone in 39%, liver in 18%, and brain in 4%. The histological diagnoses were papillary thyroid cancer (66%) and follicular thyroid cancer (34%); of those with follicular histology, 44% had Hürthle cell and 11% had clear cell subtypes. In the LENVIMA arm, 67% of patients did not demonstrate iodine uptake on any RAI scan compared to 77% in the placebo arm. Additionally, 59% of patients on the LENVIMA arm and 61% of patients on placebo arm progressed, according to RECIST 1.1, within 12 months of prior I 131 therapy; 19.2% of patients on the LENVIMA arm and 17.6% of patients on placebo arm received prior cumulative activity of >600 mCi or 22 GBq I 131, with the last dose administered at least 6 months prior to study entry. The median cumulative RAI activity administered prior to study entry was 350 mCi (12.95 GBq).
A statistically significant prolongation in PFS was demonstrated in LENVIMA-treated patients compared to those receiving placebo (Table 14 and Figure 1). Upon confirmation of progression, 83% of patients that were randomly assigned to placebo crossed over to receive open-label LENVIMA.
Table 14 : Efficacy R esults in Differentiated Thyroid Cancer in SELECT LENVIMA N=261 Placebo N=131 Progression- F ree Survival (PFS) a Number of events (%) 107 (41) 113 (86) Progressive disease 93 (36) 109 (83) Death 14 (5) 4 (3) Median PFS in months (95% CI) 18.3 (15.1, NE) 3.6 (2.2, 3.7) Hazard ratio (95% CI)b 0.21 (0.16, 0.28) p-valuec <0.001 Objective Response Rate a Objective response rate 65% 2% (95% CI) (59%, 71%) (0%, 4%) Complete response 2% 0% Partial response 63% 2% p-valued <0.001 Overall Survival (OS) e Number of deaths (%) 71 (27) 47 (36) Median OS in months (95% CI) NE (22.1, NE) NE (20.3, NE) Hazard ratio (95% CI)b 0.73 (0.50, 1.07) p-valueb 0.10 a. Independent radiologic review b. Estimated with Cox proportional hazard model stratified by region (Europe vs North America vs other), age group (≤65 years vs >65 years), and previous VEGF/VEGFR-targeted therapy (0 vs 1) c. Log-rank test stratified by region (Europe vs North America vs other), age group (≤65 years vs >65 years), and previous VEGF/VEGFR-targeted therapy (0 vs 1) d. Cochran-Mantel-Haenszel chi-square test e. NE = Not estimable
Figure 1: Kaplan-Meier Curves for P rogression- F ree Survival in SELECT
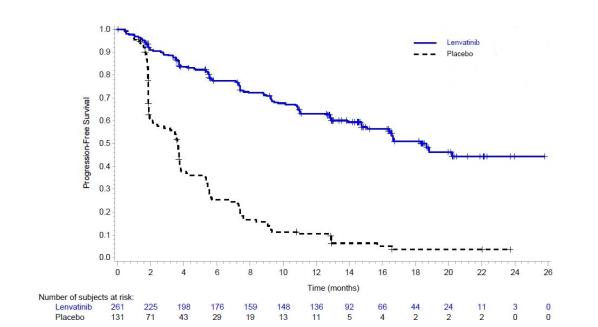
First-Line T reatment of P atients with RCC in C ombination with Pembrolizumab CLEAR
The efficacy of LENVIMA in combination with pembrolizumab was investigated in CLEAR (NCT02811861), a multicenter, open-label, randomized trial that enrolled 1069 patients with advanced RCC in the first-line setting. Patients were enrolled regardless of PD-L1 tumor expression status. Patients with active autoimmune disease or a medical condition that required immunosuppression were ineligible. Randomization was stratified by geographic region (North America and Western Europe versus “Rest of the World”) and Memorial Sloan Kettering Cancer Center (MSKCC) prognostic groups (favorable, intermediate and poor risk).
Patients were randomized (1:1:1) to one of the following treatment arms:
- LENVIMA 20 mg orally once daily in combination with pembrolizumab 200 mg intravenously every 3 weeks up to 24 months.
- LENVIMA 18 mg orally once daily in combination with everolimus 5 mg orally once daily.
- Sunitinib 50 mg orally once daily for 4 weeks then off treatment for 2 weeks.
Treatment continued until unacceptable toxicity or disease progression. Administration of LENVIMA with pembrolizumab was permitted beyond RECIST-defined disease progression if the patient was clinically stable and considered by the investigator to be deriving clinical benefit. Pembrolizumab dosing was continued for a maximum of 24 months; however, treatment with LENVIMA could be continued beyond 24 months. Assessment of tumor status was performed at baseline and then every 8 weeks.
The study population characteristics were: median age of 62 years (range: 29 to 88 years); 42% age 65 or older, 75% male; 74% White, 21% Asian, 1% Black, and 2% other races; 18% and 82% of patients had a baseline KPS of 70 to 80 and 90 to 100, respectively; patient distribution by MSKCC risk categories was 27% favorable, 64% intermediate and 9% poor. Common sites of metastases in patients were lung (68%), lymph node (45%), and bone (25%).
The major efficacy outcome measures were PFS, as assessed by independent radiologic review (IRC) according to RECIST v1.1, and OS. Additional efficacy outcome measures included confirmed ORR as assessed by IRC. At the protocol-specified interim analysis, LENVIMA in combination with pembrolizumab demonstrated statistically significant improvements in PFS, OS, and ORR compared with sunitinib. An updated OS analysis was conducted when approximately 304 deaths were observed based on the planned number of deaths for the pre-specified final analysis. Table 15 and Figures 2 and 3 summarize the efficacy results for CLEAR.
Table 15: Efficacy Results in Renal Cell Carcinoma Per IRC in CLEAR LENVIMA20 mg with Pembrolizumab 200mg N=355 Sunitinib 50mg N=357 Progression-Free Survival (PFS) Number of events, n (%) 160 (45%) 205 (57%) Progressive disease 145 (41%) 196 (55%) Death 15 (4%) 9 (3%) Median PFS in months (95% CI) 23.9 (20.8, 27.7) 9.2 (6.0, 11.0) Hazard Ratioa (95% CI) 0.39 (0.32, 0.49) p-valueb <0.0001 Overall Survival (OS) Number of deaths, n (%) 80 (23%) 101 (28%) Median OS in months (95% CI) NR (33.6, NE) NR (NE, NE) Hazard Ratioa (95% CI) 0.66 (0.49, 0.88) p-valueb 0.0049 Updated OS Number of deaths, n (%) 149 (42%) 159 (45%) Median OS in months (95% CI) 53.7 (48.7, NE) 54.3 (40.9, NE) Hazard Ratioa (95% CI) 0.79 (0.63, 0.99) Objective Response Rate (Confirmed) Objective response rate, n (%) 252 (71%) 129 (36%) (95% CI) (66, 76) (31, 41) Complete response rate 16% 4% Partial responses rate 55% 32% p-valuec <0.0001 Tumor assessments were based on RECIST 1.1; only confirmed responses are included for ORR.Data cutoff date = 28 Aug 2020, Updated OS cutoff date = 31 July 2022CI = confidence interval; NE= Not estimable; NR= Not reached a. Hazard ratio is based on a Cox Proportional Hazards Model, stratified by geographic region and MSKCC prognostic groups. b. Two-sided p-value based on stratified log-rank test. c. Two-sided p-value based upon CMH test.
