MYTESI (crofelemer 125 mg) Dailymed
Generic: crofelemer is used for the treatment of Diarrhea
IMPRINT: 125SLXP
SHAPE: oval
COLOR: white

Go PRO for all pill images
1
MYTESI is indicated for symptomatic relief of non-infectious diarrhea in adult patients with HIV/AIDS on anti-retroviral therapy.
MYTESI is an anti-diarrheal indicated for the symptomatic relief of non-infectious diarrhea in adult patients with HIV/AIDS on anti-retroviral therapy. (1 )
2
Before starting MYTESI, rule out infectious etiologies of diarrhea [see Warnings and Precautions (5.1)]. The recommended adult dosage of MYTESI is 125 mg taken orally two times a day, with or without food. Do not crush or chew MYTESI tablets. Swallow whole.
- Before starting MYTESI, rule out infectious etiologies of diarrhea. (
2 ,5.1 )- The recommended adult dosage is 125 mg taken orally twice a day, with or without food. (
2 )- Do not crush or chew the tablets. Swallow whole. (
2 )
3
Delayed-Release Tablets: 125 mg of crofelemer as a white, oval, delayed-release tablet printed on one side with 125SLXP.
Delayed-Release Tablets: 125 mg (3 )
4
None.
None (4 )
5
Risks of Treatment in Patients with Infectious Diarrhea: Consider infectious etiologies of diarrhea before starting treatment to reduce the risk of inappropriate therapy and worsening disease. (2 ,5.1 )
5. 1
Before starting MYTESI, rule out infectious etiologies of diarrhea. If infectious etiologies are not considered, and MYTESI is initiated based on a presumptive diagnosis of non-infectious diarrhea, then there is a risk that patients with infectious etiologies will not receive the appropriate treatments, and their disease may worsen. MYTESI is not indicated for the treatment of infectious diarrhea.
6
Most common adverse reactions (≥ 3%) are upper respiratory tract infection, bronchitis, cough, flatulence and increased bilirubin. (6.1 )
To report SUSPECTED ADVERSE REACTIONS, contact Napo Pharmaceuticals at 1-844-722-8256 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
6.1
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
A total of 696 HIV-positive patients in three placebo-controlled trials received MYTESI for a mean duration of 78 days. Of the total population across the three trials, 229 patients received a dosage of 125 mg twice a day for a mean duration of 141 days, and 171 patients received one of four higher than recommended dosages for a mean duration of 139 days (N=69) 14 days (N=102), 146 days (N=54), and 14 days (N=242), respectively.
Adverse reactions in patients treated with MYTESI 125 mg twice daily that occurred in at least 2% of patients and at a higher incidence than placebo are provided in Table 1.
Table 1: Common Adverse Reactions occurring in at least 2% of patients and at a higher incidence than placebo in HIV-Positive Patients in Three Placebo-Controlled TrialsAdverse Reaction MYTESI125 mg Twice DailyN = 229 n (%) Placebo N = 274 n (%) Upper respiratory tract infection 13 (6) 4 (2) Bronchitis 9 (4) 0 Cough 8 (4) 3 (1) Flatulence 7 (3) 3 (1) Increased bilirubin 7 (3) 3 (1) Nausea 6 (3) 4 (2) Back pain 6 (3) 4 (2) Arthralgia 6 (3) 0 Urinary tract infection 5 (2) 2 (1) Nasopharyngitis 5 (2) 2 (1) Musculoskeletal pain 5 (2) 1 (<1) Hemorrhoids 5 (2) 0 Giardiasis 5 (2) 0 Anxiety 5 (2) 1 (<1) Increased alanine aminotransferase 5 (2) 3 (1) Abdominal distension 5 (2) 1 (<)
Less common adverse reactions that occurred in between 1% and 2% of patients taking 125 mg twice daily of MYTESI were abdominal pain, acne, increased aspartate aminotransferase, increased conjugated bilirubin, increased unconjugated blood bilirubin, constipation, depression, dermatitis, dizziness, dry mouth, dyspepsia, gastroenteritis, herpes zoster, nephrolithiasis, pain in extremity, pollakiuria, sinusitis and decreased white blood cell count.
7
7.1
MYTESI administration did not have a clinically relevant interaction with nelfinavir, zidovudine, or lamivudine in a drug-drug interaction trial [see Clinical Pharmacology (12.3)].
8
Lactation: Women infected with HIV-1 should be instructed not to breastfeed due to the potential for HIV transmission. (8.2 )
8.1
Risk Summary
Crofelemer is minimally absorbed systemically by the oral route of administration and maternal use is not expected to result in fetal exposure to the drug [see Clinical Pharmacology (12.3)].
In pregnant rats, no adverse fetal effects were observed with oral administration of crofelemer at doses up to 177 times the recommended clinical dose during the period of organogenesis. In pregnant rabbits, an increase in fetal resorptions and abortions compared to controls were observed with crofelemer at a dose of 96 times the recommended clinical dose. However, it is not clear whether these effects in rabbits are related to the maternal toxicity (decreased body weight and decreased food consumption) observed at this dose (see Data).
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
Data
Animal Data
Crofelemer was not teratogenic and did not produce embryofetal toxicity in pregnant rats following oral administration at doses up to 738 mg/kg/day during the period of organogenesis. The 738 mg/kg/day dose is 177 times the recommended daily human dose of 4.2 mg/kg/day.
