SAPHRIS (asenapine maleate 5 mg) Dailymed
Generic: asenapine maleate is used for the treatment of Bipolar Disorder Schizophrenia Hepatic Insufficiency
IMPRINT: 5
SHAPE: round
COLOR: white
Boxed Warning
Warning: Increased Mortality In Elderly Patients With Dementia-related

Go PRO for all pill images
Warning: Increased Mortality In Elderly Patients With Dementia-related
Elderly patients with dementia-related psychosis treated with antipsychotic drugs are at an increased risk of death. SAPHRIS ® (asenapine) is not approved for the treatment of patients with dementia-related psychosis[see Warnings and Precautions ( 5.1,  5.2 )] .
WARNING: INCREASED MORTALITY IN ELDERLY PATIENTS WITH DEMENTIA-RELATED PSYCHOSIS
See full prescribing information for complete boxed warning.
Elderly patients with dementia-related psychosis treated with antipsychotic drugs are at an increased risk of death. SAPHRIS is not approved for the treatment of patients with dementia-related psychosis. (5.1 ,5.2 )
1
SAPHRIS is indicated for:
- Schizophrenia in adults [see Clinical Studies ( 14.1 ) ]
- Bipolar I disorder [see Clinical Studies ( 14.2 ) ] • Acute monotherapy of manic or mixed episodes, in adults and pediatric patients 10 to 17 years of age• Adjunctive treatment to lithium or valproate in adults• Maintenance monotherapy treatment in adults
SAPHRIS is an atypical antipsychotic indicated for (1 ):
- Schizophrenia in adults
- Bipolar I disorderâ—‹Â Acute monotherapy treatment of manic or mixed episodes, in adults and pediatric patients 10 to 17 years of ageâ—‹ Adjunctive treatment to lithium or valproate in adultsâ—‹ Maintenance monotherapy treatment in adults
2
Starting Dose Recommended Dose Maximum Dose Schizophrenia – acute treatment in adults ( 2.2 )5 mg sublingually twice daily 5 mg sublingually twice daily 10 mg sublingually twice daily Schizophrenia – maintenance treatment in adults ( 2.2 )5 mg sublingually twice daily 5-10 mg sublingually twice daily 10 mg sublingually twice daily Bipolar mania – adults: acute and maintenance monotherapy ( 2.3 )5-10 mg sublingually twice daily 5-10 mg sublingually twice daily 10 mg sublingually twice daily Bipolar mania –pediatric patients (10 to 17 years): monotherapy ( 2.3 )2.5 mg sublingually twice daily 2.5-10 mg sublingually twice daily 10 mg sublingually twice daily Bipolar mania – adults: as an adjunct to lithium or valproate ( 2.3 )5 mg sublingually twice daily 5-10 mg sublingually twice daily 10 mg sublingually twice daily
- Do not swallow tablet. SAPHRIS sublingual tablets should be placed under the tongue and left to dissolve completely. The tablet will dissolve in saliva within seconds. Eating and drinking should be avoided for 10 minutes after administration. (
2.1 ,17 )2.1
SAPHRIS is a sublingual tablet. To ensure optimal absorption, patients should be instructed to place the tablet under the tongue and allow it to dissolve completely. The tablet will dissolve in saliva within seconds. SAPHRIS sublingual tablets should not be split, crushed, chewed, or swallowed [see Clinical Pharmacology (12.3)]. Patients should be instructed to not eat or drink for 10 minutes after administration [see Clinical Pharmacology (12.3)].
2.2
The recommended dose of SAPHRIS is 5 mg given twice daily. In short-term controlled trials, there was no suggestion of added benefit with a 10 mg twice daily dose, but there was a clear increase in certain adverse reactions. If tolerated, daily dosage can be increased to 10 mg twice daily after one week. The safety of doses above 10 mg twice daily has not been evaluated in clinical studies [see Clinical Studies ( 14.1 )] .  Â
2.3
    ÂAcute Treatment of Manic or Mixed Episodes :
Monotherapy  in Adults :  The recommended starting and treatment dose of SAPHRIS is 5 mg to 10 mg twice daily. The safety of doses above 10 mg twice daily has not been evaluated in clinical trials [see Clinical Studies ( 14.2 )].Â
Monotherapy in Pediatric P atients: The recommended dose of SAPHRIS is 2.5 mg to 10 mg twice daily in pediatric patients 10 to 17 years of age, and dose may be adjusted for individual response and tolerability. The starting dose of SAPHRIS is 2.5 mg twice daily. After 3 days, the dose can be increased to 5 mg twice daily, and from 5 mg to 10 mg twice daily after 3 additional days. Pediatric patients aged 10 to 17 years appear to be more sensitive to dystonia with initial dosing with SAPHRIS when the recommended escalation schedule is not followed [see Use in Specific Populations ( 8.4 )]. The safety of doses greater than 10 mg twice daily has not been evaluated in clinical trials [see Use in Specific Populations ( 8.4 ) and Clinical Pharmacology ( 12.3 )].
Adjunctive Therapy in Adults : The recommended starting dose of SAPHRIS is 5 mg twice daily when administered as adjunctive therapy with either lithium or valproate. Depending on the clinical response and tolerability in the individual patient, the dose can be increased to 10 mg twice daily. The safety of doses above 10 mg twice daily as adjunctive therapy with lithium or valproate has not been evaluated in clinical trials.
For patients on SAPHRIS, whether used as monotherapy or as adjunctive therapy with lithium or valproate, it is generally recommended that responding patients continue treatment beyond the acute episode.
    ÂMaintenance Trea tment of Bipolar I Disorder:
Monotherapy in Adults: Continue on the SAPHRIS dose that the patient received during stabilization (5 mg to 10 mg twice daily). Depending on the clinical response and tolerability in the individual patient, a dose of 10 mg twice daily can be decreased to 5 mg twice daily. The safety of doses above 10 mg twice daily has not been evaluated in clinical trials [see Clinical Studies ( 14.2 )].
Dosage Forms & Strengths Section
- SAPHRIS 2.5Â mg tablets, black cherry flavor, are round, white to off-white sublingual tablets, with a hexagon on one side.
- SAPHRIS 5 mg tablets, black cherry flavor, are round, white to off-white sublingual tablets, with “5” on one side within a circle.
- SAPHRIS 10 mg tablets, black cherry flavor, are round, white to off-white sublingual tablets, with “10” on one side within a circle.
Sublingual tablets, black cherry flavor: 2.5 mg, 5 mg and 10 mg (3 )
Contraindications Section
SAPHRIS is contraindicated in patients with:
- Severe hepatic impairment (Child-Pugh C)Â [see Specific Populations ( 8.7 ), Clinical Pharmacology ( 12.3 )].
- A history of hypersensitivity reactions to asenapine. Reactions have included anaphylaxis, angioedema, hypotension, tachycardia, swollen tongue, dyspnea, wheezing and rash [see Warnings and Precautions ( 5.6 ), Adverse Reactions ( 6 )].
- Severe hepatic impairment (Child-Pugh C). (
8.7 ,12.3 )- Known hypersensitivity to SAPHRIS (asenapine), or to any components in the formulation. (
4 ,5.6 ,17 )
Warnings And Precautions Section
- Cerebrovascular Adverse Reactions in Elderly Patients with Dementia-Related Psychosis : Increased incidence of cerebrovascular adverse reactions (e.g., stroke, transient ischemic attack). (
5.2 )- Neuroleptic Malignant Syndrome: Manage with immediate discontinuation and close monitoring. (
5.3 )- Tardive Dyskinesia : Â Discontinue if clinically appropriate.
( 5.4 ) - Metabolic Changes :  Monitor for hyperglycemia/diabetes mellitus, dyslipidemia, and weight gain.
( 5.5 ) Â - Orthostatic Hypotension:Â Monitor heart rate and blood pressure and warn patients with known cardiovascular or cerebrovascular disease, and risk of dehydration or syncope. (
5.7 )- Leukopenia, Neutropenia, and Agranulocytosis : Â Perform complete blood counts (CBC) in patients with pre-existing low white blood cell count (WBC) or history of leukopenia or neutropenia. Consider discontinuing SAPHRIS if a clinically significant decline in WBC occurs in absence of other causative factors. (
5.9 )- QT Prolongation: Increases in QT interval; avoid use with drugs that also increase the QT interval and in patients with risk factors for prolonged QT interval. (
5.10 )- Seizures: Use cautiously in patients with a history of seizures or with conditions that lower the seizure threshold. (
5.12 )- Potential for Cognitive and Motor Impairment: Use caution when operating machinery. (
5.13 )5.1
Elderly patients with dementia-related psychosis treated with antipsychotic drugs are at an increased risk of death. Analyses of 17 placebo-controlled trials (modal duration of 10Â weeks), largely in patients taking atypical antipsychotic drugs, revealed a risk of death in drug-treated patients of between 1.6 to 1.7 times the risk of death in placebo-treated patients. Over the course of a typical 10-week controlled trial, the rate of death in drug-treated patients was about 4.5%, compared to a rate of about 2.6% in the placebo group.
Although the causes of death were varied, most of the deaths appeared to be either cardiovascular (e.g., heart failure, sudden death) or infectious (e.g., pneumonia) in nature. SAPHRIS is not approved for the treatment of patients with dementia-related psychosis [ see Boxed Warning  and Warnings and Precautions ( 5.2 ) ].
5.2
In placebo-controlled trials in elderly subjects with dementia, patients randomized to risperidone, aripiprazole, and olanzapine had a higher incidence of stroke and transient ischemic attack, including fatal stroke. SAPHRISÂ is not approved for the treatment of patients with dementia-related psychosis [see Boxed Warning , Warnings and Precautions ( 5.1 )].
5.3
A potentially fatal symptom complex sometimes referred to as Neuroleptic Malignant Syndrome (NMS) has been reported in association with administration of antipsychotic drugs. Clinical manifestations of NMS are hyperpyrexia, muscle rigidity, delirium, and autonomic instability. Additional signs may include elevated creatine phosphokinase, myoglobinuria (rhabdomyolysis), and acute renal failure. If NMS is suspected, immediately discontinue SAPHRIS and provide intensive symptomatic treatment and monitoring.
5.4
Tardive dyskinesia, a syndrome consisting of potentially irreversible, involuntary, dyskinetic movements, may develop in patients treated with antipsychotic drugs, including SAPHRIS. The risk appears to be highest among the elderly, especially elderly women, but it is not possible to predict which patients are likely to develop the syndrome. Whether antipsychotic drug products differ in their potential to cause tardive dyskinesia is unknown.
The risk of tardive dyskinesia and the likelihood that it will become irreversible increase with the duration of treatment and the cumulative dose. The syndrome can develop after a relatively brief treatment period, even at low doses. It may also occur after discontinuation of treatment.
There is no known treatment for tardive dyskinesia, although the syndrome may remit, partially or completely, if antipsychotic treatment is discontinued. Antipsychotic treatment itself, however, may suppress (or partially suppress) the signs and symptoms of the syndrome, possibly masking the underlying process. The effect that symptomatic suppression has upon the long-term course of tardive dyskinesia is unknown.
Given these considerations, SAPHRIS should be prescribed in a manner most likely to reduce the risk of tardive dyskinesia. Chronic antipsychotic treatment should generally be reserved for patients: 1) who suffer from a chronic illness that is known to respond to antipsychotic drugs; and 2) for whom alternative, effective, but potentially less harmful treatments are not available or appropriate. In patients who do require chronic treatment, use the lowest dose and the shortest duration of treatment producing a satisfactory clinical response should be sought. Periodically reassess the need for continued treatment.
If signs and symptoms of TD appear in a patient on SAPHRIS, drug discontinuation should be considered. However, some patients may require treatment with SAPHRIS despite the presence of the syndrome.
Atypical antipsychotic drugs, including SAPHRIS, have caused metabolic changes, including hyperglycemia, diabetes mellitus, dyslipidemia, and body weight gain. Although all of the drugs in the class to date have been shown to produce some metabolic changes, each drug has its own specific risk profile.
Hyperglycemia and Diabetes Mellitus
Hyperglycemia, in some cases extreme and associated with ketoacidosis or hyperosmolar coma or death, has been reported in patients treated with atypical antipsychotics. There have been reports of hyperglycemia in patients treated with SAPHRIS. Assess fasting plasma glucose before or soon after initiation of antipsychotic medication, and monitor periodically during long-term treatment.
Adult Patients:Â Pooled data from the short-term placebo-controlled schizophrenia and bipolar mania trials are presented in Table 1 .
TABLE 1: Changes in Fasting Glucose in Adult Patients Schizophrenia (6-weeks) Bipolar I Disorder (3-weeks)        Placebo SAPHRIS Placebo SAPHRIS 5 mg twice daily 10 mg twice daily 5 or 10 mgtwice daily§ 5 mg twice daily 10 mg twice daily 5 or 10 mgtwice daily†Mean Change from Baseline in Fasting Glucose at Endpoint                   Change from Baseline (mg/dL) (N*) -0.2(232) 3.8(158) 1.1 (153) 3.2 (377)  0 (174) 4.1(84) 3.5(81)  1.7 (321) Proportion of Patients with Shifts from Baseline to Endpoint Normal to High<100 to ≥126 mg/dL 4.1% 4.5% 4.5% 5.0% 2.4% 0% 1.7% 1.8% (n/N**) (7/170) (5/111) (5/111) (13/262) (3/126) (0/53) (1/60) (4/224) Borderline to High≥100 and <126 to ≥126 mg/dL 5.9% 6.8% 6.3% 10.5% 0% 12.5% 15.8% 12.8% (n/N**) (3/51) (3/44) (2/32) (10/95) (0/39) (3/24) (3/19) (10/78)
N* = Number of patients who had assessments at both Baseline and Endpoint.
N** = Number of patients at risk at Baseline with assessments at both Baseline and Endpoint.