Figure 2: Kaplan-Meier Curves for Progression-Free Survival in CLEAR
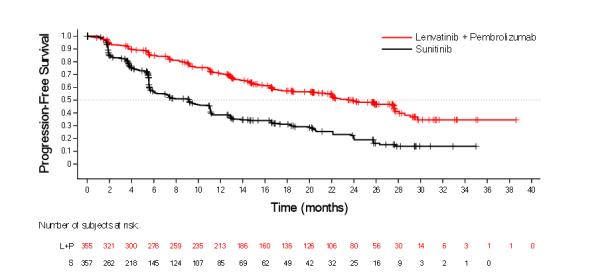
L + P = LENVIMA + Pembrolizumab; S = Sunitinib.
Figure 3: Kaplan-Meier Curves for Updated Overall Survival in CLEAR
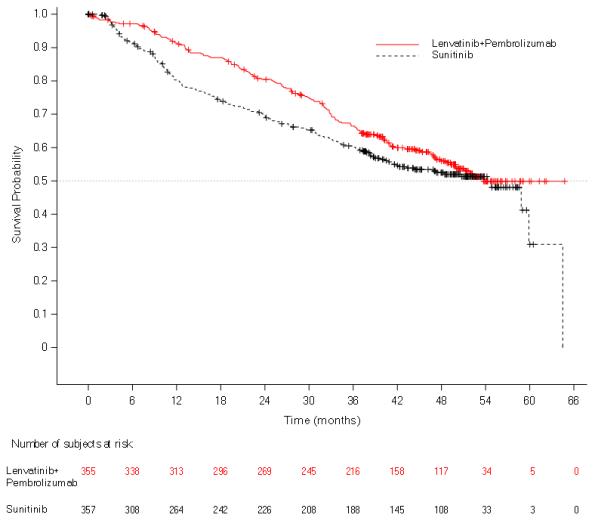
L + P = LENVIMA + Pembrolizumab; S = Sunitinib.
KEYNOTE-B61
The efficacy of LENVIMA in combination with pembrolizumab was investigated in a multicenter, single-arm study, KEYNOTE-B61 (NCT04704219), that enrolled 160 patients with advanced/metastatic non-clear cell RCC in the first-line setting. Patients with active autoimmune disease or a medical condition that required immunosuppression were ineligible.
Patients received LENVIMA 20 mg orally once daily in combination with pembrolizumab 400 mg every 6 weeks for up to 24 months or until unacceptable toxicity or disease progression. LENVIMA could be continued as a single agent beyond 24 months until unacceptable toxicity or disease progression. Administration of LENVIMA with pembrolizumab was permitted beyond RECIST-defined disease progression if the patient was considered by the investigator to be deriving clinical benefit.
Among the 158 treated patients, the baseline characteristics were: median age of 60 years (range: 24 to 87 years); 71% male; 86% White, 8% Asian, and 3% Black; <1% Hispanic or Latino; 22% and 78% of patients had a baseline KPS of 70 to 80 and 90 to 100, respectively; histologic subtypes were 59% papillary, 18% chromophobe, 4% translocation, <1% medullary, 13% unclassified, and 6% other; patient distribution by IMDC risk categories was 35% favorable, 54% intermediate, and 10% poor. Common sites of metastases in patients were lymph node (65%), lung (35%), bone (30%), and liver (21%).
The major efficacy outcome measure was ORR as assessed by BICR using RECIST 1.1. Additional efficacy outcome measures included DOR as assessed by BICR using RECIST 1.1. Efficacy results are summarized in Table 16.
Table 16: Efficacy Results in Study ( NCT04704219)
Endpoint LENVIMA 20 mg and pembrolizumab 400 mg every 6 weeks n=158 Objective Response Rate (Confirmed) ORR, (95% CI) 51% (43, 59) Complete response 8% Partial response 42% Duration of Response* Median in months (range) 19.5 (1.5+, 23.5+) CI = confidence interval * Based on Kaplan-Meier estimates+ Denotes ongoing response
Previously T reated RCC in Combination w ith E verolimus (Study 205)
The efficacy was evaluated in a multicenter, randomized (1:1:1) study (Study 205: NCT01136733), in which 153 patients with advanced or metastatic renal cell carcinoma who have previously received anti-angiogenic therapy received LENVIMA 18 mg orally once daily with everolimus 5 mg orally once daily, LENVIMA 24 mg orally once daily, or everolimus 10 mg orally once daily. Patients were required to have histological confirmation of clear cell RCC and ECOG PS of 0 or 1. Patients were stratified by hemoglobin level (≤ or >13 g/dL for males and ≤ or >11.5 g/dL for females) and corrected serum calcium (≥10 mg/dL vs. <10 mg/dL). The major efficacy outcome measure was investigator-assessed PFS evaluated according to RECIST 1.1.
Of the 101 patients randomized to the LENVIMA with everolimus arm or everolimus arm, 72% were male, the median age was 60 years, 31% were older than 65 years, and 96% were White. Metastases were present in 95% of the patients and unresectable advanced disease was present in 5%. All patients had a baseline ECOG PS of either 0 (54%) or 1 (46%) with similar distribution across these two treatment arms. MSKCC favorable, intermediate, and poor risk categories were observed, respectively, in 24%, 37%, and 39% of patients in the LENVIMA with everolimus arm and 24%, 38%, and 38% of patients in the everolimus arm.
Efficacy results from Study 205 are summarized in Table 17 and Figures 4 and 5. The treatment effect of the combination on PFS was supported by a retrospective independent review of radiographs with an observed hazard ratio (HR) of 0.43 (95% CI: 0.24, 0.75) compared with the everolimus arm.
Table 1 7 : Efficacy Results in Renal Cell Carcinoma Per Investigator Assessment in Study 2 05 LENVIMA 18 mg with Everolimus 5 mg N=51 Everolimus 10 mg N=50 Progression-Free Survival (PFS) a Number of events, n (%) 26 (51) 37 (74) Progressive disease 21 (41) 35 (70) Death 5 (10) 2 (4) Median PFS in months (95% CI) 14.6 (5.9, 20.1) 5.5 (3.5, 7.1) Hazard Ratio (95% CI)b LENVIMA with Everolimus vs Everolimus 0.37 (0.22, 0.62) - Overall Survival (OS) c Number of deaths, n (%) 32 (63) 37 (74) Median OS in months (95% CI) 25.5 (16.4, 32.1) 15.4 (11.8, 20.6) Hazard Ratio (95% CI)b LENVIMA with Everolimus vs Everolimus 0.67 (0.42, 1.08) - Objective Response Rate (Confirmed) Objective response rate, n (%) 19 (37) 3 (6) (95% CI) (24, 52) (1, 17) Number of complete responses, n (%) 1 (2) 0 Number of partial responses (%) 18 (35) 3 (6) Tumor assessments were based on RECIST v1.1 criteria for progression but only confirmed responses are included for ORR. Data cutoff date = 13 Jun 2014CI = confidence interval a. Point estimates are based on Kaplan-Meier method and 95% CIs are based on the Greenwood formula using log-log transformation. b. Hazard ratio is based on a stratified Cox regression model including treatment as a covariate factor and hemoglobin and corrected serum calcium as strata. c. Data cutoff date = 31 Jul 2015
Figure 4 : Kaplan-Meier Curves for Progression-Free Survival
in Study 2 05
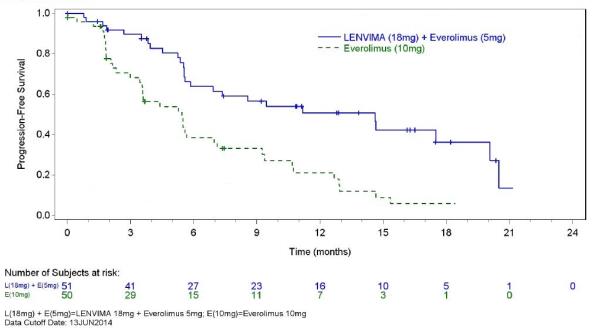
Figure 5 : Kaplan-Meier Curves for Overall Survival in Study 2 05
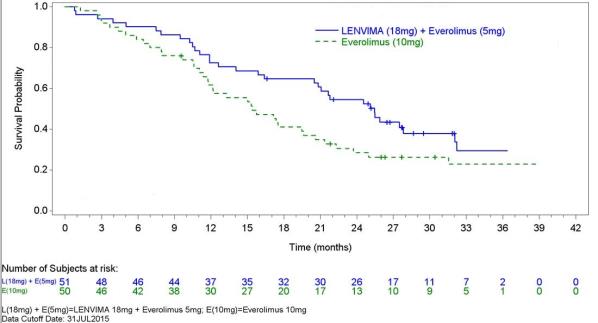
14.3
The efficacy of LENVIMA was evaluated in a randomized, open-label, multicenter, international study (REFLECT; NCT01761266) conducted in patients with previously untreated unresectable hepatocellular carcinoma (HCC). The study enrolled adults with Child-Pugh A and Barcelona Clinic Liver Cancer (BCLC) Stage C or B HCC who were ineligible for local liver-directed therapy; had an ECOG PS of 0 or 1; had received no prior systemic therapy for HCC; and had at least one measurable target lesion according to modified RECIST for HCC.
Patients were randomized (1:1) to receive LENVIMA (12 mg for baseline body weight ≥60 kg or 8 mg for baseline body weight <60 kg) orally once daily or sorafenib 400 mg orally twice daily until radiological disease progression or unacceptable toxicity. Randomization was stratified by region (Western vs Asia Pacific), presence of macroscopic portal vein invasion or extrahepatic spread (yes vs no), ECOG PS (0 vs 1), and body weight (<60 kg vs ≥60 kg). The major efficacy outcome measure was overall survival (OS). REFLECT was designed to show the non-inferiority of LENVIMA to sorafenib for OS. Additional efficacy outcome measures were progression-free survival (PFS) and objective response rate (ORR) according to modified RECIST for HCC.
A total of 954 patients were randomized, 478 to the LENVIMA arm and 476 to the sorafenib arm. The demographics of the study population were: median age of 62 years (range: 20 to 88 years); 84% male; 69% Asian and 29% White; 63% ECOG PS of 0; and 69% weighed ≥60 kg. Of the 590 (62%) patients with at least one site of documented distant metastatic disease, 52% had lung metastasis, 45% had lymph node metastasis, and 16% had bone metastasis.
Macroscopic portal vein invasion, extra-hepatic spread, or both were present in 70% of patients. HCC was categorized as Child-Pugh A and BCLC Stage C in 79% and Child-Pugh A and BCLC Stage B in 21% of patients. Seventy-five percent (75%) of patients had radiographic evidence of cirrhosis at baseline. Investigator-documented primary risk factors for the development of HCC were hepatitis B (50%), hepatitis C (23%), alcohol use (6%), other (7%), and unknown (14%).
REFLECT demonstrated that LENVIMA was non-inferior to sorafenib for OS. REFLECT did not demonstrate a statistically significant improvement in OS for patients randomized to LENVIMA as compared to those in the sorafenib arm. LENVIMA was statistically significantly superior to sorafenib for PFS and ORR. Efficacy results are summarized in Table 18 and Figure 6.
Table 1 8 : Efficacy Results in Hepatocellular Carcinoma in REFLECT LENVIMA N= 47 8 Sorafenib N=47 6 Overall Survival Number of deaths (%) 351 (73) 350 (74) Median OS in months (95% CI) 13.6 (12.1, 14.9) 12.3 (10.4, 13.9) Hazard Ratio (95% CI)a 0.92 (0.79, 1.06) Progression-Free Survival b ( mRECIST) Number of Events (%) 311 (65) 323 (68) Median PFS in months (95% CI) 7.3 (5.6, 7.5) 3.6 (3.6, 3.7) Hazard Ratio (95% CI) 0.64 (0.55, 0.75) p-value <0.001 Objective Response Rate b (mRECIST) Objective response rate 41% 12% Complete responses, n (%) 10 (2.1) 4 (0.8) Partial responses, n (%) 184 (38.5) 55 (11.6) 95% CI (36%, 45%) (10%, 16%) p-value <0.001 Progression-Free Survival b (RECIST 1.1) Number of Events (%) 307 (64) 320 (67) Median PFS in months (95% CI) 7.3 (5.6, 7.5) 3.6 (3.6, 3.9) Hazard Ratio (95% CI) 0.65 (0.56, 0.77) Objective Response Rate b (RECIST 1.1) Objective response rate 19% 7% Complete responses, n (%) 2 (0.4) 1 (0.2) Partial responses, n (%) 88 (18.4) 30 (6.3) 95% CI (15%, 22%) (4%, 9%) CI = confidence interval; ECOG PS = Eastern Cooperative Oncology Group Performance Status; HR = hazard ratio; OS = overall survival. a. Based on stratified Cox-model. Non-inferiority margin for HR (LENVIMA vs sorafenib) is 1.08. b. Per independent radiology review.
Figure 6 : Kaplan-Meier Curves for Overall Survival in REFLECT
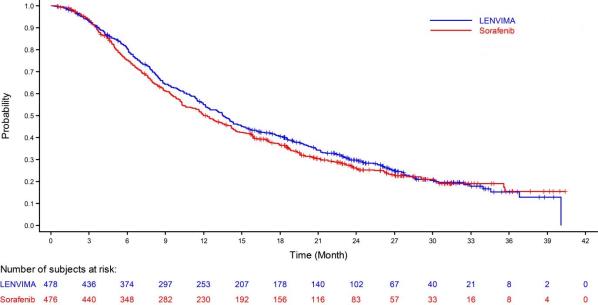
14.4
The efficacy of LENVIMA in combination with pembrolizumab was investigated in Study 309 (NCT03517449), a multicenter, open-label, randomized, active-controlled trial that enrolled 827 patients with advanced endometrial carcinoma who had been previously treated with at least one prior platinum-based chemotherapy regimen in any setting, including in the neoadjuvant and adjuvant settings. Patients with endometrial sarcoma, including carcinosarcoma, or patients who had active autoimmune disease or a medical condition that required immunosuppression were ineligible. Patients with endometrial carcinoma that were pMMR (using the VENTANA MMR RxDx Panel test) or not MSI-H were stratified by ECOG performance status, geographic region, and history of pelvic radiation. Patients were randomized (1:1) to one of the following treatment arms:
- LENVIMA 20 mg orally once daily in combination with pembrolizumab 200 mg intravenously every 3 weeks.
- Investigator’s choice consisting of either doxorubicin 60 mg/m2 every 3 weeks, or paclitaxel 80 mg/m2 given weekly, 3 weeks on/1 week off.
Treatment with LENVIMA and pembrolizumab continued until RECIST v1.1-defined progression of disease as verified by BICR, unacceptable toxicity, or for pembrolizumab, a maximum of 24 months. Treatment was permitted beyond RECIST v1.1-defined disease progression if the treating investigator considered the patient to be deriving clinical benefit and the treatment was tolerated. Assessment of tumor status was performed every 8 weeks. The major efficacy outcome measures were OS and PFS as assessed by BICR according to RECIST v1.1, modified to follow a maximum of 10 target lesions and a maximum of 5 target lesions per organ. Additional efficacy outcome measures included ORR and DOR, as assessed by BICR.
Among the 697 pMMR or not MSI-H patients, 346 patients were randomized to LENVIMA in combination with pembrolizumab, and 351 patients were randomized to investigator’s choice of doxorubicin (n=254) or paclitaxel (n=97). The population characteristics of these patients were: median age of 65 years (range: 30 to 86), 52% age 65 or older; 62% White, 22% Asian, and 3% Black; 60% ECOG PS of 0 and 40% ECOG PS of 1. The histologic subtypes were endometrioid carcinoma (55%), serous (30%), clear cell carcinoma (7%), mixed (4%), and other (3%). All 697 of these patients received prior systemic therapy for endometrial carcinoma: 67% had one, 30% had two, and 3% had three or more prior systemic therapies. Thirty-seven percent of patients received only prior neoadjuvant or adjuvant therapy.