§ Includes patients treated with flexible dose of SAPHRIS 5 or 10 mg twice daily (N=90).
†Includes patients treated with flexible dose of SAPHRIS 5 or 10 mg twice daily (N=379).
In a 52-week, double-blind, comparator-controlled trial that included primarily patients with schizophrenia, the mean increase from baseline of fasting glucose was 2.4 mg/dL.
Pediatric Patients:Â Data from the short-term, placebo-controlled trial in pediatric patients with bipolar I disorder are shown in Table 2.
TABLE 2: Changes in Fasting Glucose in Pediatric Subjects   Bipolar I Disorder (3-weeks) Placebo SAPHRIS 2.5 mgtwice daily SAPHRIS 5 mg twice daily SAPHRIS 10 mg twice daily Mean Change from Baseline in Fasting Glucose at Endpoint Change from Baseline (mg/dL) (N*) -2.24(56) 1.43(51) -0.45(57) 0.34(52) Proportion of Subjects with Shifts from Baseline to Endpoint Normal to High>45 & <100 to ≥126 mg/dL 0% 0% 1.8% 0% (n/N*) (0/56) (0/51) (1/57) (0/52)
N* = Number of subjects who had assessments at both Baseline and Endpoint.
Dyslipidemia Â
Atypical antipsychotics cause adverse alterations in lipids. Before or soon after initiation of antipsychotic medication, obtain a fasting lipid profile at baseline and monitor periodically during treatment.
Adult Patients:Â Pooled data from the short-term, placebo-controlled schizophrenia and bipolar mania trials are presented in Table 3 .
TABLE 3: Changes in Lipids in Adult Patients Schizophrenia (6-weeks) Bipolar I Disorder (3-weeks) Placebo SAPHRIS Placebo SAPHRIS 5 mg twice daily 10 mg twice daily 5 or 10 mg twice daily§ 5 mg twice daily 10 mg twice daily 5 or 10 mg twice daily†Mean Change from Baseline (mg/dL) Total cholesterol (N*) -2.2(351) -2.4(258) 3.3(199) 0.4 (539) -1.6(278) -1.6(108) -4.7(95) -0.5(525) LDL (N*) 0.1(285) -0.2(195) 2.6(195) 1.3 (465)  1.4(271) -2.5(101) -4.1(94)  -0.3(499) HDL (N*) 0.5(290) 0.4(199) 1.0(199) 0.5 (480) 0.2(278) 0.1(108) 0.7(95) 0.7(525) Fasting triglycerides (N*) -7.6(233) -1.9(159) 0.1(154) 3.8 (380)  -16.9(222) 3.9(89) -8.5(85)  -3.0(411) Proportion of Patients with Shifts from Baseline to Endpoint Total cholesterolNormal to High <200 to ≥240 (mg/dL) (n/N*) 1.3%(3/225) 0.6%(1/161) 2.2%(3/134) 1.7%(6/343)  1.2%(2/174) 3.0%(2/66) 0(0/63)  2.1% (7/333) LDL Normal to High <100 to ≥160(mg/dL) (n/N*) 1.7%(2/117) 0.0%(0/80) 1.2%(1/86) 1.0%(2/196) 1.9%(2/108) 2.4%(1/41) 0(0/41) 0.5% (1/223) HDL Normal to Low ≥40 to <40 (mg/dL) (n/N*) 10.7%(21/196) 13.3%(18/135) 14.7%(20/136) 14.0%(45/322) 7.4%(16/215) 4.1%(4/97) 5.1%(4/78) 7.0%(29/417) Fasting triglycerides Normal to High <150 to ≥200 (mg/dL) (n/N*) 2.4%(4/167) 7.0%(8/115) 8.3%(9/108) 7.7%(20/260) 4.6% (7/153) 8.2%(5/61) 1.6%(1/64)  6.2% (17/273)
N* = Number of subjects who had assessments at both Baseline and Endpoint.
§ Includes subjects treated with flexible dose of SAPHRIS 5 or 10 mg twice daily (N=90).
†Includes patients treated with flexible dose of SAPHRIS 5 or 10 mg twice daily (N=379)
In short-term schizophrenia trials, the proportion of patients with total cholesterol elevations ≥240 mg/dL (at Endpoint) was 8.3% for SAPHRIS-treated patients versus 7% for placebo-treated patients. The proportion of patients with elevations in triglycerides ≥200 mg/dL (at Endpoint) was 13.2% for SAPHRIS-treated patients versus 10.5% for placebo-treated patients. In short-term, placebo-controlled bipolar mania trials, the proportion of patients with total cholesterol elevations ≥240 mg/dL (at Endpoint) was 7.8% for SAPHRIS-treated patients versus 7.9% for placebo-treated patients. The proportion of patients with elevations in triglycerides ≥200 mg/dL (at Endpoint) was 13.1% for SAPHRIS-treated patients versus 8.6% for placebo-treated patients.
Pediatric Patients: Data from the short-term, placebo-controlled bipolar mania trial are presented in Table 4.
TABLE 4: Changes in Fasting Lipids in Pediatric Subjects Bipolar I Disorder (3-weeks) Placebo SAPHRIS2.5 mgtwice daily SAPHRIS5 mgtwice daily SAPHRIS10 mgtwice daily Mean Change from Baseline (mg/dL) Total fasting cholesterol (N*) -2.3(57) 3.7(50) 7.2(57) 9.3(52) Fasting LDL (N*) -2.5(57) -0.2(50) 3.0(57) 4.9(51) Fasting HDL (N*) 1.6(57) 2.3(50) 1.5(57) 1.7(52) Fasting triglycerides (N*) -6.6(57) 8.7(50) 13.4(57) 14.7(52) Proportion of Subjects with Shifts from Baseline to Endpoint Total fasting cholesterolNormal to High<170 to >=200 (mg/dL) (n/N*) 1.8%(1/57) 0%(0/50) 1.8%(1/57) 0%(0/52) Fasting LDL Normal to High<110 to >=130(n/N*) 1.8%(1/57) 2.0%(1/50) 1.8%(1/57) 0%(0/51) Fasting HDL Normal to Low≥40 to <40 (mg/dL) (n/N*) 3.5%(2/57) 6.0%(3/50) 3.5%(2/57) 9.6%(5/52) Fasting triglycerides Normal to High<150 to ≥200 (mg/dL) (n/N*) 0%(0/57) 4.0%(2/50) 3.5%(2/57) 1.9%(1/52)
N* = Number of patients who had assessments at both Baseline and Endpoint
Weight Gain
Weight gain has been observed in patients treated with atypical antipsychotics, including SAPHRIS. Monitor weight at baseline and frequently thereafter.
Adult Patients: Pooled data on mean changes in body weight and the proportion of subjects meeting a weight gain criterion of ≥7% of body weight from the short-term, placebo-controlled schizophrenia and bipolar mania trials are presented in Table 5 .Â
Table 5: Change in Body Weight in Adult Patients from Baseline Schizophrenia (6-weeks) Bipolar I Disorder (3-weeks) Placebo SAPHRIS Placebo SAPHRIS 5 mg twice daily 10 mg twice daily 5 or 10 mgtwice daily§ 5 mg twice daily 10 mg twice daily 5 or 10 mgtwice daily†Change from Baseline (kg) (N*) 0.0(348) 1.0(251) 0.9(200) 1.1(532) 0.2(288) 1.4(110) 1.3(98) 1.3(544) Proportion of Patients with a ≥7% Increase in Body Weight % with ≥7% increase in body weight 1.6% 4.4% 4.8% 4.9% 0.4% 6.4% 1.0% 5.5%
N* = Number of subjects who had assessments at both Baseline and Endpoint.
§ Includes subjects treated with flexible dose of SAPHRIS 5 or 10 mg twice daily (N=90).
† Includes patients treated with flexible dose of SAPHRIS 5 or 10 mg twice daily (N=379).
Adult Patients: In a 52-week, double-blind, comparator-controlled adult trial that included primarily patients with schizophrenia, the mean weight gain from baseline was 0.9 kg. The proportion of patients with a ≥7% increase in body weight (at Endpoint) was 14.7%. Table 6 provides the mean weight change from baseline and the proportion of patients with a weight gain of ≥7% categorized by Body Mass Index (BMI) at baseline.
Table 6: Weight Change Results Categorized by BMI at Baseline: Comparator-Controlled 52-Week Study in Adults with Schizophrenia BMI <23 SAPHRIS N=295 BMI 23 - ≤27 SAPHRIS N=290 BMI >27 SAPHRIS N=302 Mean change from Baseline (kg) 1.7 1 0 % with ≥7% increase in body weight 22% 13% 9%
Pediatric Patients: Data on mean changes in body weight and the proportion of pediatric patients meeting a weight gain criterion of ≥7% of body weight from the short-term, placebo-controlled bipolar mania trial are presented in Table 7. To adjust for normal growth, z-scores were derived (measured in standard deviations [SD]), which normalize for the natural growth of pediatric patients by comparisons to age- and sex-matched population standards.
The distance of a z-score from 0 represents the distance of a percentile from the median, measured in standard deviations (SD). After adjusting for age and sex, the mean change from baseline to endpoint in weight z-score for SAPHRIS 2.5 mg, 5 mg, and 10 mg twice daily, was 0.11, 0.08 and 0.09 SD versus 0.02 SD for placebo, respectively.
When treating pediatric patients, weight gain should be monitored and assessed against that expected for normal growth.
Table 7: Change in Body Weight in Pediatric Subjects from Baseline   Bipolar I Disorder (3-weeks) Placebo SAPHRIS 2.5 mgtwice daily SAPHRIS 5 mg twice daily SAPHRIS 10 mg twice daily Change from Baseline (kg) (N*)  0.5(89) 1.7 (92) 1.6(90) 1.4(87) Proportion of Subjects with a ≥7% Increase in Body Weight % with ≥7% increase in body weight 1.1% 12.0% 8.9% 8.0%
N* = Number of subjects who had assessments at both Baseline and Endpoint.
5.
Hypersensitivity reactions have been observed in patients treated with SAPHRIS. In several cases, these reactions occurred after the first dose. These hypersensitivity reactions included: anaphylaxis, angioedema, hypotension, tachycardia, swollen tongue, dyspnea, wheezing and rash.
5.
Atypical antipsychotics cause orthostatic hypotension and syncope. Generally, the risk is greatest during initial dose titration and when increasing the dose. In short-term schizophrenia adult trials, syncope was reported in 0.2% (1/572) of patients treated with therapeutic doses (5 mg or 10 mg twice daily) of SAPHRIS, compared to 0.3% (1/378) of patients treated with placebo. In short-term bipolar mania adult trials, syncope was reported in 0.2% (1/620) of patients treated with therapeutic doses (5 mg or 10 mg twice daily) of SAPHRIS, compared to 0% (0/329) of patients treated with placebo. During adult pre-marketing clinical trials with SAPHRIS, including long-term trials without comparison to placebo, syncope was reported in 0.6% (11/1953) of patients treated with SAPHRIS. In a 3-week, bipolar mania pediatric trial, syncope was reported in 1% (1/104) of patients treated with SAPHRIS 2.5 mg twice daily, 1% (1/99) of patients treated with SAPHRIS 5Â mg twice daily, and 0% (0/99) for patients treated with SAPHRIS 10 mg twice daily compared to 0% (0/101) for patients treated with placebo.
Orthostatic vital signs should be monitored in patients who are vulnerable to hypotension (elderly patients, patients with dehydration, hypovolemia, concomitant treatment with antihypertensive medications, patients with known cardiovascular disease (history of myocardial infarction or ischemic heart disease, heart failure, or conduction abnormalities), and patients with cerebrovascular disease. SAPHRIS should be used cautiously when treating patients who receive treatment with other drugs that can induce hypotension, bradycardia, respiratory or central nervous system depression [ see Drug Interactions ( 7.1 )]. Monitoring of orthostatic vital signs should be considered in all such patients, and a dose reduction should be considered if hypotension occurs.
5.8
SAPHRIS may cause somnolence, postural hypotension, motor and sensory instability, which may lead to falls and, consequently, fractures or other injuries. For patients with diseases, conditions, or medications that could exacerbate these effects, complete fall risk assessments when initiating antipsychotic treatment and recurrently for patients on long-term antipsychotic therapy.
5.
In clinical trial and postmarketing experience, leukopenia and neutropenia have been reported temporally related to antipsychotic agents, including SAPHRIS. Agranulocytosis (including fatal cases) has been reported with other agents in the class.
Possible risk factors for leukopenia/neutropenia include pre-existing low white blood cell count (WBC) or absolute neutrophil count (ANC) and history of drug induced leukopenia/neutropenia. In patients with a pre-existing low WBC or ANC or a history of drug-induced leukopenia or neutropenia, perform a complete blood count (CBC) during the first few months of therapy. In such patients, consider discontinuation of SAPHRIS at the first sign of a clinically significant decline in WBC in the absence of other causative factors.
Monitor patients with clinically significant neutropenia for fever or other symptoms or signs of infection and treat promptly if such symptoms or signs occur. Discontinue SAPHRIS in patients with severe neutropenia (absolute neutrophil count <1000/mm3) and follow their WBC until recovery.
5.
The effects of SAPHRIS on the QT/QTc interval were evaluated in a dedicated adult QT study. This trial involved SAPHRIS doses of 5 mg, 10 mg, 15 mg, and 20 mg twice daily, and placebo, and was conducted in 151 clinically stable patients with schizophrenia, with electrocardiographic assessments throughout the dosing interval at baseline and steady state. At these doses, SAPHRIS was associated with increases in QTc interval ranging from 2 to 5 msec compared to placebo. No patients treated with SAPHRIS experienced QTc increases ≥60 msec from baseline measurements, nor did any patient experience a QTc of ≥500 msec.
Electrocardiogram (ECG) measurements were taken at various time points during the SAPHRIS clinical trial program (5Â mg or 10Â mg twice daily doses). Post-baseline QT prolongations exceeding 500 msec were reported at comparable rates for SAPHRIS and placebo in these short-term trials. There were no reports of Torsade de Pointes or any other adverse reactions associated with delayed ventricular repolarization.
The use of SAPHRIS should be avoided in combination with other drugs known to prolong QTc including Class 1A antiarrhythmics (e.g., quinidine, procainamide) or Class 3 antiarrhythmics (e.g., amiodarone, sotalol), antipsychotic medications (e.g., ziprasidone, chlorpromazine, thioridazine), and antibiotics (e.g., gatifloxacin, moxifloxacin). SAPHRIS should also be avoided in patients with a history of cardiac arrhythmias and in other circumstances that may increase the risk of the occurrence of torsade de pointes and/or sudden death in association with the use of drugs that prolong the QTc interval, including bradycardia; hypokalemia or hypomagnesemia; and presence of congenital prolongation of the QT interval.
5.1
Like other drugs that antagonize dopamine D2 receptors, SAPHRIS can elevate prolactin levels, and the elevation can persist during chronic administration. Hyperprolactinemia may suppress hypothalamic GnRH, resulting in reduced pituitary gonadotropin secretion. This, in turn, may inhibit reproductive function by impairing gonadal steroidogenesis in both female and male patients. Galactorrhea, amenorrhea, gynecomastia, and impotence have been reported in patients receiving prolactin-elevating compounds. Long-standing hyperprolactinemia when associated with hypogonadism may lead to decreased bone density in both female and male subjects. In SAPHRIS adult pre-marketing clinical trials, the incidences of adverse events related to abnormal prolactin levels were 0.4% versus 0% for placebo. In a 3-week, bipolar mania pediatric trial, the incidence of adverse events related to abnormal prolactin levels were 0% in the SAPHRIS 2.5 mg twice daily treatment group, 2% in the SAPHRIS 5 mg twice daily treatment group, and 1% in the SAPHRIS 10 mg twice daily treatment group versus to 1% for patients treated with placebo [see Adverse Reactions ( 6.1 )].
Tissue culture experiments indicate that approximately one-third of human breast cancers are prolactin-dependent in vitro, a factor of potential importance if the prescription of these drugs is considered in a patient with previously-detected breast cancer. Neither clinical studies nor epidemiologic studies conducted to date have shown an association between chronic administration of this class of drugs and tumorigenesis in humans, but the available evidence is too limited to be conclusive.
5.1
Seizures were reported in 0% and 0.3% (0/572, 1/379) of adult patients treated with doses of 5 mg and 10 mg twice daily of SAPHRIS, respectively, compared to 0% (0/503, 0/203) of patients treated with placebo in pre-marketing short-term schizophrenia and bipolar mania trials, respectively. During adult pre-marketing clinical trials with SAPHRIS, including long-term trials without comparison to placebo, seizures were reported in 0.3% (5/1953) of patients treated with SAPHRIS. There were no reports of seizures in pediatric patients treated with SAPHRIS in a 3-week-term, bipolar mania trial.
As with other antipsychotic drugs, SAPHRIS should be used with caution in patients with a history of seizures or with conditions that potentially lower the seizure threshold. Conditions that lower the seizure threshold may be more prevalent in patients 65 years or older.
5.1
Somnolence was reported in patients treated with SAPHRIS. It was usually transient with the highest incidence reported during the first week of treatment. In short-term, fixed-dose, placebo-controlled schizophrenia adult trials, somnolence was reported in 15% (41/274) of patients on SAPHRIS 5 mg twice daily and in 13% (26/208) of patients on SAPHRIS 10 mg twice daily compared to 7% (26/378) of placebo patients. In short-term, placebo-controlled bipolar mania adult trials of therapeutic doses (5-10 mg twice daily), somnolence was reported in 23% (145/620) of patients on SAPHRIS compared to 5% (18/329) of placebo patients.  In the 3-week fixed-dose study, somnolence occurred at a lower rate in the 5mg twice daily dose 20% (24/122) versus the 10mg twice daily dose 26% (31/119) compared to 4% (5/126) in placebo patients. During adult pre-marketing clinical trials with SAPHRIS, including long-term trials without comparison to placebo, somnolence was reported in 18% (358/1953) of patients treated with SAPHRIS. Somnolence led to discontinuation in 0.6% (12/1953) of patients in short-term, placebo-controlled trials.Â
In a 3-week, placebo-controlled, bipolar I pediatric trial, the incidence of somnolence (including sedation and hypersomnia)Â for placebo, SAPHRIS 2.5 mg twice daily, 5 mg twice daily, and 10 mg twice daily, was 12% (12/101), 46% (48/104), 53% (52/99), and 49% (49/99), respectively. Somnolence led to discontinuation in 0%, 3%, 1%, and 2% of patients treated with placebo, and SAPHRIS 2.5 mg twice daily, 5 mg twice daily, and 10 mg twice daily, respectively.
Patients should be cautioned about operating hazardous machinery, including motor vehicles, until they are reasonably certain that SAPHRIS therapy does not affect them adversely.
5.1
Atypical antipsychotics may disrupt the body’s ability to reduce core body temperature. In the pre-marketing short-term placebo-controlled trials for both schizophrenia and acute bipolar I disorder, the incidence of adverse reactions suggestive of body temperature increases was low (≤1%) and comparable to placebo (0%). During pre-marketing clinical trials with SAPHRIS, including long-term trials without comparison to placebo, the incidence of adverse reactions suggestive of body temperature increases (pyrexia and feeling hot) was ≤1%.
Strenuous exercise, exposure to extreme heat, dehydration, and anticholinergic medications may contribute to an elevation in core body temperature; use SAPHRIS with caution in patient who may experience these conditions.
5.1
Esophageal dysmotility and aspiration have been associated with antipsychotic drug use. Dysphagia has been reported with SAPHRIS. SAPHRIS and other antipsychotic drugs should be used cautiously in patients at risk for aspiration.Â
Adverse Reactions Section
The following adverse reactions are discussed in more detail in other sections of the labeling:
- Use in Elderly Patients with Dementia-Related Psychosis [see Boxed Warning and Warnings and Precautions ( 5.1 and 5.2 )]
- Neuroleptic Malignant Syndrome [see Warnings and Precautions ( 5.3 )]
- Tardive Dyskinesia [see Warnings and Precautions ( 5.4 )]
- Metabolic Changes [see Warnings and Precautions ( 5.5 )]
- Hypersensitivity Reactions [see Contraindications, Warnings and Precautions ( 5.6 )]
- Orthostatic Hypotension, Syncope, and other Hemodynamic Effects [see Warnings and Precautions ( 5.7 )]
- Falls [see Warnings and Precautions ( 5.8 )]
- Leukopenia, Neutropenia, and Agranulocytosis [see Warnings and Precautions ( 5.9 )]
- QT Interval Prolongation [see Warnings and Precautions ( 5.10 )]
- Hyperprolactinemia [see Warnings and Precautions ( 5.11 )]
- Seizures [see Warnings and Precautions ( 5.12 )]
- Potential for Cognitive and Motor Impairment [see Warnings and Precautions ( 5.13 )]
- Body Temperature Regulation [see Warnings and Precautions ( 5.14 )]
- Dysphagia [see Warnings and Precautions ( 5.15 )]
The most common adverse reactions (≥5% and at least twice the rate of placebo) reported with acute treatment in adults with schizophrenia were akathisia, oral hypoesthesia, and somnolence. The safety profile of SAPHRIS in the maintenance treatment of schizophrenia in adults was similar to that seen with acute treatment.
The most common adverse reactions (≥5% and at least twice the rate of placebo) reported with acute monotherapy treatment of manic or mixed episodes associated with bipolar I disorder in adults were somnolence, oral hypoesthesia dizziness, extrapyramidal symptoms (excluding akathisia) and akathisia; and during the adjunctive therapy trial in bipolar I disorder in adults were somnolence and oral hypoesthesia. The rates were lower at the 5mg twice daily dose than the 10mg twice daily dose for all of these most common adverse reactions. The safety profile of SAPHRIS in the maintenance treatment of manic or mixed episodes associated with bipolar I disorder in adults was similar to that seen with acute treatment.
The adult information below is derived from a clinical trial database for SAPHRIS consisting of over 5355 patients and/or healthy subjects exposed to one or more sublingual doses of SAPHRIS. A total of 1427 SAPHRIS-treated patients were treated for at least 24 weeks and 785 SAPHRIS-treated patients had at least 52 weeks of exposure at therapeutic doses.
In a 3-week monotherapy trial, the most common adverse reactions (≥5% and at least twice the rate of placebo) reported in pediatric patients with bipolar I disorder treated with SAPHRIS were somnolence, dizziness, dysgeusia, oral hypoesthesia, nausea, increased appetite, fatigue, and increased weight. No new major safety findings were reported from a 50-week, open-label, uncontrolled safety trial.
A total of 651 pediatric patients were treated with SAPHRIS. Of these patients, 352 pediatric patients were treated with SAPHRIS for at least 180 days and 58 pediatric patients treated with SAPHRIS had at least 1 year of exposure. The safety of SAPHRIS was evaluated in 403 pediatric patients with bipolar I disorder who participated in a 3-week, placebo-controlled, double-blind trial, of whom 302 patients received SAPHRIS at fixed doses ranging from 2.5 mg to 10 mg twice daily.
The stated frequencies of adverse reactions represent the proportion of individuals who experienced a treatment-emergent adverse event of the type uled. A reaction was considered treatment emergent if it occurred for the first time or worsened while receiving therapy following baseline evaluation.
The most commonly observed adverse reactions (incidence ≥5% and at least twice that for placebo) were (6.1 ):
- Schizophrenia  Adults : akathisia, oral hypoesthesia, somnolence.
- Bipolar I Disorder Adults (Monotherapy): somnolence, oral hypoesthesia, dizziness, extrapyramidal symptoms (excluding akathisia) and akathisia.
- Bipolar I Disorder Pediatric Patients (Monotherapy): somnolence, dizziness, dysgeusia, oral paresthesia, nausea, increased appetite, fatigue, increased weight.
- Bipolar I Disorder Adults (Adjunctive): somnolence, oral hypoesthesia.
To report SUSPECTED ADVERSE REACTIONS, contact Allergan at 1-800-678-1605 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
6.
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Adult Patients with Schizophrenia: The following findings are based on the short-term placebo-controlled pre-marketing trials for schizophrenia (a pool of three 6-week fixed-dose trials and one 6-week flexible-dose trial) in which sublingual SAPHRIS was administered in doses ranging from 5 to 10 mg twice daily.
Adverse Reactions Associated with Discontinuation of Treatment:  A total of 9% of SAPHRIS-treated patients and 10% of placebo-treated patients discontinued due to adverse reactions. There were no drug-related adverse reactions associated with discontinuation in patients treated with SAPHRIS at the rate of at least 1% and at least twice the placebo rate.
Adverse Reactions Occurring at an Incidence of 2% or More in SAPHRIS-Treated Patients with Schizophrenia : Adverse reactions associated with the use of SAPHRIS (incidence of 2% or greater, rounded to the nearest percent, and SAPHRIS incidence greater than placebo) that occurred during acute therapy (up to 6-weeks in patients with schizophrenia) are shown in Table 8.
Table 8: Adverse Reactions Reported in 2% or More of Adult Patients in Any SAPHRIS Dose Group and Which Occurred at Greater Incidence Than in the Placebo Group in 6-Week Schizophrenia Trials System Organ Class/ Preferred Term Placebo N=378 % SAPHRIS 5 mg twice daily N=274 % SAPHRIS 10 mg twice daily N=208 % All SAPHRIS § 5 mg or 10 mg twice daily N=572 % Gastrointestinal disorders    Constipation 6 7 4 5    Dry mouth 1 3 1 2    Oral hypoesthesia 1 6 7 5    Salivary hypersecretion 0 <1 4 2    Stomach discomfort 1 <1 3 2    Vomiting 5 4 7 5 General disorders    Fatigue 3 4 3 3    Irritability <1 2 1 2 Investigations    Increased weight <1 2 2 3 Metabolism disorders    Increased appetite <1 3 0 2 Nervous system disorders    Akathisia* 3 4 11 6    Dizziness 4 7 3 5    Extrapyramidal symptoms    7 9 12 10    (excluding akathisia)†   Somnolence‡ 7 15 13 13 Psychiatric disorders    Insomnia 13 16 15 15 Vascular disorders    Hypertension 2 2 3 2
*Â Akathisia includes: akathisia and hyperkinesia.
† Extrapyramidal symptoms included dystonia, oculogyration, dyskinesia, tardive dyskinesia, muscle rigidity, parkinsonism, tremor, and extrapyramidal disorder (excluding akathisia).
‡  Somnolence includes the following events: somnolence, sedation, and hypersomnia.
§  Also includes the Flexible-dose trial (N=90).
Dose-Related Adverse Reactions: Â In the short term schizophrenia trials the incidence of akathisia appeared to be dose-related (see Table 8).
Monotherapy in Adult Patients with Bipolar Mania:Â The following findings are based on the short-term placebo-controlled trials for bipolar mania (a pool of two 3-week flexible-dose trials and one 3-week fixed-dose trial) in which sublingual SAPHRIS was administered in doses of 5 mg or 10 mg twice daily.
Adverse Reactions Associated with Discontinuation of Treatment: Â Approximately 10% (61/620) of SAPHRIS-treated patients in short-term, placebo-controlled trials discontinued treatment due to an adverse reaction, compared with about 7% (22/329) on placebo. There were no adverse reactions associated with discontinuation in patients treated with SAPHRIS at the rate of at least 1% and at least twice the placebo rateÂ
Adverse Reactions Occurring at an Incidence of 2% or More Among SAPHRIS-Treated (Monotherapy ) patients with Bipolar I Disorder : Â Adverse reactions associated with the use of SAPHRIS (incidence of 2% or greater, rounded to the nearest percent, and SAPHRIS incidence greater than placebo) that occurred during acute monotherapy (up to 3-weeks in patients with bipolar mania) are shown in Table 9.