Efficacy results for these patients are summarized in Table 19 and Figures 7 and 8.
Table 1 9 : Efficacy Results in Endometrial Carcinoma in Study 309 Endometrial Carcinoma ( pMMR or not MSI-H) Endpoint LENVIMA with pembrolizumab N=346 Doxorubicin or Paclitaxel N=351 OS Number (%) of patients with event 165 (48%) 203 (58%) Median in months (95% CI) 17.4 (14.2, 19.9) 12.0 (10.8, 13.3) Hazard ratioa (95% CI) 0.68 (0.56, 0.84) p-valueb 0.0001 PFS c Number (%) of patients with event 247 (71%) 238 (68%) Median in months (95% CI) 6.6 (5.6, 7.4) 3.8 (3.6, 5.0) Hazard ratioa (95% CI) 0.60 (0.50, 0.72) p-valueb <0.0001 Objective Response Rate ORRc (95% CI) 30% (26, 36) 15% (12,19) Complete response 5% 3% Partial response 25% 13% p-valued <0.0001 Duration of Response N=105 N=53 Median in months (range) 9.2 (1.6+, 23.7+) 5.7 (0.0+, 24.2+) a Based on the stratified Cox regression model b Based on stratified log-rank test c Per independent radiology review d Based on Miettinen and Nurminen method stratified by ECOG performance status, geographic region, and history of pelvic radiation
Figure 7 : Kaplan-Meier Curves for Overall Survival in Study 309
( pMMR or not MSI-H)
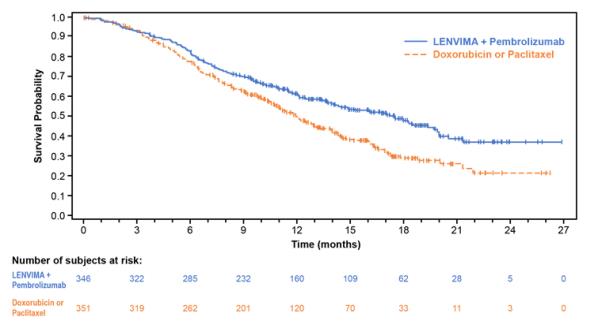
Figure 8 : Kaplan-Meier Curves for Progression-Free Survival in Study 309
( pMMR or not MSI-H)
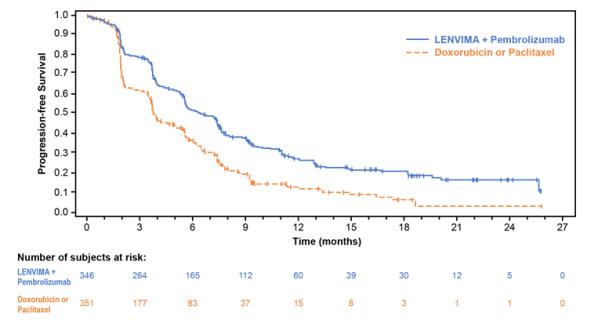
How Supplied Section
LENVIMA 4 mg capsules are supplied as hard hypromellose capsules with yellowish-red body and yellowish-red cap, marked in black ink with “Є” on the cap and “LENV 4 mg” on the body.
LENVIMA 10 mg capsules are supplied as hard hypromellose capsules with yellow body and yellowish-red cap, marked in black ink with “Є” on the cap and “LENV 10 mg” on the body.
LENVIMA capsules are supplied in cartons of 6 cards. Each card is a 5-day buler card as follows:
- NDC 62856-724-30: 24 mg, carton with 6 cards NDC 62856-724-05 (ten 10 mg capsules and five 4 mg capsules per card).
- NDC 62856-720-30: 20 mg, carton with 6 cards NDC 62856-720-05 (ten 10 mg capsules per card).
- NDC 62856-718-30: 18 mg, carton with 6 cards NDC 62856-718-05 (five 10 mg capsules and ten 4 mg capsules per card).
- NDC 62856-714-30: 14 mg, carton with 6 cards NDC 62856-714-05 (five 10 mg capsules and five 4 mg capsules per card).
- NDC 62856-712-30: 12 mg, carton with 6 cards NDC 62856-712-05 (fifteen 4 mg capsules per card).
- NDC 62856-710-30: 10 mg, carton with 6 cards NDC 62856-710-05 (five 10 mg capsules per card).
- NDC 62856-708-30: 8 mg, carton with 6 cards NDC 62856-708-05 (ten 4 mg capsules per card).
- NDC 62856-704-30: 4 mg, carton with 6 cards NDC 62856-704-05 (five 4 mg capsules per card).
Store at 20°C to 25°C (68°F to 77°F); excursions permitted to 15°C to 30°C (59°F to 86°F) [see USP Controlled Room Temperature].
Information For Patients Section
Advise the patient to read the FDA-approved patient labeling (Patient Information).
Hypertension
Advise patients to undergo regular blood pressure monitoring and to contact their health care provider if blood pressure is elevated [see Warnings and Precautions ( 5.1 )] .
Cardiac Dysfunction
Advise patients that LENVIMA can cause cardiac dysfunction and to immediately contact their healthcare provider if they experience any clinical symptoms of cardiac dysfunction [see Warnings and Precautions ( 5.2 )].
Arterial Thrombotic Events
Advise patients to seek immediate medical attention for new onset chest pain or acute neurologic symptoms consistent with myocardial infarction or stroke [see Warnings and Precautions ( 5.3 )].
Hepatotoxicity
Advise patients that they will need to undergo laboratory tests to monitor liver function and to report any new symptoms indicating hepatic toxicity or failure [see Warnings and Precautions ( 5.4 )].
Proteinuria and Renal Failure/Impairment
Advise patients that they will need to undergo regular laboratory tests to monitor kidney function and protein in the urine [see Warnings and Precautions ( 5.5 , 5.6 )].
Diarrhea
Advise patients when to start standard anti-diarrheal therapy and to maintain adequate hydration. Advise patients to contact their healthcare provider if they are unable to maintain adequate hydration [see Warnings and Precautions ( 5.7 )].
F istula F ormation and Gastrointestinal Perforation
Advise patients that LENVIMA can increase the risk of fistula formation or gastrointestinal perforation and to seek immediate medical attention for severe abdominal pain [see Warnings and Precautions ( 5.8 )].
QT c Interval Prolongation
Advise patients who are at risk for QTc prolongation that they will need to undergo regular ECGs. Advise all patients that they will need to undergo laboratory tests to monitor electrolytes [see Warnings and Precautions ( 5.9 )].
Hypocalcemia
Advise patients of the risks of hypocalcemia, that they will need to undergo laboratory tests to monitor calcium levels, and the potential requirement for calcium supplementation [see Warnings and Precautions ( 5.10 )] .
Reversible P osterior L eukoencephalopathy S yndrome (RPLS)
Advise patients of the signs and symptoms of RPLS and to contact their healthcare provider for new onset or worsening neurological function [see Warnings and Precautions ( 5.11 )] .
Hemorrhagic Events
Advise patients that LENVIMA can increase the risk for bleeding and to contact their healthcare provider for bleeding or symptoms of severe bleeding [see Warnings and Precautions ( 5.12 )] .
Impairment of Thyroid Stimulating Hormone Suppression/Thyroid Dysfunction
Advise patients that LENVIMA can cause hypothyroidism and that their thyroid function should be monitored regularly during treatment [see Warnings and Precautions ( 5.13 )].