Table 9: Adverse Reactions Reported in 2% or More of Adult Patients in Any SAPHRIS Dose Group and Which Occurred at Greater Incidence Than in the Respective Placebo Group in 3-Week Bipolar Mania Fixed and Flexible Dose Trials System Organ Class/Preferred  Term (Fixed Dose Study) All Placebo a All SAPHRIS 5 mg or 10 mg twice daily b Placebo SAPHRIS 5 mg twice daily SAPHRIS 10 mg twice daily N= 126 % N= 122 % N=119 % N= 329 % N=620 % Gastrointestinal disorders     Oral Hypoesthesiac 2 13 24 1 10    Nausea 3 4 5 5 5    Constipation 2 4 3 4 4    Dyspepsiah 6 4 5 4 4    Vomiting 2 1 3 3 3    Abdominal Paind 0 2 3 3 3    Dry Mouth 5 3 1 2 3    Toothache 1 2 2 2 3 General disorders    Fatiguee      2 2 5 2 4 Infections and Infestations    Nasopharyngitisi 2 1 5 2 3 Investigations    Weight Increase 1 0 1 1 3    Alanine Aminotransferase 0 0 3 0 1    Increase Metabolism disorders    Increased appetite 2 1 6 2 4 Musculoskeletal and connective tissue disorders    Arthralgia 1 1 2 1 2 Nervous system disorders    Somnolencef 4 20 26 5 23    Dizziness 5 3 5 4 8    Extrapyramidal symptoms 7 7 11 4 8    (excluding akathisia)g    Akathisia 1 4 15 2 6    Dysgeusia 0 3 9 <1 4 Psychiatric Disorders    Bipolar Disorder/Mania j 3 8 3 5 6    Agitation 1 4 3 3 4    Anxiety 3 0 3 2 3
a Includes fixed and flexible dose trials
b  SAPHRIS 5 mg to 10 mg twice daily with fixed and flexible dosing.
c Oral Hypoesthesia includes the preferred terms: oral hypoesthesia, oral paresthesia, and oral dysaesthesia.
d Abdominal pain includes the preferred terms: abdominal pain, abdominal pain upper, abdominal pain lower, and abdominal discomfort.
e  Fatigue includes the preferred terms: fatigue and lethargy.
f  Somnolence includes the preferred terms: somnolence, sedation, and hypersomnia.
g  Extrapyramidal symptoms (excluding akathisia) includes the preferred terms: dyskinesia, dystonia, resting tremor, tremor, oromandibular dystonia, myoclonus, muscle spasms, muscle rigidity, musculoskeletal stiffness, muscle contractions involuntary, blepharospasm, tongue disorder, and Parkinsonism. Â
h  Dyspepsia includes the preferred terms: dyspepsia and gastroesophageal reflux disease.
i  Nasopharyngitis includes the preferred terms: nasopharyngitis and upper respiratory tract infection.  Â
j   Bipolar Disorder/Mania includes the preferred terms: bipolar disorder, bipolar I disorder and mania.
Monotherapy in Pediatric Patients with Bipolar Mania: Â The following findings are based on a 3-week , placebo-controlled trial for bipolar mania in which SAPHRIS was administered at doses of 2.5 mg, 5 mg, or 10 mg twice daily.
Adverse Reactions Leading to Discontinuation of Treatment:  A total of 6.7% (7/104) of patients treated with SAPHRIS 2.5 mg twice daily, 5.1% (5/99) of patients treated with SAPHRIS 5 mg twice daily, and 5.1% (5/99) of patients treated with SAPHRIS 10 mg twice daily discontinued treatment due to adverse reactions compared to 4% (4/101) on placebo. The most common adverse reactions that led to discontinuation in pediatric patients treated with SAPHRIS (rates at least 2% in any SAPHRIS arm and at least twice the placebo rate) were somnolence (3% in the 2.5mg twice daily group, 1% in the 5mg twice daily group, and 2% in the 10mg twice daily group), abdominal pain (2% in the 10mg twice daily group), and nausea (2% in the 10mg twice daily group) No placebo-treated patients dropped out for these events.
Adverse Reactions Occurring with SAPHRIS at an Incidence of 2% or More in SAPHRIS-treated Bipolar I Patients: Adverse reactions associated with the use of SAPHRIS (incidence of ≥2% in any SAPHRIS dose group and greater than placebo) that occurred during acute therapy are shown in Table 10.
Table 10: Adverse Reactions Reported in 2% or More of Pediatric Patients (Ages 10 to 17 Years) in Any SAPHRIS Dose Group and Which Occurred at Greater Incidence Than in the Placebo Group in a 3-Week Bipolar Mania Trial System Organ Class/ AE Preferred Term Placebo SAPHRIS 2.5 mg twice daily SAPHRIS 5 mg twice daily SAPHRIS 10 mg twice daily All SAPHRIS 2.5, 5, and 10 mg N=101 % N=104 % N=99 % N=99 % N=302 % Cardiac Disorders      Tachycardia1 0 3 0 1 1 Gastrointestinal Disorders      Oral hypoesthesia2 4 25 25 30 27      Nausea 3 6 6 6 6    Vomiting 3 4 4 4 4      Abdominal pain3 7 9 3 5 6      Glossodynia 0 0 2 0 1 General Disorders and Administrative Site Disorders      Fatigue4 5 4 8 14 9      Irritability 1 1 1 2 1 Injury, Poisoning, and Procedural Complications      Muscle strain 0 0 0 2 1 Investigations      Increased weight 0 6 2 2 3      Hyperinsulinemia5 0 1 3 1 2      ALT increased 0 0 0 2 1      AST increased 0 0 0 2 1 Metabolism and Nutrition Disorders      Increased appetite 2 10 9 6 8      Dehydration 1 0 2 0 1 Musculoskeletal and Connective Tissue Disorders      Myalgia 0 0 2 1 1 Nervous System Disorders      Somnolence6 12 46 53 49 49      Headache 6 8 11 9 9      Dizziness 3 6 10 5 7      Dysgeusia 2 4 5 9 6      Akathisia 0 2 2 1 2      Parkinsonism 0 1 0 2 1 Psychiatric Disorders      Insomnia 3 3 4 3 3      Suicidal ideation 1 4 1 3 3      Anger 0 0 0 2 1 Reproductive System and Breast Disorders      Dysmenorrhea 1 0 2 0 1 Respiratory, Thoracic, and Mediastinal Disorders      Oropharyngeal pain 2 0 3 1 1      Nasal congestion 1 0 2 0 1      Dyspnea 0 0 2 0 1 Skin and Subcutaneous Tissue Disorders      Rash 1 0 1 2 1
1 Includes the preferred terms tachycardia and heart rate increased.
2 Includes the preferred terms oral hypoesthesia, oral paresthesia, and oral dysesthesia.
3 Includes the preferred terms abdominal pain, abdominal pain upper, abdominal pain lower, and abdominal discomfort.
4 Includes the preferred terms fatigue and lethargy.
5 Includes the preferred terms hyperinsulinemia and blood insulin increased.
6 Includes the preferred terms somnolence, sedation, and hypersomnia.
Dose-Related Adverse Reactions: Â In the short term pediatric bipolar I trial the incidence of fatigue appeared to be dose-related (see Table 10).
Adjunctive Therapy in Adult Patients with Bipolar Mania: Â The following findings are based on a 12 week placebo-controlled trial (with a 3 week efficacy endpoint) in adult patients with bipolar mania in which sublingual SAPHRIS was administered in doses of 5 mg or 10 mg twice daily as adjunctive therapy with lithium or valproate.
Adverse Reactions Associated with Discontinuation of Treatment: Â Approximately 16% (25/158) of SAPHRIS-treated patients discontinued treatment due to an adverse reaction, compared with about 11% (18/166) on placebo. The most common adverse reactions associated with discontinuation in subjects treated with SAPHRIS (rates at least 1% and at least twice the placebo rate) were depression (2.5%), suicidal ideation (2.5%), bipolar I disorder (1.9%), insomnia (1.9%) and depressive symptoms (1.3%).
Adverse Reactions Occurring at an Incidence of 2% or More Among SAPHRIS-Treated (Adjunctive) Bipolar I Patients: Â Adverse reactions associated with the use of SAPHRIS (incidence of 2% or greater, rounded to the nearest percent, and SAPHRIS incidence greater than placebo) that occurred during acute adjunctive therapy at 3 weeks, a time when most of the patients were still participating in the trial, are shown in Table 11.
Table 11: Adverse Reactions Reported in 2% or More of Adult Patients In Any SAPHRIS-Dose Group and Which Occurred at Greater Incidence Than in the Placebo Group at 3 Weeks in Adjunctive Bipolar Mania Trials System Organ Class/Preferred Term Placebo N=166 % SAPHRIS 5 mg or 10 mg twice daily* N=158 % Gastrointestinal disorders     Dyspepsia 2 3     Oral hypoesthesia 0 5 General disorders     Fatigue 2 4     Edema peripheral <1 3 Investigations     Increased weight 0 3 Nervous system disorders     Dizziness 2 4     Other extrapyramidal symptoms (excluding akathisia)†5 6     Somnolence‡ 10 22 Psychiatric disorders     Insomnia 8 10 Vascular disorders     Hypertension <1 3
* Â Â Â Â Â SAPHRIS 5 mg to 10 mg twice daily with flexible dosing.
†     Extrapyramidal symptoms included: dystonia, parkinsonism, oculogyration, and tremor (excluding akathisia).
‡      Somnolence includes the following events: somnolence and sedation.
Dystonia: Symptoms of dystonia, prolonged abnormal contractions of muscle groups, may occur in susceptible individuals during the first few days of treatment. Dystonic symptoms include: spasm of the neck muscles, sometimes progressing to tightness of the throat, swallowing difficulty, difficulty breathing, and/or protrusion of the tongue. While these symptoms can occur at low doses, they occur more frequently and with greater severity with high potency and at higher doses of first generation antipsychotic drugs. An elevated risk of acute dystonia is observed in males and younger age groups [see Dosage and Administration ( 2.3 ), Use in Specific Populations ( 8.4 ), and Clinical Pharmacology ( 12.3 )].
Extrapyramidal Symptoms: In the short-term, placebo-controlled schizophrenia and bipolar mania adult trials, data was objectively collected on the Simpson Angus Rating Scale for extrapyramidal symptoms (EPS), the Barnes Akathisia Scale (for akathisia) and the Assessments of Involuntary Movement Scales (for dyskinesias). The mean change from baseline for the all-SAPHRIS 5 mg or 10 mg twice daily treated group was comparable to placebo in each of the rating scale scores.
In the short-term, placebo-controlled schizophrenia adult trials, the incidence of reported EPS-related events, excluding events related to akathisia, for SAPHRIS-treated patients was 10% versus 7% for placebo; and the incidence of akathisia-related events for SAPHRIS-treated patients was 6% versus 3% for placebo. In short-term placebo-controlled bipolar mania adult trials, the incidence of EPS-related events, excluding events related to akathisia, for SAPHRIS-treated patients was 8% versus 4% for placebo; and the incidence of akathisia-related events for SAPHRIS-treated patients was 7% versus 3% for placebo. The incidence rates of all EPS events (including akathisia) were lower at the 5mg twice daily dose (11% of N=122) than the 10mg twice daily dose (25% of N=119) in a fixed-dose study.
In a 3-week, placebo-controlled pediatric trial with bipolar I disorder, the incidences of EPS-related events, excluding events related to akathisia, were 4%, 3%, and 5% for patients treated with SAPHRIS 2.5 mg, 5 mg, and 10 mg twice daily, respectively, as compared to 3% for placebo-treated patients. EPS-related events include: bradykinesia, dyskinesia, dystonia, oromandibular dystonia, muscle contractions involuntary, muscle twitching, musculoskeletal stiffness, parkinsonism, protrusion tongue, resting tremor, and tremor.
For events of akathisia, incidences were 2%, 2%, and 1% for pediatric patients treated with SAPHRIS 2.5 mg, 5 mg, and 10 mg twice daily, respectively, as compared to 0% for placebo-treated patients.
Other Findings:Â Oral hypoesthesia and/or oral paresthesia may occur directly after administration of SAPHRISÂ and usually resolves within 1 hour.
Laboratory Test Abnormalities:
Transaminases:  Transient elevations in serum transaminases (primarily ALT) in the short-term schizophrenia and bipolar mania adult trials were more common in treated patients. In short-term, placebo-controlled schizophrenia adult trials, the mean increase in transaminase levels for SAPHRIS-treated patients was 1.6 units/L compared to a decrease of 0.4 units/L for placebo-treated patients. The proportion of patients with transaminase elevations ≥3 times ULN (at Endpoint) was 0.9% for SAPHRIS-treated patients versus 1.3% for placebo-treated patients. In short-term, placebo-controlled bipolar mania adult trials, the mean increase in transaminase levels for SAPHRIS-treated patients was 6.1units/L compared to a decrease of 3.9units/L in placebo-treated patients. The proportion of patients with transaminase elevations ≥3 times upper limit of normal (ULN) (at Endpoint) was 2.1% for SAPHRIS-treated patients versus 0.7% for placebo-treated patients. The incidence rate of transaminase elevations ≥3 times ULN is 3% of N=95 for 10mg twice daily dose, and 0% of N=108 for the 5mg twice daily dose and 0% of N=115 for placebo in a fixed-dose study.
In a 52-week, double-blind, comparator-controlled trial that included primarily adult patients with schizophrenia, the mean increase from baseline of ALT was 1.7 units/L.