Impaired Wound Healing
Advise patients that LENVIMA may impair wound healing. Advise patients to inform their healthcare provider of any planned surgical procedure [see Warnings and Precautions ( 5.14 )].
Osteonecrosis of the Jaw (ONJ)
Advise patients regarding good oral hygiene practices and to have preventive dentistry performed prior to treatment with LENVIMA and throughout treatment with LENVIMA. Inform patients being treated with LENVIMA, particularly those who are at high risk for ONJ, to avoid invasive dental procedures, if possible, and to inform their healthcare provider of any planned dental procedures [see Warnings and Precautions ( 5.15 )]. Advise patients to immediately contact their healthcare provider for signs or symptoms associated with ONJ.
Embryo - F etal Toxicity
Advise females of reproductive potential of the potential risk to a fetus and to inform their healthcare provider of a known or suspected pregnancy [see Warnings and Precautions ( 5.16 ), Use in Specific Populations ( 8.1 )] .
Advise females of reproductive potential to use effective contraception during treatment with LENVIMA and for 30 days after the last dose [see Use in Specific Populations ( 8.3 )].
Lactation
Advise women to discontinue breastfeeding during treatment with LENVIMA and for 1 week after the last dose [see Use in Specific Populations ( 8.2 )].
Distributed by:
Eisai Inc.
Nutley, NJ 07110
LENVIMA® is a registered trademark of Eisai R&D Management Co., Ltd. and is licensed to Eisai Inc.
© 2015-2024 Eisai Inc.
Spl Patient Package Insert Section
PATIENT INFORMATION LENVIMA ® (lehn-veema) (lenvatinib) capsules What is LENVIMA? LENVIMA is a prescription medicine that is used to treat people with certain kinds of cancer. The safety and efficacy of LENVIMA have not been established in children.
- LENVIMA is used by itself to treat differentiated thyroid cancer (DTC), a type of thyroid cancer that can no longer be treated with radioactive iodine and is progressing.
- LENVIMA is used to treat adults with a type of kidney cancer called advanced renal cell carcinoma (RCC): ○ along with the medicine pembrolizumab as your first treatment when your kidney cancer has spread or cannot be removed by surgery.○ along with the medicine everolimus after one course of treatment with another anti-cancer medicine (anti-angiogenic therapy).
- LENVIMA is used by itself as the first treatment for a type of liver cancer called hepatocellular carcinoma (HCC) when it cannot be removed by surgery.
- LENVIMA is used along with another medicine called pembrolizumab to treat advanced endometrial carcinoma (EC), a type of uterine cancer:○ when a laboratory test shows that your tumor is mismatch repair proficient (pMMR) or not microsatellite instability-high (MSI-H), and ○ you have received anti-cancer treatment, and it is no longer working, and ○ your cancer cannot be cured by surgery or radiation.
Before you take LENVIMA, tell your healthcare provider about all of your medical conditions, including if you: Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. Especially tell your healthcare provider if you are taking, or have taken, an osteoporosis medicine.Know the medicines you take. Keep a ul of your medicines to show to your healthcare provider and pharmacist when you get a new medicine.
- have high blood pressure
- have heart problems
- have a history of blood clots in your arteries (type of blood vessel), including stroke, heart attack, or change in vision
- have or have had liver or kidney problems
- have a history of a tear (perforation) in your stomach or intestine, or an abnormal connection between two or more body parts (fistula)
- have headaches, seizures, or vision problems
- have any bleeding problems
- plan to have surgery, a dental procedure, or have had a recent surgery. You should stop taking LENVIMA at least 1 week before planned surgery. See “What are the possible side effects of LENVIMA?”
- are pregnant or plan to become pregnant. LENVIMA can harm your unborn baby. Females who are able to become pregnant : ○ Your healthcare provider should do a pregnancy test before you start treatment with LENVIMA. ○ You should use an effective method of birth control (contraception) during treatment with LENVIMA and for 30 days after the last dose of LENVIMA. Talk with your healthcare provider about birth control methods you can use during this time. Tell your healthcare provider right away if you become pregnant or think you are pregnant during treatment with LENVIMA.
- are breastfeeding or plan to breastfeed. It is not known if LENVIMA passes into your breast milk. Do not breastfeed during treatment with LENVIMA and for 1 week after the last dose.
How should I take LENVIMA? How to take LENVIMA by mouth if you cannot swallow whole capsules:
- Take LENVIMA exactly as your healthcare provider tells you to take it.
- Your healthcare provider will tell you how much LENVIMA to take and when to take it. Your healthcare provider may change your dose during treatment, stop treatment for some time, or completely stop treatment with LENVIMA if you have side effects.
- Take LENVIMA 1 time each day at the same time, with or without food.
- If you miss a dose of LENVIMA, take it as soon as you remember. If your next dose is due within 12 hours, skip the missed dose and take the next dose at your regular time.
- Swallow LENVIMA capsules whole. Do not crush or chew the LENVIMA capsules.
- If you cannot swallow LENVIMA capsules whole, LENVIMA capsules can be mixed with water or apple juice,then taken by mouth, or mixed with water and given through a feeding tube.
- If you take too much LENVIMA, call your healthcare provider or go to the nearest hospital emergency room right away.
How to give LENVIMA through a feeding tube:
- Place your daily dose, up to 5 capsules, in a small container or oral syringe (approximately 20 mL capacity).
- Add 3 mL of water or apple juice to the container or oral syringe. Wait 10 minutes for the capsule shell (outer surface) to dissolve completely, then stir or shake the mixture for 3 minutes until capsules are fully dissolved. Do not break or crush the capsules.
- Drink the liquid mixture or use an oral syringe to take directly into the mouth.
- Next, using a second syringe, add an additional 2 mL of liquid to the container or oral syringe (cap the first oral syringe before adding the additional water) then swirl or shake and take the liquid mixture. Repeat this step at least one time and until you cannot see any of the LENVIMA mixture left in the container or oral syringe to make sure all of the medicine is taken.
- If 6 capsules are required for your daily dose, follow the above instructions using 3 capsules at a time.
- LENVIMA should be given in feeding tubes of at least 5 French diameter (polyvinyl chloride or polyurethane tube) and at least 6 French diameter (silicone tube).○ Place your daily dose, up to 5 capsules, in a syringe (20 mL capacity).○ Add 3 mL of water to the syringe. Wait 10 minutes for the capsule shell (outer surface) to dissolve completely, then stir or shake the mixture for 3 minutes until capsules are fully dissolved. Do not break or crush the capsules.○ Give the mixture through a feeding tube. ○ Next, cap the syringe and remove the plunger. Use a second syringe and add an additional 2 mL of liquid to the syringe. Swirl or shake and give the mixture in the feeding tube. Repeat this step at least one time and until you cannot see any of the LENVIMA mixture left in the syringe to make sure all of the medicine is taken. ○ If 6 capsules are required for your daily dose, follow the above instructions using 3 capsules at a time.
- LENVIMA mixture may be stored in a covered container in the refrigerator at 36ºF to 46ºF (2ºC to 8ºC) for a maximum of 24 hours. Throw away the LENVIMA mixture if not used within 24 hours of mixing.
What are the possible side effects of LENVIMA? LENVIMA may cause serious side effects, including:
- H igh blood pressure (hypertension). High blood pressure is a common side effect of LENVIMA and can be serious. Your blood pressure should be well controlled before you start taking LENVIMA. Your healthcare provider should check your blood pressure regularly during treatment with LENVIMA. If you develop blood pressure problems, your healthcare provider may prescribe medicine to treat your high blood pressure.
- H eart problems. LENVIMA can cause serious heart problems that may lead to death. Call your healthcare provider right away if you get symptoms of heart problems, such as shortness of breath or swelling of your ankles.