In a 3-week, placebo-controlled pediatric trial with bipolar I disorder, transient elevations in serum transaminases (primarily ALT) were more common in treated patients. The proportion of pediatric patients with ALT elevations ≥3 times upper limit of normal (ULN) was 2.4% for patients treated with SAPHRIS 10 mg twice daily versus none for the other SAPHRIS dose groups and placebo-treated patients. Â
Prolactin: In short-term, placebo-controlled adult schizophrenia trials, the mean decreases in prolactin levels were 6.5 ng/mL for SAPHRIS-treated patients compared to 10.7 ng/mL for placebo-treated patients. The proportion of patients with prolactin elevations ≥4 times ULN (at Endpoint) were 2.6% for SAPHRIS-treated patients versus 0.6% for placebo-treated patients. In short-term, placebo-controlled bipolar mania adult trials, the mean increase in prolactin levels was 6.7 ng/mL for SAPHRIS-treated patients compared to a decrease of 1.0 ng/mL for placebo-treated patients. The proportion of patients with prolactin elevations ≥4 times ULN (at Endpoint) were 2.0% for SAPHRIS-treated patients versus 0.8% for placebo-treated patients.
In a long-term (52-week), double-blind, comparator-controlled adult trial that included primarily patients with schizophrenia, the mean decrease in prolactin from baseline for SAPHRIS-treated patients was 26.9 ng/mL.
In a 3-week, placebo-controlled pediatric trial with bipolar I disorder, the mean increases (at Endpoint) in prolactin levels were 3.2 ng/mL for patients treated with SAPHRIS 2.5 mg twice daily, 2.1 ng/mL for patients treated with SAPHRIS 5 mg twice daily, and 6.4 ng/mL for patients treated with SAPHRIS 10 mg twice daily compared to an increase of 2.5 ng/mL for placebo-treated patients. There were no reports of prolactin elevations ≥4 times ULN (at Endpoint) for patients treated with SAPHRIS or placebo. Galactorrhea or dysmenorrhea were reported in 0% of patients treated with SAPHRIS 2.5 mg twice daily, 2% of patients treated with SAPHRIS 5 mg twice daily, and 1% of patients treated with SAPHRIS 10 mg twice daily compared to 1% of placebo-treated patients. There were no reports of gynecomastia in this trial.
Creatine Kinase (CK): The proportion of adult patients with CK elevations >3 times ULN at any time were 6.4% and 11.1% for patients treated with SAPHRIS 5 mg twice daily and 10 mg twice daily, respectively, as compared to 6.7% for placebo-treated patients in pre-marketing short-term, fixed-dose trials in schizophrenia and bipolar mania. The clinical relevance of this finding is unknown.
The proportion of patients with CK elevations ≥3 times ULN during a 3-week trial in pediatric bipolar I disorder at any time were 1%, 0%, and 1% for patients treated with SAPHRIS 2.5 mg, 5 mg, and 10 mg twice daily, respectively, versus 3% for placebo-treated patients.
O ther Adverse Reactions Observed During the Premarketing Evaluation of SAPHRIS:  Following is a ul of MedDRA terms that reflect adverse reactions reported by patients treated with sublingual SAPHRIS at multiple doses of ≥5 mg twice daily during any phase of a trial within the database of adult patients. The reactions uled are those that could be of clinical importance, as well as reactions that are plausibly drug-related on pharmacologic or other grounds. Reactions already uled for either adults or pediatric patients in other parts of Adverse Reactions ( 6 ), or those considered in Contraindications ( 4 ), Warnings and Precautions ( 5 ) or Overdosage ( 10 ) are not included. Reactions are further categorized by MedDRA system organ class and uled in order of decreasing frequency according to the following definitions: those occurring in at least 1/100 patients (frequent) (only those not already uled in the tabulated results from placebo-controlled trials appear in this uling); those occurring in 1/100 to 1/1000 patients (infrequent); and those occurring in fewer than 1/1000 patients (rare).
    ÂBlood and lymphatic disorders:   infrequent : anemia; rare: thrombocytopeniaÂ
    ÂCardiac disorders:  infrequent : temporary bundle branch block
    ÂEye disorders:  infrequent : accommodation disorder
    ÂGastrointestinal disorders:  infrequent :  swollen tongue
    ÂGeneral disorders: rare : idiosyncratic drug reaction
    ÂInvestigations:  infrequent : hyponatremia
    ÂNervous system disorders: infrequent : dysarthria
Following is a ul of MedDRA terms not already uled either for adults or pediatric patients in other parts of Adverse Reactions ( 6 ), or those considered in Contraindications ( 4 ), Warnings and Precautions ( 5 ) or Overdosage ( 10 ) that reflect adverse reactions reported by pediatric patients (Ages 10 to 17 years) treated with sublingual SAPHRIS at doses of 2.5 mg, 5 mg, or 10 mg twice daily during any phase of a trial within the database of pediatric patients.
    ÂEye disorders: infrequent: diplopia, vision blurred
    ÂGastrointestinal disorders: infrequent: gastroesophageal reflux disease
    ÂInjury, Poisoning, and Procedural Complication s :  infrequent: fall     Â
    ÂSkin and subcutaneous tissue disorders:  infrequent : photosensitivity reaction
    ÂRenal and urinary disorders: infrequent:  enuresis
POSTMARKETING EXPERIENCE SECTION
The following adverse reactions have been identified during post-approval use of SAPHRIS. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to establish a causal relationship to drug exposure. In many cases, the occurrence of these adverse reactions led to discontinuation of therapy.
     • Application site reactions, primarily in the sublingual area, have been reported. These application site reactions included oral ulcers, bulers, peeling/sloughing, and inflammation.
     • Choking has been reported by patients, some of whom may have also experienced oropharyngeal muscular dysfunction or hypoesthesia.
Drug Interactions Section
- Antihypertensive Drugs: SAPHRIS may cause hypotension. (
5.7 ,7.1 ,12.3 )- Paroxetine (CYP2D6 substrate and inhibitor): Reduce paroxetine by half when used in combination with SAPHRIS. (
7.1 ,12.3 )
Table 12: Clinically Important Drug Interactions with SAPHRIS Concomitant Drug Name or Drug Class Clinical Rationale Clinical Recommendation Antihypertensive Drugs Because of its α1-adrenergic antagonism with potential for inducing hypotension, SAPHRIS may enhance the effects of certain antihypertensive agents [see Warnings and Precautions ( 5.7 )]. Monitor blood pressure and adjust dosage of antihypertensive drug accordingly. Strong CYP1A2 Inhibitors (e.g., Fluvoxamine) SAPHRIS is metabolized by CYP1A2. Marginal increase of asenapine exposure was observed when SAPHRIS is used with fluvoxamine at 25 mg administered twice daily [ see Clinical Pharmacology ( 12.3 )]. However, the tested fluvoxamine dose was suboptimal. Full therapeutic dose of fluvoxamine is expected to cause a greater increase in asenapine exposure.   Dosage reduction for SAPHRIS based on clinical response may be necessary. CYP2D6 substrates and inhibitors (e.g., paroxetine) SAPHRIS may enhance the inhibitory effects of paroxetine on its own metabolism. Concomitant use of paroxetine with SAPHRIS increased the paroxetine exposure by 2-fold as compared to use paroxetine alone [see Clinical Pharmacology ( 12.3 )]. Reduce paroxetine dose by half when paroxetine is used in combination with SAPHRIS.
No dosage adjustment of SAPHRIS is necessary when administered concomitantly with paroxetine (see Table 12 in Drug Interactions (7.1) for paroxetine dosage adjustment), imipramine, cimetidine, valproate, lithium, or a CYP3A4 inducer (e.g., carbamazepine, phenytoin, rifampin).
In addition, valproic acid and lithium pre-dose serum concentrations collected from an adjunctive therapy study were comparable between asenapine-treated patients and placebo-treated patients indicating a lack of effect of asenapine on valproic and lithium plasma levels.
Use In Specific Populations Section
- Pregnancy: May cause extrapyramidal and/or withdrawal symptoms in neonates with third trimester exposure.
 ( 8.1 )- Pediatric Use: Safety and efficacy in the treatment of bipolar I disorder in patients less than 10 years of age, and patients with schizophrenia ages less than 12 years have not been evaluated.
 ( 8.4 )8.1
Pregnancy Exposure Registry
There is a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to SAPHRIS during pregnancy. For more information contact the National Pregnancy Registry for Atypical Antipsychotics at 1-866-961-2388 or visit http://womensmentalhealth.org/clinical-and-research-programs/pregnancyregistry/.
Risk Summary
Neonates exposed to antipsychotic drugs during the third trimester of pregnancy are at risk for extrapyramidal and/or withdrawal symptoms. Studies have not been conducted with SAPHRIS in pregnant women. There are no available human data informing the drug-associated risk. The background risk of major birth defects and miscarriage for the indicated populations are unknown. However, the background risk in the U.S. general population of major birth defects is 2-4% and of miscarriage is 15-20% of clinically recognized pregnancies.  No teratogenicity was observed in animal reproduction studies with intravenous administration of asenapine to rats and rabbits during organogenesis at doses 0.7 and 0.4 times, respectively, the maximum recommended human dose (MRHD) of 10 mg sublingually twice daily. In a pre-and post-natal study in rats, intravenous administration of asenapine at doses up to 0.7 times the MRHD produced increases in post-implantation loss and early pup deaths, and decreases in subsequent pup survival and weight gain [see Data]. Advise pregnant women of the potential risk to a fetus.Â
Clinical Considerations
Fetal/Neonatal Adverse Reactions
Extrapyramidal and/or withdrawal symptoms, including agitation, hypertonia, hypotonia, tremor, somnolence, respiratory distress and feeding disorder have been reported in neonates who were exposed to antipsychotic drugs during the third trimester of pregnancy. These symptoms have varied in severity. Some neonates recovered within hours or days without specific treatment; others required prolonged hospitalization. Monitor neonates for extrapyramidal and/or withdrawal symptoms and manage symptoms appropriately.
Data
Animal Data
In animal studies, asenapine increased post-implantation loss and decreased pup weight and survival at doses similar to or less than recommended clinical doses. In these studies there was no increase in the incidence of structural abnormalities caused by asenapine.
Asenapine was not teratogenic in reproduction studies in rats and rabbits at intravenous doses up to 1.5 mg/kg in rats and 0.44 mg/kg in rabbits administered during organogenesis. These doses are 0.7 and 0.4 times, respectively, the maximum recommended human dose (MRHD) of 10 mg twice daily given sublingually on a mg/m2 basis. Plasma levels of asenapine were measured in the rabbit study, and the area under the curve (AUC) at the highest dose tested was 2 times that in humans receiving the MRHD.
In a study in which rats were treated from day 6 of gestation through day 21 postpartum with intravenous doses of asenapine of 0.3, 0.9, and 1.5 mg/kg/day (0.15, 0.4, and 0.7 times the MRHD of 10 mg twice daily given sublingually on a mg/m2 basis), increases in post-implantation loss and early pup deaths were seen at all doses, and decreases in subsequent pup survival and weight gain were seen at the two higher doses. A cross-fostering study indicated that the decreases in pup survival were largely due to prenatal drug effects. Increases in post-implantation loss and decreases in pup weight and survival were also seen when pregnant rats were dosed orally with asenapine.
LACTATION SECTION
Risk Summary
Lactation studies have not been conducted to assess the presence of asenapine in human milk, the effects of asenapine on the breastfed infant, or the effects of asenapine on milk production. Asenapine is excreted in rat milk. The development and health benefits of breastfeeding should be considered along with the mother’s clinical need for SAPHRIS and any potential adverse effects on the breastfed infant from SAPHRIS or from the underlying maternal condition.
8.4
Safety and efficacy of SAPHRIS in pediatric patients below the age of 10 years of age have not been evaluated.
Bipolar I Disorder
The safety and efficacy of SAPHRIS as monotherapy in the treatment of bipolar I disorder were established in a 3-week, placebo-controlled, double-blind trial of 403 pediatric patients 10 to 17 years of age, of whom 302 patients received SAPHRIS at fixed doses ranging from 2.5 mg to 10 mg twice daily [see Dosage and Administration ( 2.3 ), Adverse Reactions ( 6.1 ), Clinical Pharmacology ( 12.3 ), and Clinical Studies ( 14.2 )]. In a Phase 1 study, pediatric patients aged 10 to 17 years appeared to be more sensitive to dystonia with initial dosing with asenapine when the recommended dose escalation schedule was not followed. Similar safety findings were reported from a 50-week, open-label, uncontrolled safety trial in pediatric patients with bipolar I disorder treated with SAPHRIS monotherapy. The safety and efficacy of SAPHRIS as adjunctive therapy in the treatment of bipolar I disorder have not been established in the pediatric population. In general, the pharmacokinetics of asenapine in pediatric patients (10 to 17 years) and adults are similar [see Clinical Pharmacology ( 12.3 )].
Schizophrenia
Efficacy of SAPHRIS was not demonstrated in an 8-week, placebo-controlled, double-blind trial, in 306 adolescent patients aged 12 to 17 years with schizophrenia at doses of 2.5 and 5 mg twice daily. The most common adverse reactions (proportion of patients equal or greater than 5% and at least twice placebo) reported were somnolence, akathisia, dizziness, and oral hypoesthesia or paresthesia. The proportion of patients with an equal or greater than 7% increase in body weight at endpoint compared to baseline for placebo, SAPHRIS 2.5 mg twice daily, and SAPHRIS 5 mg twice daily was 3%, 10%, and 10%, respectively.
The clinically relevant adverse reactions identified in the pediatric schizophrenia trial were generally similar to those observed in the pediatric bipolar I and adult bipolar I and schizophrenia trials. No new major safety findings were reported from a 26-week, open-label, uncontrolled safety trial in pediatric patients with schizophrenia treated with SAPHRIS monotherapy.