- P roblem with blood clots in your blood vessels (arteries). Get emergency medical help right away if you get any of the following symptoms:
- severe chest pain or pressure
- pain in your arms, back, neck or jaw
- shortness of breath
- numbness or weakness on one side of your body
- trouble talking
- sudden severe headache
- sudden vision changes
- L iver problems. LENVIMA may cause liver problems that may lead to liver failure and death. Your healthcare provider will check your liver function before and during treatment with LENVIMA. Tell your healthcare provider right away if you develop any of the following symptoms:○ your skin or the white part of your eyes turns yellow (jaundice) ○ dark “tea colored” urine○ light-colored bowel movements (stools)○ feeling drowsy, confused or loss of consciousness
- K idney problems. Kidney failure, which can lead to death, has happened with LENVIMA treatment. Your healthcare provider should do regular blood tests to check your kidneys.
- I ncreased protein in your urine (proteinuria). Proteinuria is a common side effect of LENVIMA and can be serious. Your healthcare provider should check your urine for protein before and during your treatment with LENVIMA.
- D iarrhea. Diarrhea is a common side effect of LENVIMA and can be serious. If you get diarrhea, ask your healthcare provider about what medicines you can take to treat your diarrhea. It is important to drink more water when you get diarrhea. Tell your healthcare provider or go to the emergency room if you are unable to drink enough liquids and your diarrhea is not able to be controlled.
- A n opening in the wall of your stomach or intestines (perforation) or an abnormal connection between two or more body parts (fistula). Get emergency medical help right away if you develop severe stomach (abdomen) pain.
- C hanges in the electrical activity of your heart called QT prolongation. QT prolongation can cause irregular heartbeats that can be life threatening. Your healthcare provider will do blood tests before and during your treatment with LENVIMA to check the levels of potassium, magnesium, and calcium in your blood, and may check the electrical activity of your heart with an electrocardiogram (ECG).
- L ow levels of blood calcium (hypocalcemia). Your healthcare provider will check your blood calcium levels during treatment with LENVIMA and may tell you to take a calcium supplement if your calcium levels are low.
- A condition called Reversible Posterior Leukoencephalopathy Syndrome (RPLS). Call your healthcare provider right away if you get severe headache, seizures, weakness, confusion, or blindness or change in vision.
- B leeding. LENVIMA may cause serious bleeding problems that may lead to death. Tell your healthcare provider if you develop any signs or symptoms of bleeding during treatment with LENVIMA, including:
- severe and persistent nose bleeds
- vomiting blood
- red or black (looks like tar) stools
- blood in your urine
- coughing up blood or blood clots
- heavy or new onset vaginal bleeding
- C hange in thyroid hormone levels. Your healthcare provider should check your thyroid hormone levels before starting and every month during treatment with LENVIMA.
- W ound healing problems. Wound healing problems have happened in some people who take LENVIMA. Tell your healthcare provider if you plan to have any surgery before or during treatment with LENVIMA. ○ You should stop taking LENVIMA at least 1 week before planned surgery. ○ Your healthcare provider should tell you when you may start taking LENVIMA again after surgery.
- S evere jawbone problems (osteonecrosis). Severe jawbone problems have happened in some people who take LENVIMA. Certain risk factors such as taking a bisphosphonate medicine or the medicine denosumab, having dental disease, or an invasive dental procedure may increase your risk of getting jawbone problems. Your healthcare provider should examine your mouth before you start and during treatment with LENVIMA. Tell your dentist that you are taking LENVIMA. It is important for you to practice good mouth care during treatment with LENVIMA. Tell your healthcare provider right away if you get signs or symptoms of jawbone problems during treatment with LENVIMA, including jaw pain, toothache, or sores on your gums. Tell your healthcare provider if you plan to have any dental procedures before or during treatment with LENVIMA. You should avoid having invasive dental procedures if possible, during treatment with LENVIMA. Stopping your bisphosphonate medicine before an invasive dental procedure may help decrease your risk of getting these jaw problems.○ You should stop taking LENVIMA at least 1 week before planned dental surgery or invasive dental procedures.○ Your healthcare provider should tell you when you may start taking LENVIMA again after dental procedures.
The most common side effects of LENVIMA in people treated for thyroid cancer include:
- tiredness
- joint and muscle pain
- decreased appetite
- weight loss
- nausea
- mouth sores
- headache
- vomiting
- rash, redness, itching, or peeling of your skin on your hands and feet
- stomach (abdomen) pain
- hoarseness
The most common side effects of LENVIMA in people treated for kidney cancer in combination with everolimus include:
- tiredness
- joint and muscle pain
- decreased appetite
- vomiting
- nausea
- mouth sores
- swelling in your arms and legs
- cough
- stomach (abdomen) pain
- trouble breathing
- rash
- weight loss
- bleeding
The most common side effects of LENVIMA in people treated for liver cancer include:
- tiredness
- decreased appetite
- joint and muscle pain
- weight loss
- stomach (abdomen) pain
- rash, redness, itching, or peeling of your skin on your hands and feet
- hoarseness
- bleeding
- decrease in thyroid hormone levels
- nausea
The most common side effects of LENVIMA when given in combination with pembrolizumab include:
- decrease in thyroid hormone levels
- tiredness
- joint and muscle pain
- nausea
- decreased appetite
- vomiting
- mouth sores
- weight loss
- stomach-area (abdomen) pain
- urinary tract infection
- constipation
- headache
- bleeding
- rash, redness, itching, or peeling of your skin on your hands and feet
- hoarseness
- rash
LENVIMA may cause fertility problems in males and females. Talk to your healthcare provider if this is a concern for you. Your healthcare provider may need to reduce your dose of LENVIMA, or delay or completely stop treatment, if you have certain side effects. These are not all the possible side effects of LENVIMA. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. H ow should I store LENVIMA?
- Store LENVIMA at room temperature, between 68°F to 77°F (20°C to 25°C).
- Keep LENVIMA and all medicines out of the reach of children.
General information about the safe and effective use of LENVIMA. Medicines are sometimes prescribed for purposes other than those uled in a Patient Information leaflet. Do not use LENVIMA for a condition for which it was not prescribed. Do not give LENVIMA to other people, even if they have the same symptoms you have. It may harm them. You can ask your healthcare provider or pharmacist for information about LENVIMA that is written for health professionals. What are the ingredients in LENVIMA? Active ingredient: lenvatinib Inactive ingredients: calcium carbonate, hydroxypropyl cellulose, low-substituted hydroxypropylcellulose, mannitol, microcrystalline cellulose, and talc. The capsule shell contains: ferric oxide red, ferric oxide yellow, hypromellose, and titanium dioxide. The printing ink contains black iron oxide, potassium hydroxide, propylene glycol and shellac.Distributed by: Eisai Inc., Nutley, NJ 07110LENVIMA® is a registered trademark of Eisai R&D Management Co., Ltd. and is licensed to Eisai Inc.For more information, call 1-877-873-4724 or go to www.LENVIMA.com.© 2015-2024 Eisai Inc.
This Patient Information has been approved by the U.S. Food and Drug Administration. Revised: 04/2024
Package Label.principal Display Panel
PRINCIPAL DISPLAY PANEL
NDC 62856-704-30Lenvima(lenvatinib) capsules4 mg daily dose
Package Label.principal Display Panel
PRINCIPAL DISPLAY PANEL
NDC 62856-708-30Lenvima(lenvatinib) capsules8 mg daily dose
Package Label.principal Display Panel
PRINCIPAL DISPLAY PANEL
NDC 62856-710-30Lenvima(lenvatinib) capsules10 mg daily dose30 day supply10 mg daily-dose carton containing 6 cards
Package Label.principal Display Panel
PRINCIPAL DISPLAY PANEL
NDC 62856-712-30Lenvima(lenvatinib) capsules12 mg daily dose
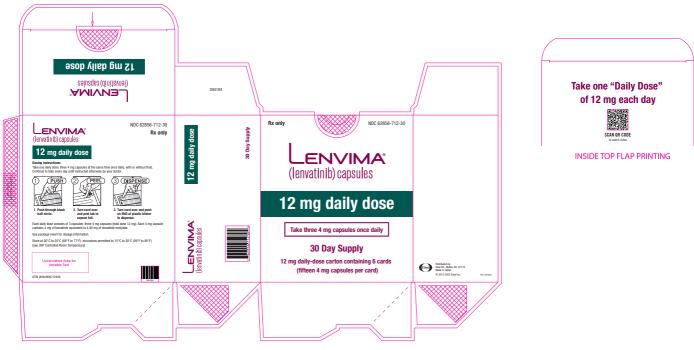
Package Label.principal Display Panel
PRINCIPAL DISPLAY PANEL
NDC 62856-714-30Lenvima(lenvatinib) capsules14 mg daily dose30 day supply14 mg daily-dose carton containing 6 cards
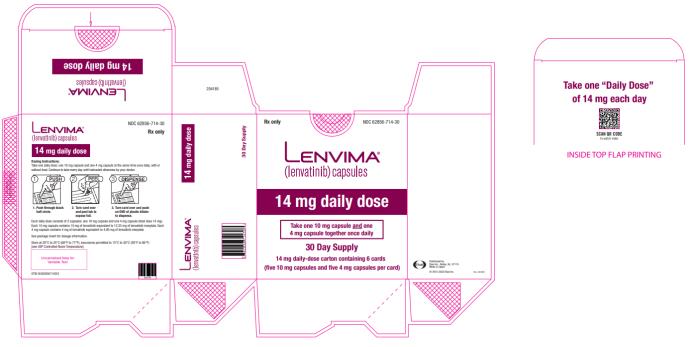
Package Label.principal Display Panel
PRINCIPAL DISPLAY PANEL
NDC 62856-718-30Lenvima(lenvatinib) capsules18 mg daily dose
30 day supply18 mg daily-dose carton containing 6 cards
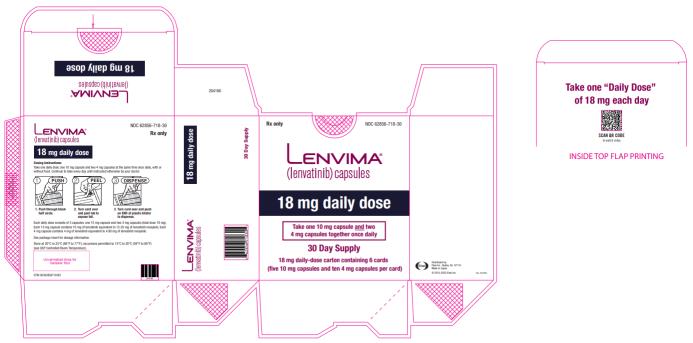
Package Label.principal Display Panel
PRINCIPAL DISPLAY PANEL
NDC 62856-720-30Lenvima(lenvatinib) capsules20 mg daily dose30 day supply20 mg daily-dose carton containing 6 cards
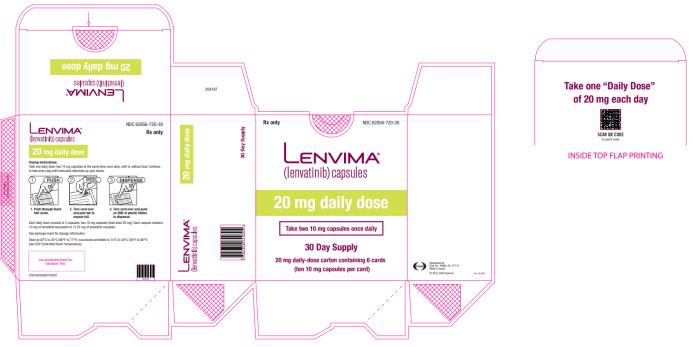
Package Label.principal Display Panel
PRINCIPAL DISPLAY PANEL
NDC 62856-724-30Lenvima(lenvatinib) capsules24 mg daily dose30 day supply24 mg daily- dose carton containing 6 cards
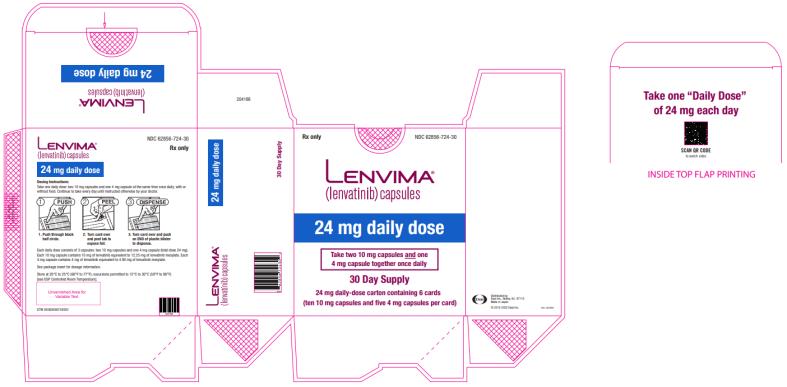
Package Label.principal Display Panel
PRINCIPAL DISPLAY PANEL
NDC 62856-704-05Lenvima(lenvatinib) capsules4 mg daily dose
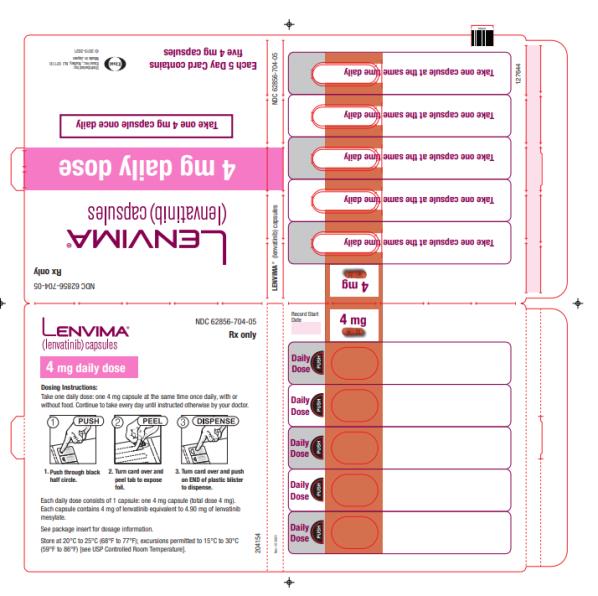
Package Label.principal Display Panel
PRINCIPAL DISPLAY PANEL
NDC 62856-708-05Lenvima(lenvatinib) capsules8 mg daily dose