Juvenile Animal Data
Subcutaneous administration of asenapine to juvenile rats for 56 days from day 14 of age to day 69 of age at 0.4, 1.2, and 3.2 mg/kg/day (0.2, 0.6 and 1.5 times the maximum recommended human dose of 10 mg twice daily given sublingually on a mg/m2 basis) resulted in significant reduction in body weight gain in animals of both sexes at all dose levels from the start of dosing until weaning. Body weight gain remained reduced in males to the end of treatment, however, recovery was observed once treatment ended. Neurobehavioral assessment indicated increased motor activity in animals at all dose levels following the completion of treatment, with the evidence of recovery in males. There was no recovery after the end of treatment in female activity pattern as late as day 30 following the completion of treatment (last retesting). Therefore, a No Observed Adverse Effect Level (NOAEL) for the juvenile animal toxicity of asenapine could not be determined. There were no treatment-related effects on the startle response, learning/memory, organ weights, microscopic evaluations of the brain and, reproductive performance (except for minimally reduced conception rate and fertility index in males and females administered 1.2 and 3.2 mg/kg/day).
8.5
Clinical studies of SAPHRIS in the treatment of schizophrenia and bipolar mania did not include sufficient numbers of patients aged 65 and over to determine whether or not they respond differently than younger patients. Of the approximately 2250 patients in pre-marketing clinical studies of SAPHRIS, 1.1% (25) were 65 years of age or over. Multiple factors that might increase the pharmacodynamic response to SAPHRIS, causing poorer tolerance or orthostasis, could be present in elderly patients, and these patients should be monitored carefully. Based on a pharmacokinetic study in elderly patients, dosage adjustments are not recommended based on age alone [see Clinical Pharmacology ( 12.3 )].
Elderly patients with dementia-related psychosis treated with SAPHRIS are at an increased risk of death compared to placebo. SAPHRIS is not approved for the treatment of patients with dementia-related psychosis [ see Boxed Warning ].
8.6
No dosage adjustment for SAPHRIS is required on the basis of a patient’s renal function (mild to severe renal impairment, glomerular filtration rate between 15 and 90 mL/minute). The exposure of asenapine was similar among subjects with varying degrees of renal impairment and subjects with normal renal function [ see Clinical Pharmacology ( 12.3 )]. The effect of renal function on the excretion of other metabolites and the effect of dialysis on the pharmacokinetics of asenapine has not been studied.
8.7
SAPHRIS is contraindicated in patients with severe hepatic impairment (Child-Pugh C)Â because asenapine exposure is 7-fold higher in subjects with severe hepatic impairment than the exposure observed in subjects with normal hepatic function.
No dosage adjustment for SAPHRIS is required in patients with mild to moderate hepatic impairment (Child-Pugh A and B) because asenapine exposure is similar to that in subjects with normal hepatic function [ see Contraindications ( 4 ) and Clinical Pharmacology ( 12.3 )].
No dosage adjustment for SAPHRIS is required on the basis of a patient’s sex, race (Caucasian and Japanese), or smoking status [see Clinical Pharmacology ( 12.3 )].
Drug Abuse And Dependence Section
9.1
SAPHRIS is not a controlled substance.
9.2
SAPHRIS has not been systematically studied in animals or humans for its abuse potential or its ability to induce tolerance or physical dependence. Thus, it is not possible to predict the extent to which a CNS-active drug will be misused, diverted and/or abused once it is marketed. Patients should be evaluated carefully for a history of drug abuse, and such patients should be observed carefully for signs that they are misusing or abusing SAPHRIS (e.g., drug-seeking behavior, increases in dose).
Overdosage Section
Human Experience: In adult pre-marketing clinical studies involving more than 3350 patients and/or healthy subjects, accidental or intentional acute overdosage of SAPHRIS was identified in 3 patients. Among these few reported cases of overdose, the highest estimated ingestion of SAPHRIS was 400 mg. Reported adverse reactions at the highest dosage included agitation and confusion.
Management of Overdosage: There is no specific antidote to SAPHRIS. The possibility of multiple drug involvement should be considered. An electrocardiogram should be obtained and management of overdose should concentrate on supportive therapy, maintaining an adequate airway, oxygenation and ventilation, and management of symptoms. Consult with a Certified Poison Control Center for up-to-date guidance and advice on the management of overdosage (1-800-222-1222.)
Hypotension and circulatory collapse should be treated with appropriate measures, such as intravenous fluids and/or sympathomimetic agents (epinephrine and dopamine should not be used, since beta stimulation may worsen hypotension in the setting of SAPHRIS-induced alpha blockade). In case of severe extrapyramidal symptoms, anticholinergic medication should be administered. Close medical supervision and monitoring should continue until the patient recovers.
Description Section
SAPHRIS contains asenapine maleate which is an atypical antipsychotic that is available for sublingual administration. Asenapine belongs to the class dibenzo-oxepino pyrroles. The chemical designation is (3aRS,12bRS)-5-Chloro-2-methyl-2,3,3a,12b-tetrahydro-1Hdibenzo[2,3:6,7]oxepino[4,5-c]pyrrole (2Z)-2-butenedioate (1:1). Its molecular formula is C17H16ClNO•C4H4O4 and its molecular weight is 401.84 (free base: 285.8). The chemical structure is:
    Â
Asenapine maleate is a white to off-white powder.
SAPHRIS, black cherry flavor, is supplied for sublingual administration in tablets containing 2.5Â mg, 5Â mg or 10Â mg asenapine; inactive ingredients include gelatin, mannitol, sucralose, and black cherry flavor.
Clinical Pharmacology Section
12.1
The mechanism of action of asenapine, in schizophrenia and bipolar I disorder, is unknown. It has been suggested that the efficacy of asenapine in schizophrenia could be mediated through a combination of antagonist activity at D2 and 5-HT2A receptors.
12.2
Asenapine exhibits high affinity for serotonin 5-HT1A, 5-HT1B, 5-HT2A, 5-HT2B, 5-HT2C, 5-HT5A, 5-HT6, and 5-HT7 receptors (Ki values of 2.5, 2.7, 0.07, 0.18, 0.03, 1.6, 0.25, and 0.11 nM, respectively), dopamine D2A, D2B, D3, D4, and D1 receptors (Ki values of 1.3, 1.4, 0.42, 1.1, and 1.4 nM, respectively), α1A, α2A, α2B, and α2C -adrenergic receptors (Ki values of 1.2, 1.2, 0.33 and 1.2 nM, respectively), and histamine H1 receptors (Ki value 1.0 nM), and moderate affinity for H2 receptors (Ki value of 6.2 nM). In in vitro assays asenapine acts as an antagonist at these receptors. Asenapine has no appreciable affinity for muscarinic cholinergic receptors (e.g., Ki value of 8128 nM for M1).
12.3
Following a single 5Â mg dose of SAPHRIS, the mean Cmax was approximately 4 ng/mL and was observed at a mean tmax of 1 hour. Elimination of asenapine is primarily through direct glucuronidation by UGT1A4 and oxidative metabolism by cytochrome P450 isoenzymes (predominantly CYP1A2). Following an initial more rapid distribution phase, the mean terminal half-life is approximately 24 hrs. With multiple-dose twice-daily dosing, steady-state is attained within 3 days. Overall, steady-state asenapine pharmacokinetics are similar to single-dose pharmacokinetics.
Absorption:Â Following sublingual administration, asenapine is rapidly absorbed with peak plasma concentrations occurring within 0.5 to 1.5 hours. The absolute bioavailability of sublingual asenapine at 5 mg is 35%. Increasing the dose from 5 mg to 10 mg twice daily (a two-fold increase) results in less than linear (1.7 times) increases in both the extent of exposure and maximum concentration. The absolute bioavailability of asenapine when swallowed is low (<2% with an oral tablet formulation).
The intake of water several (2 or 5) minutes after asenapine administration resulted in decreased asenapine exposure. Therefore, eating and drinking should be avoided for 10 minutes after administration [see Dosage and Administration ( 2.1 )].
Distribution: Asenapine is rapidly distributed and has a large volume of distribution (approximately 20 - 25 L/kg), indicating extensive extravascular distribution. Asenapine is highly bound (95%) to plasma proteins, including albumin and α1-acid glycoprotein.
Metabolism and Elimination:Â Direct glucuronidation by UGT1A4 and oxidative metabolism by cytochrome P450 isoenzymes (predominantly CYP1A2) are the primary metabolic pathways for asenapine.
Asenapine is a high clearance drug with a clearance after intravenous administration of 52 L/h. In this circumstance, hepatic clearance is influenced primarily by changes in liver blood flow rather than by changes in the intrinsic clearance, i.e., the metabolizing enzymatic activity. Following an initial more rapid distribution phase, the terminal half-life of asenapine is approximately 24 hours. Steady-state concentrations of asenapine are reached within 3 days of twice daily dosing.
After administration of a single dose of [14C]-labeled asenapine, about 90% of the dose was recovered; approximately 50% was recovered in urine, and 40% recovered in feces. About 50% of the circulating species in plasma have been identified. The predominant species was asenapine N+-glucuronide; others included N-desmethylasenapine, N-desmethylasenapine N-carbamoyl glucuronide, and unchanged asenapine in smaller amounts. SAPHRIS activity is primarily due to the parent drug.
In vitro studies indicate that asenapine is a substrate for UGT1A4, CYP1A2 and to a lesser extent CYP3A4 and CYP2D6. Asenapine is a weak inhibitor of CYP2D6. Asenapine does not cause induction of CYP1A2 or CYP3A4 activities in cultured human hepatocytes. Coadministration of asenapine with known inhibitors, inducers or substrates of these metabolic pathways has been studied in a number of drug-drug interaction studies [ see Drug Interactions ( 7.1 )].
Food: A crossover study in 26 healthy adult male subjects was performed to evaluate the effect of food on the pharmacokinetics of a single 5Â mg dose of asenapine. Consumption of food immediately prior to sublingual administration decreased asenapine exposure by 20%; consumption of food 4 hours after sublingual administration decreased asenapine exposure by about 10%. These effects are probably due to increased hepatic blood flow.
In clinical trials establishing the efficacy and safety of SAPHRIS, patients were instructed to avoid eating for 10 minutes following sublingual dosing. There were no other restrictions with regard to the timing of meals in these trials [ see Dosage and Administration ( 2.1 ) ].
Water: In clinical trials establishing the efficacy and safety of SAPHRIS, patients were instructed to avoid drinking for 10 minutes following sublingual dosing. The effect of water administration following 10 mg sublingual SAPHRIS dosing was studied at different time points of 2, 5, 10, and 30 minutes in 15 healthy adult male subjects. The exposure of asenapine following administration of water 10 minutes after sublingual dosing was equivalent to that when water was administered 30 minutes after dosing. Reduced exposure to asenapine was observed following water administration at 2 minutes (19% decrease) and 5 minutes (10% decrease) [ see Dosage and Administration ( 2.1 ) ].
Drug Interaction Studies:
Effects of other drugs on the exposure of asenapine are summarized in Figure 1. In addition, a population pharmacokinetic analysis indicated that the concomitant administration of lithium had no effect on the pharmacokinetics of asenapine.
Figure 1: Effect of Other Drugs on Asenapine Pharmacokinetics
    Â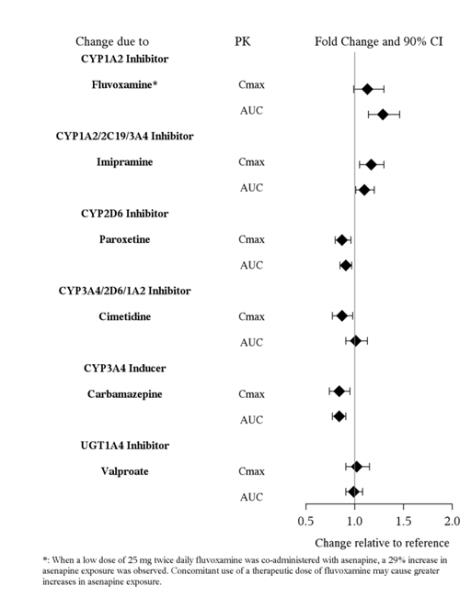
The effects of asenapine on the pharmacokinetics of other co-administered drugs are summarized in Figure 2.
Coadministration of paroxetine with SAPHRIS caused a two-fold increase in the maximum plasma concentrations and systemic exposure of paroxetine. Asenapine enhances the inhibitory effects of paroxetine on its own metabolism by CYP2D6.
Figure 2: Effect of Asenapine on Other Drug Pharmacokinetics
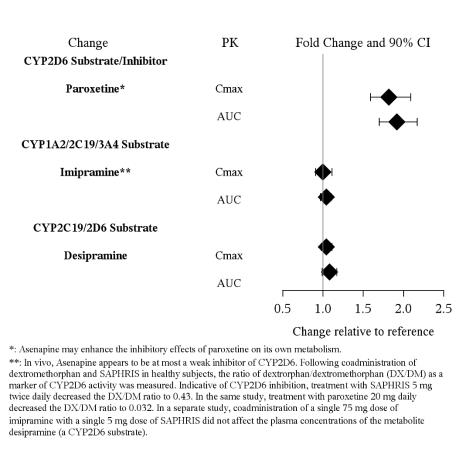
Studies in Special Populations:
Exposures of asenapine in special populations are summarized in Figure 3. Additionally, based on population pharmacokinetic analysis, no effects of sex, race, BMI, and smoking status on asenapine exposure were observed. Exposure in elderly patients is 30-40% higher as compared to adults. Â
Figure 3: Effect of Intrinsic Factors on Asenapine Pharmacokinetics
    Â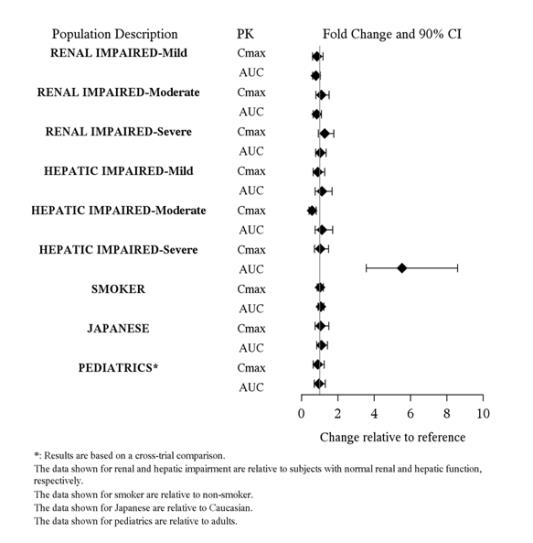
Nonclinical Toxicology Section
13.1
Carcinogenesis: In a lifetime carcinogenicity study in CD-1 mice asenapine was administered subcutaneously at doses up to those resulting in plasma levels (AUC) estimated to be 5 times those in humans receiving the MRHD of 10 mg twice daily. The incidence of malignant lymphomas was increased in female mice, with a no-effect dose resulting in plasma levels estimated to be 1.5 times those in humans receiving the MRHD. The mouse strain used has a high and variable incidence of malignant lymphomas, and the significance of these results to humans is unknown. There were no increases in other tumor types in female mice. In male mice, there were no increases in any tumor type.
In a lifetime carcinogenicity study in Sprague-Dawley rats, asenapine did not cause any increases in tumors when administered subcutaneously at doses up to those resulting in plasma levels (AUC) estimated to be 5 times those in humans receiving the MRHD.
Mutagenesis: No evidence for genotoxic potential of asenapine was found in the in vitro bacterial reverse mutation assay, the in vitro forward gene mutation assay in mouse lymphoma cells, the in vitro chromosomal aberration assays in human lymphocytes, the in vitro sister chromatid exchange assay in rabbit lymphocytes, or the in vivo micronucleus assay in rats.
Impairment of Fertility: Asenapine did not impair fertility in rats when tested at doses up to 11 mg/kg twice daily given orally. This dose is 10 times the maximum recommended human dose of 10 mg twice daily given sublingually on a mg/m2 basis.
Clinical Studies Section
Efficacy of SAPHRIS was established in the following trials:
- Two fixed-dose, short-term trials and one flexible-dose, maintenance trial in adult patients with schizophrenia as monotherapy [see Clinical Studies ( 14.1 )]
- One fixed-dose and two flexible-dose, short-term trials of monotherapy in adults with manic or mixed episodes associated with bipolar I disorder [see Clinical Studies ( 14.2 )]
- One flexible-dose, maintenance trial of monotherapy in adults with bipolar I disorder [see Clinical Studies ( 14.2 )]
- One fixed-dose, short term trial of monotherapy in children (10 to 17 years) with manic or mixed episodes associated with bipolar I disorder [see Clinical Studies ( 14.2 )]
- One flexible-dose, short-term trial in adult patients with manic or mixed episode associated with bipolar I disorder as adjunctive treatment to lithium or valproate [see Clinical Studies ( 14.2 )]
14.1
The efficacy of SAPHRIS in the treatment of schizophrenia in adults was evaluated in three fixed-dose, short-term (6Â week), randomized, double-blind, placebo-controlled, and active-controlled (haloperidol, risperidone, and olanzapine) trials of adult patients who met DSM-IV criteria for schizophrenia and were having an acute exacerbation of their schizophrenic illness. In two of the three trials SAPHRIS demonstrated superior efficacy to placebo. In a third trial, SAPHRIS could not be distinguished from placebo; however, an active control in that trial was superior to placebo.
In the two positive trials for SAPHRIS, the primary efficacy rating scale was the Positive and Negative Syndrome Scale (PANSS). The PANSS is a 30 li scale that measures positive symptoms of schizophrenia (7 lis), negative symptoms of schizophrenia (7 lis), and general psychopathology (16 lis), each rated on a scale of 1 (absent) to 7 (extreme); total PANSS scores range from 30 to 210. The primary endpoint was change from baseline to endpoint on the PANSS total score. The results of the SAPHRIS trials in schizophrenia follow:
In trial 1, a 6-week trial (n=174), comparing SAPHRIS (5 mg twice daily) to placebo, SAPHRIS 5 mg twice daily was statistically superior to placebo on the PANSS total score (Trial 1 in Table 13).
In trial 2, a 6-week trial (n=448), comparing two fixed doses of SAPHRIS (5 mg and 10 mg twice daily) to placebo, SAPHRIS 5 mg twice daily was statistically superior to placebo on the PANSS total score. SAPHRIS 10 mg twice daily showed no added benefit compared to 5 mg twice daily and was not significantly different from placebo (Trial 2 in Table 13).
An examination of population subgroups did not reveal any clear evidence of differential responsiveness on the basis of age, sex or race.
Table 1 3 : Short-Term Schizophrenia Trials Establishing Efficacy in Adults Trial Number Treatment Group Primary Efficacy Measure: PANSS Total Score Mean Baseline Score (SD) LS Mean Change from Baseline (SE) Placebo-subtracted Difference a  (95% CI) Trial 1 SAPHRIS 5 mg* twice daily 96.5 (16.4) -14.4 (2.6) -9.7 (-17.6, -1.8) Placebo 92.4 (14.9) -4.6 (2.5) -- Trial 2 SAPHRIS 5 mg* twice daily 89.2 (12.0) -16.2 (1.7) -5.5 (-10.7, -0.2) SAPHRIS 10 mg twice daily 89.1 (12.9) -14.9 (1.7) -4.1 (-9.4, 1.2) Placebo 88.9 (11.7) -10.7 (1.6) -- SD: standard deviation; SE: standard error; LS Mean: least-squares mean; CI: confidence interval, not adjusted for multiple comparisons. a Difference (drug minus placebo) in least-squares mean change from baseline. * Doses that are demonstrated to be effective.
Maintenance of efficacy has been demonstrated in a placebo-controlled, double-blind, multicenter, flexible dose (5 mg or 10 mg twice daily based on tolerability) clinical trial with a randomized withdrawal design. All patients were initially administered 5 mg twice daily for 1 week and then titrated up to 10 mg twice daily. A total of 700 patients entered open-label treatment with SAPHRIS for a period of 26 weeks. Of these, a total of 386 patients who met pre-specified criteria for continued stability (mean length of stabilization was 22 weeks) were randomized to a double-blind, placebo-controlled, randomized withdrawal phase. SAPHRIS was statistically superior to placebo in time to relapse or impending relapse defined as increase in PANSS ≥20% from baseline and a Clinical Global Impression–Severity of Illness (CGI-S) score ≥4 (at least 2 days within 1 week) or PANSS score ≥5 on “hostility” or “uncooperativeness” lis and CGI-S score ≥4 (≥2 days within a week), or PANSS score ≥5 on any two of the following lis: “unusual thought content,” “conceptual disorganization,” or “hallucinatory behavior” lis, and CGI-S score ≥4 (≥2 days within 1 week) or investigator judgment of worsening symptoms or increased risk of violence to self (including suicide) or other persons. The Kaplan-Meier curves of the time to relapse or impending relapse during the double-blind, placebo-controlled, randomized withdrawal phase of this trial for SAPHRIS and placebo are shown in Figure 4.
Figure 4: Kaplan-Meier Estimation of Percent Relapse/Impending Relapse for SAPHRIS and placebo
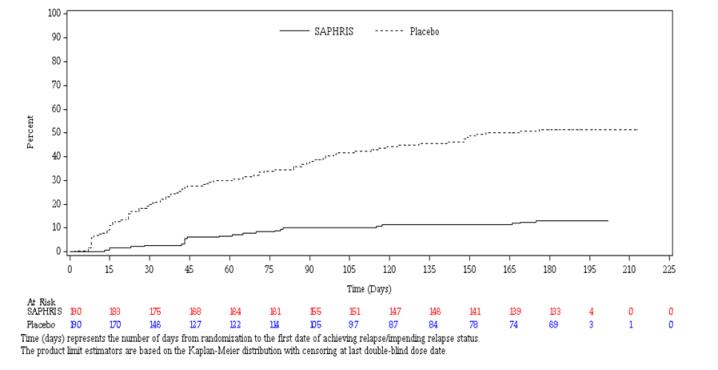
14.2
Monotherapy
Adults: Â The efficacy of SAPHRIS in the treatment of acute mania was established in two similarly designed 3-week, randomized, double-blind, placebo-controlled, and active-controlled (olanzapine) trials of adult patients who met DSM-IV criteria for Bipolar I Disorder with an acute manic or mixed episode with or without psychotic features.
The primary rating instrument used for assessing manic symptoms in these trials was the Young Mania Rating Scale (YMRS), an 11-li clinician-rated scale traditionally used to assess the degree of manic symptomatology in a range from 0 (no manic features) to 60 (maximum score). Patients were also assessed on the Clinical Global Impression – Bipolar (CGI-BP) scale. In both trials, all patients randomized to SAPHRIS were initially administered 10 mg twice daily, and the dose could be adjusted within the dose range of 5 to 10 mg twice daily from Day 2 onward based on efficacy and tolerability. Ninety percent of patients remained on the 10 mg twice daily dose. SAPHRIS was statistically superior to placebo on the YMRS total score and the CGI-BP Severity of Illness score (mania) in both studies (Trials 1 and 2 in Table 14).
In another 3-week, randomized, double-blind, placebo-controlled trial (n=359), comparing two fixed doses of SAPHRIS (5 mg and 10 mg twice daily) to placebo, both doses were statistically superior to placebo on the YMRS total score and CGI-BP Severity of Illness overall score. (Trial 3 in Table 14).Â
An examination of subgroups did not reveal any clear evidence of differential responsiveness on the basis of age, sex, or race.
Maintenance of efficacy has been demonstrated in a placebo-controlled, double-blind, multicenter, flexible dose (5 mg or 10 mg twice daily based on tolerability) clinical trial with a randomized withdrawal design. All patients were initially administered 5 or 10 mg twice daily, and the option to titrate down to 5 mg twice daily was provided based on tolerability. A total of 549 patients entered open-label treatment with SAPHRIS for a period of 12 to 16 weeks. Of these, a total of 252 patients who met pre-specified criteria for continued stability were randomized to and treated in a double-blind, placebo-controlled, randomized withdrawal phase. SAPHRIS was statistically superior to placebo in time to relapse defined as 1) YMRS or MADRS score ≥ 16; 2) requirement or initiation of any non-study medication to treat mixed, manic, or depressive symptoms, including an antipsychotic, antidepressant, or mood-stabilizing agent; 3) requirement or initiation of psychiatric hospitalization;  4) investigator judgment to discontinue the study due to a mood event. The Kaplan-Meier curves of the time to relapse during the double-blind, placebo-controlled, randomized withdrawal phase of this trial for SAPHRIS and placebo are shown in Figure 5.
Figure 5: Kaplan-Meier Estimation of Percent Relapse for SAPHRIS and Placebo
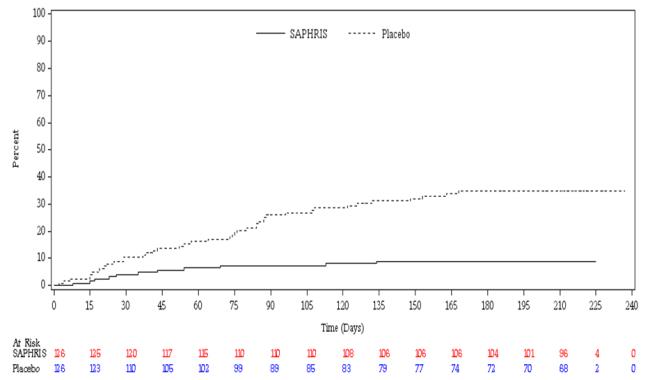
     Time (days) represents the number of days from randomization in the double-blind period to the first date of achieving relapse status.
     The product limit estimators are based on the Kaplan-Meier distribution with censoring at last contact date.
Pediatric patients: The efficacy of SAPHRIS in the treatment of acute mania was established in a single, 3-week, placebo-controlled, double-blind trial of 403 pediatric patients 10 to 17 years of age, of whom 302 patients received SAPHRIS at fixed doses of 2.5 mg, 5 mg and 10 mg twice daily. All patients were started on 2.5 mg twice daily. For those assigned to 5 mg twice daily, the dose was increased to 5 mg twice daily after 3 days. For those assigned to 10 mg twice daily, the dose was increased from 2.5 to 5 mg twice daily after 3 days, and then to 10 mg twice daily after 3 additional days.Â
SAPHRIS was statistically superior to placebo in improving YMRS total score and the CGI-BP Severity of Illness overall score as measured by the change from baseline to week 3 (Trial 3 Pediatric in Table 14). An examination of subgroups did not reveal any clear evidence of differential responsiveness on the basis of age, sex, and race.
Adjunctive Therapy: The efficacy of SAPHRIS as an adjunctive therapy in acute mania was established in a 12-week, placebo-controlled trial with a 3-week primary efficacy endpoint involving 326 adult patients with a manic or mixed episode of Bipolar I Disorder, with or without psychotic features, who were partially responsive to lithium or valproate monotherapy after at least 2 weeks of treatment. All patients randomized to SAPHRIS were initially administered 5 mg twice daily, and the dose could be adjusted within the dose range of 5 to 10 mg twice daily from Day 2 onward based on efficacy and tolerability. SAPHRIS was statistically superior to placebo in the reduction of manic symptoms (measured by the YMRS total score) as an adjunctive therapy to lithium or valproate monotherapy at Week 3 (Trial 5 Adjunctive in Table 14).