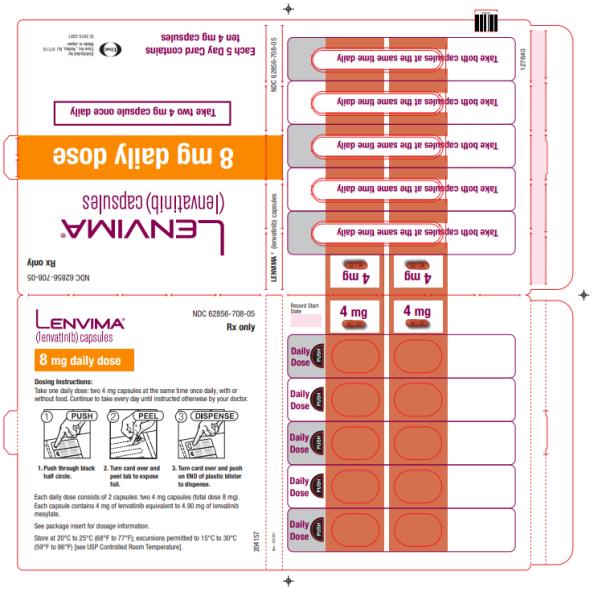
Package Label.principal Display Panel
PRINCIPAL DISPLAY PANEL
NDC 62856-710-05Lenvima(lenvatinib) capsules10 mg daily dose
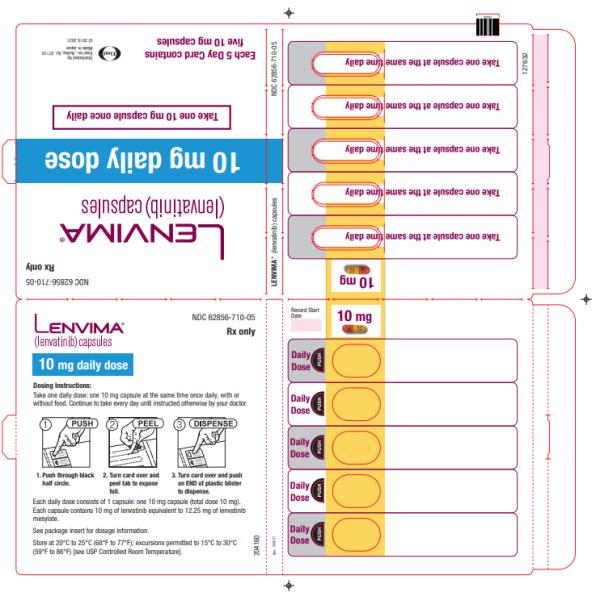
Package Label.principal Display Panel
PRINCIPAL DISPLAY PANEL
NDC 62856-712-05Lenvima(lenvatinib) capsules12 mg daily dose
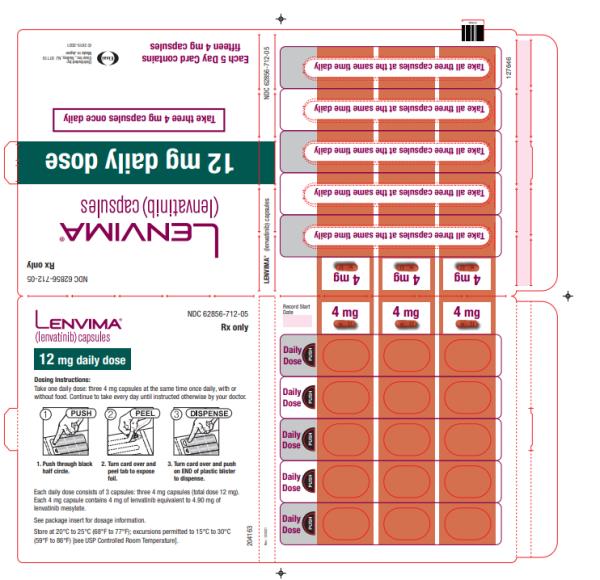
Package Label.principal Display Panel
PRINCIPAL DISPLAY PANEL
NDC 62856-714-05Lenvima(lenvatinib) capsules14 mg daily dose
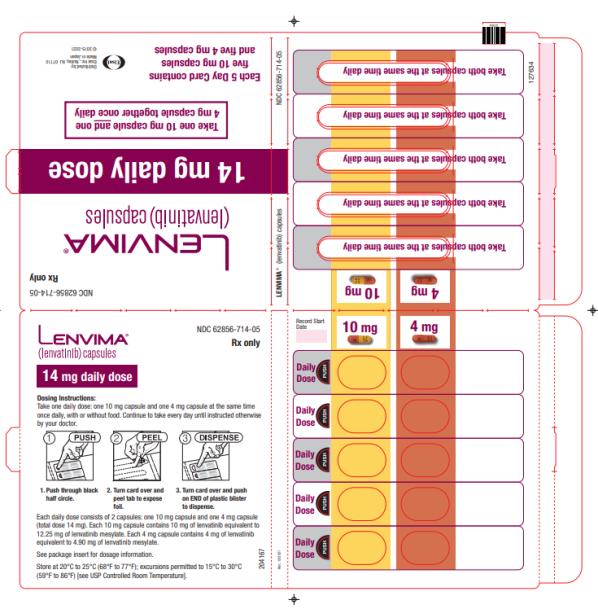
Package Label.principal Display Panel
PRINCIPAL DISPLAY PANEL
NDC 62856-718-05Lenvima(lenvatinib) capsules18 mg daily dose

Package Label.principal Display Panel
PRINCIPAL DISPLAY PANEL
NDC 62856-720-05Lenvima(lenvatinib) capsules20 mg daily dose
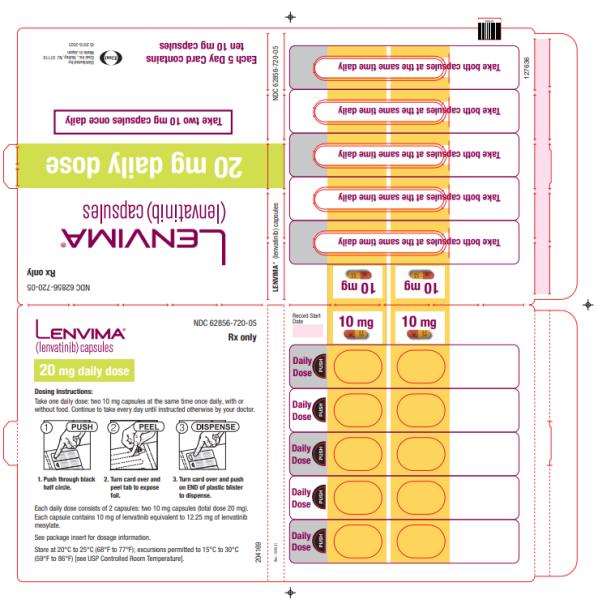
Package Label.principal Display Panel
PRINCIPAL DISPLAY PANEL
NDC 62856-724-05Lenvima(lenvatinib) capsules24 mg daily dose
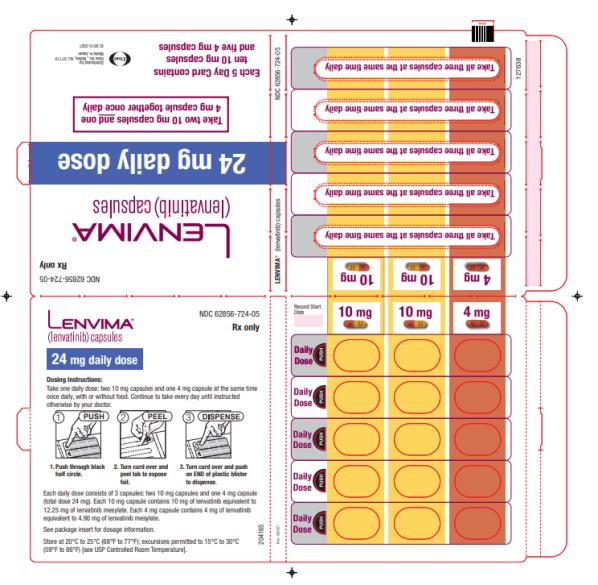
DISCLAIMER:
"This tool does not provide medical advice, and is for informational and educational purposes only, and is not a substitute for professional medical advice, treatment or diagnosis. Call your doctor to receive medical advice. If you think you may have a medical emergency, please dial 911."
"Do not rely on openFDA to make decisions regarding medical care. While we make every effort to ensure that data is accurate, you should assume all results are unvalidated. We may limit or otherwise restrict your access to the API in line with our Terms of Service."
"This product uses publicly available data from the U.S. National Library of Medicine (NLM), National Institutes of Health, Department of Health and Human Services; NLM is not responsible for the product and does not endorse or recommend this or any other product."
PillSync may earn a commission via links on our site