Table 1 4 : Acute Bipolar I Trials Establishing Efficacy in Adults and Pediatric Patients 10 to 17 Years Study Number Treatment Group Primary Efficacy Measure: YMRS Total Score Mean Baseline Score (SD) LS Mean Change from Baseline (SE) Placebo-subtracted Difference a (95% CI) Trial 1 SAPHRIS 5-10 mg* twice daily 29.4 (6.7) -11.5 (0.8) -3.7 (-6.6, -0.7) Placebo 28.3 (6.3) -7.8 (1.1) -- Trial 2 SAPHRIS 5-10 mg* twice daily 28.3 (5.5) -10.8 (0.8) -5.3 (-8.0, -2.5) Placebo 29.0 (6.1) -5.5 (1.0) -- Trial 3 SAPHRIS 5 mg* twice dailySAPHRIS 10 mg* twice dailyPlacebo 29.7 (5.9)30.2 (5.4)30.0 (5.6) -14.4 (1.0)-14.9 (1.0)-10.9 (1.0) -3.5 (-6.3, -0.7)-4.0 (-6.9, -1.2)-- Trial 4(Pediatric 10 to 17 years) SAPHRIS 2.5 mg* twice daily 29.5 (5.7) -12.8 (0.8) -3.2 (-5.6, -0.8) SAPHRIS 5 mg* twice daily 30.4 (5.9) -14.9 (0.8) -5.3 (-7.7, -2.9) SAPHRIS 10 mg* twice daily 30.1 (5.7) -15.8 (0.9) -6.2 (-8.6, -3.8) Placebo 30.1 (5.7) - 9.6 (0.9) -- Trial 5 (Adjunctive) SAPHRIS 5-10 mg* twice daily + lithium/ Valproate 28.0 (5.6) -10.3 (0.8) -2.4 (-4.4, -0.3) Lithium/Valproate 28.2 (5.8) -7.9 (0.8) -- SD: standard deviation; SE: standard error; LS Mean: least-squares mean; CI: confidence interval, not adjusted for multiple comparisons. a  Difference (drug minus placebo) in least-squares mean change from baseline.   * Doses that are demonstrated to be effective.
How Supplied Section
SAPHRIS (asenapine) sublingual tablets are supplied as:
2.5 Â mg Tablets, black cherry flavor
Round, white to off-white sublingual tablets, with a hexagon on one side.
Child-resistant packaging
     Box of 60      6 bulers with 10 tablets                NDC 0456-2402-60
Hospital Unit Dose
     Box of 100      10 bulers with 10 tablets           NDC 0456-2402-63
5 Â mg Tablets, black cherry flavor
Round, white to off-white sublingual tablets, with “5” on one side within a circle.
Child-resistant packaging
     Box of 60      6 bulers with 10 tablets           NDC 0456-2405-60
Hospital Unit Dose
     Box of 100      10 bulers with 10 tablets      NDC 0456-2405-63
10 Â mg Tablets, black cherry flavor
Round, white to off-white sublingual tablets, with “10” on one side within a circle.
Child-resistant packaging
     Box of 60      6 bulers with 10 tablets           NDC 0456-2410-60
Hospital Unit Dose
     Box of 100      10 bulers with 10 tablets      NDC 0456-2410-63
Storage
Store at 20 º C to 25 º C  (68 º F to 77 º F) ; excursions permitted between 15 º C  and 30 º C (59 º F  and 86 º F) [see USP Controlled Room Temperature].
Information For Patients Section
Advise the patient to read the FDA-approved patient labeling ( Instructions for Use ).
Dosage and Administration
Counsel patients on proper sublingual administration of SAPHRIS and advise them to read the FDA-approved patient labeling (Instructions for Use). When initiating treatment with SAPHRIS, provide dosage escalation instructions [see Dosage and Administration ( 2 )].Â
Hypersensitivity Reactions
Counsel patients on the signs and symptoms of a serious allergic reaction (e.g., difficulty breathing, itching, swelling of the face, tongue or throat, feeling lightheaded etc.) and to seek immediate emergency assistance if they develop any of these signs and symptoms [see Contraindications ( 4 ), Warnings and Precautions ( 5.6 ) and Adverse Reactions ( 6 )].
Application Site Reactions
Inform patients that application site reactions, primarily in the sublingual area, including oral ulcers, bulers, peeling/sloughing and inflammation have been reported. Instruct patients to monitor for these reactions [see Adverse Reactions ( 6.2 )] . Inform patients that numbness or tingling of the mouth or throat may occur directly after administration of SAPHRIS and usually resolves within 1 hour [ see Adverse R eactions ( 6.1 ) ].
Neuroleptic Malignant Syndrome
Counsel patients about a potentially fatal symptom complex sometimes referred to as Neuroleptic Malignant Syndrome (NMS) that has been reported in association with administration of antipsychotic drugs. Patients should contact their health care provider or report to the emergency room if they experience the following signs and symptoms of NMS, including hyperpyrexia, muscle rigidity, altered mental status, and evidence of autonomic instability (irregular pulse or blood pressure, tachycardia, diaphoresis, and cardiac dysrhythmia) [see Warnings and Precautions ( 5.3 )].
Tardive Dyskinesia
Counsel patients on the signs and symptoms of tardive dyskinesia and to contact their health care provider if these abnormal movements occur [ see Warnings and Precautions ( 5.4 ) ] .Â
Metabolic Changes ( Hyperglycemia and Diabetes Mellitus , Dyslipidemia, and Weight Gain)
Educate patients about the risk of metabolic changes, how to recognize symptoms of hyperglycemia (high blood sugar) and diabetes mellitus, and the need for specific monitoring, including blood glucose, lipids, and weight [see Warnings and Precautions ( 5.5 )].
Orthostatic Hypotension
Educate patients about the risk of orthostatic hypotension (symptoms include feeling dizzy or lightheaded upon standing) especially early in treatment, and also at times of re-initiating treatment or increases in dose [ see Warnings and Precautions ( 5.7 ) ].
Leukopenia/Neutropenia
Advise patients with a pre-existing low WBC or a history of drug induced leukopenia/neutropenia they should have their CBC monitored while taking SAPHRIS [s ee Warnings and Precautions ( 5.9 )].
H yperprolactinemia
Counsel patients on the signs and symptoms of hyperprolactinemia and to contact their health care provider if these abnormalities occur [ see Warnings and Precautions ( 5.11 ) ] .
Interference with Cognitive and Motor Performance
Caution patients about performing activities requiring mental alertness, such as operating hazardous machinery or operating a motor vehicle, until they are reasonably certain that SAPHRIS therapy does not affect them adversely [see Warnings and Precautions ( 5.13 )].
Heat Exposure and Dehydration
Counsel patients regarding appropriate care in avoiding overheating and dehydration [see Warnings and Precautions ( 5.14 )].
Concomitant Medication s Â
Advise patients to inform their health care provider if they are taking, or plan to take, any prescription or over-the-counter medications since there is a potential for interactions [see Drug Interactions ( 7.1 )].
Pregnancy
Advise patients that SAPHRIS may cause fetal harm as well as extrapyramidal and/or withdrawal symptoms in a neonate.  Advise patients to notify their healthcare provider with a known or suspected pregnancy [see Use in Specific Populations ( 8.1 )].
Pregnancy Registry
Advise patients that there is a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to SAPHRIS during pregnancy [see Use in Specific Populations ( 8.1 )].
Distributed by:Allergan USA, Inc.,Madison, NJ 07940
© 2021 N. V. Organon; used by AbbVie under license.© 2021 AbbVie. All rights reserved.
The trademarks SAPHRIS, SAPHRIS & Star Design, and Star Design are used by AbbVie under license from N. V. Organon.
v1.0USPI2402
Instructions For Use Section
INSTRUCTIONS FOR USE
SAPHRIS® (asenapine)
sublingual tablets
Read these Instructions for Use before you start using SAPHRIS and each time you get a refill. There may be new information. This leaflet does not take the place of talking to your doctor about your medical condition or your treatment.
IMPORTANT:
- For  sublingu al (under your tongue)  use only
- Do not remove tablet until ready to administer.
- Use dry hands when handling tablet.
Your SAPHRIS tablets
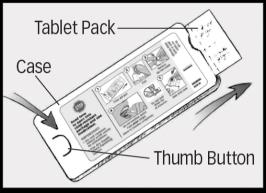
Directions for Taking your SAPHRIS Tablet s :
Step 1. Firmly press and hold thumb button, then pull out the tablet pack (see Figure A). Do not push tablet through the tablet pack. Do not cut or tear the tablet pack.
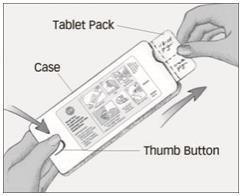
Figure A
Step 2. Peel back the colored tab (see Figure B).
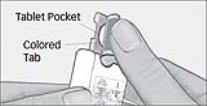
Figure B
Step 3. Gently remove the tablet (see Figure C). Do not split, cut or crush the tablet.
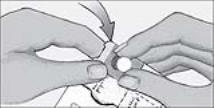
Figure C
Step 4. Place the whole tablet under tongue and allow it to dissolve completely (see Figure D).

Figure D
Do not chew or swallow the tablet.
Do not eat or drink for 10 minutes (See Figure E).
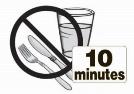
Figure E
Step 5. Slide the tablet pack back into case until it clicks (see Figure F).

Figure F
Stor ing SAPHRIS tablets :
Store SAPHRIS tablets at room temperature between 68°F to 77°F (20°C to 25°C). Â
These Instructions for Use have been approved by the U.S. Food and Drug Administration.
Distributed by: Â Allergan USA, Inc.,Madison, NJ 07940
© 2021 N. V. Organon; used by AbbVie under license.© 2021 AbbVie. All rights reserved.
The trademarks SAPHRIS, SAPHRIS & Star Design, and Star Design are used by AbbVie under license from N. V. Organon.
Revised: October 2021
v1.0IFU2402
Principal Display Panel
NDC 0456-2402-06Rx only 10 Tablets Saphris ® 2.5 mg (asenapine) sublingual tablets For opening and use instructions, see other side. Black Cherry Flavor

Principal Display Panel
Rx onlyNDC 0456-2402-60 Saphris ® 2.5 mg (asenapine) sublingual tablets 60 Tablets Black CherryFlavor
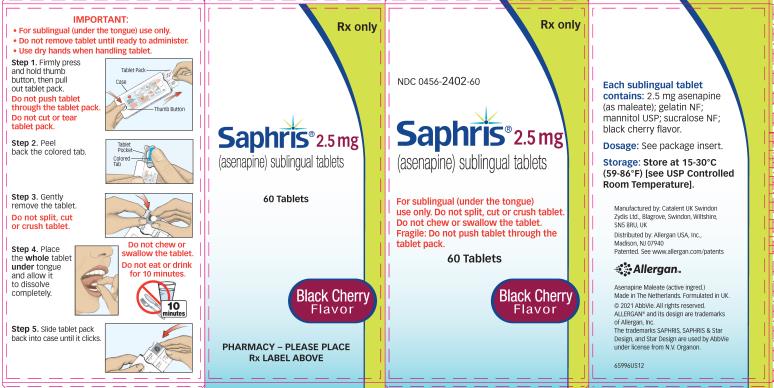
Principal Display Panel
Rx onlySaphris 5 mgBlack Cherry100 Tablets

Principal Display Panel
Rx only
NDC 0456-2405-60 Saphris ® 5 mg (asenapine) sublingual tablets 60 Tablets Black CherryFlavor
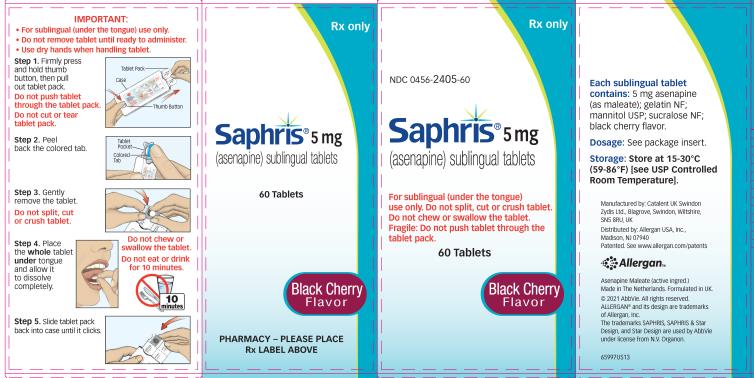
Principal Display Panel
Rx onlyNDC 0456-2410-60 Saphris ® 10 mg (asenapine) sublingual tablets 60 Tablets Black CherryFlavor
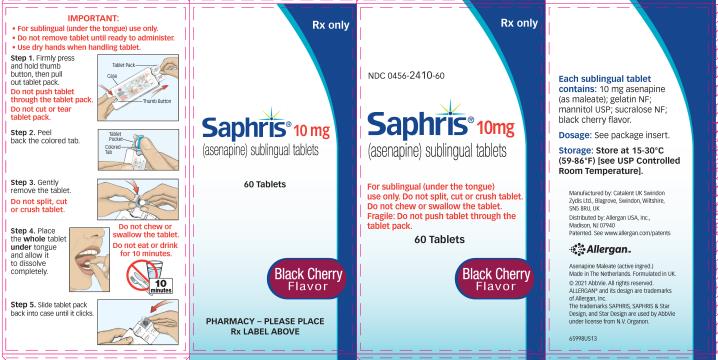
Principal Display Panel
Rx onlyNDC 0456-2410-63Saphris® 10 mg(asenapine) sublingual tabletsBlack CherryFlavor100 TabletsHOSPITAL UNIT DOSE

DISCLAIMER:
"This tool does not provide medical advice, and is for informational and educational purposes only, and is not a substitute for professional medical advice, treatment or diagnosis. Call your doctor to receive medical advice. If you think you may have a medical emergency, please dial 911."
"Do not rely on openFDA to make decisions regarding medical care. While we make every effort to ensure that data is accurate, you should assume all results are unvalidated. We may limit or otherwise restrict your access to the API in line with our Terms of Service."
"This product uses publicly available data from the U.S. National Library of Medicine (NLM), National Institutes of Health, Department of Health and Human Services; NLM is not responsible for the product and does not endorse or recommend this or any other product."
PillSync may earn a commission via links on our site