Crofelemer was not teratogenic in pregnant rabbits following oral administration at doses up to 400 mg/kg/day during the period of organogenesis. At a dose level of 400 mg/kg/day, which is 96 times the recommended daily human dose of 4.2 mg/kg/day, crofelemer produced an increase in fetal resorptions and abortions compared to controls. However, it is not clear whether these effects are related to the maternal toxicity (decreased body weight and decreased food consumption) observed.
8.2
Risk Summary
The Centers for Disease Control and Prevention recommend that HIV-1 infected mothers not breastfeed their infants to avoid risking postnatal transmission of HIV-1.
There are no data on the presence of crofelemer in human milk, the effects on the breastfed infant, or the effects on milk production. Because of the potential for HIV transmission and adverse effects on a breastfed infant, instruct mothers not to breastfeed if they are taking MYTESI.
8.4
The safety and effectiveness of MYTESI have not been established in pediatric patients.
8.5
Clinical studies with MYTESI did not include sufficient numbers of patients aged 65 and over to determine whether they respond differently than younger patients.
8.6
No dose modifications are recommended with respect to CD4 cell count and HIV viral load, based on the findings in subgroups of patients defined by CD4 cell count and HIV viral load.
The safety profile of MYTESI was similar in patients with baseline CD4 cell count less than 404 cells/microL (lower limit of normal range) (N=388) and patients with baseline CD4 cell counts greater than or equal to 404 cells/microL (N=289).
The safety profile of crofelemer was similar in patients with baseline HIV viral loads less than 400 copies/mL (N = 412) and patients with baseline HIV viral loads greater than or equal to 400 copies/mL (N = 278).
11
MYTESI (crofelemer) delayed-release tablets is an anti-diarrheal, enteric-coated drug product for oral administration. It contains 125 mg of crofelemer, a botanical drug substance that is derived from the red latex of Croton lechleri Müll. Arg. Crofelemer is an oligomeric proanthocyanidin mixture primarily composed of (+)–catechin, (–)–epicatechin, (+)–gallocatechin, and (–)–epigallocatechin monomer units linked in random sequence, as represented below. The average degree of polymerization for the oligomers ranges between 5 and 7.5, as determined by phloroglucinol degradation.
    Â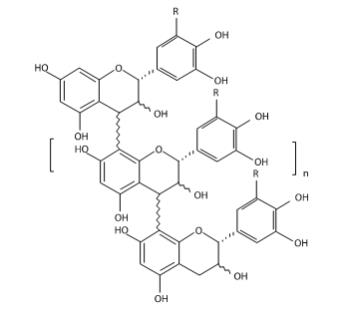
    Â
    Â
R = H or OH range n = 3 to 5.5
Inactive ingredients: colloidal silicon dioxide, croscarmellose sodium, magnesium stearate, and microcrystalline cellulose.
Coating ingredients: ethylacrylate and methylacrylate copolymer dispersion, talc, triethyl citrate, and white dispersion which contains xanthan gum, titanium dioxide, propyl paraben, and methyl paraben.
12
12.1
Crofelemer is an inhibitor of both the cyclic adenosine monophosphate (cAMP)-stimulated cystic fibrosis transmembrane conductance regulator (CFTR) chloride ion (ClÂŻ) channel, and the calcium-activated ClÂŻ channels (CaCC) at the luminal membrane of enterocytes. The CFTR ClÂŻ channel and CaCC regulate ClÂŻ and fluid secretion by intestinal epithelial cells. Crofelemer acts by blocking ClÂŻ secretion and accompanying high volume water loss in diarrhea, normalizing the flow of ClÂŻ and water in the gastrointestinal tract.
12.2
Consistent with the mechanism of action of crofelemer (i.e., inhibition of CFTR and CaCC in the gastrointestinal lumen), data suggest stool chloride concentrations decreased in patients treated with crofelemer 500 mg four times daily (8-times the recommended daily dosage) (n=25) for four days relative to placebo (n=24); stool chloride concentrations decreased in both African American patients treated with crofelemer (n=3) relative to placebo (n=5) and non-African American patients treated with Mytesi (n=22) relative to placebo (n=19).
Cardiac Electrophysiology
At a dose 10 times the maximum recommended dose, crofelemer does not prolong the QTc interval to any clinically relevant extent.
12.3
Absorption
The absorption of crofelemer is minimal following oral dosing in healthy adults and HIV–positive patients and concentrations of crofelemer in plasma are below the level of quantitation (50 ng/mL). Therefore, standard pharmacokinetic parameters such as area under the curve, maximum concentration, and half-life cannot be estimated.
Effect of Food
Administration of crofelemer with a high-fat meal was not associated with an increase in systemic exposure of crofelemer in healthy subjects. In the clinical trial, a single 500 mg dose of crofelemer (4-times the recommended dose) was administered one-half hour before the morning and evening meals [see Dosage and Administration (2)].
Drug Interaction Studies
In vitro studies have shown that crofelemer has the potential to inhibit transporters MRP2 and OATP1A2 but not P-gp and BCRP at concentrations expected in the gut.
Due to the minimal absorption of crofelemer, crofelemer is unlikely to inhibit cytochrome P450 isoenzymes 1A2, 2A6, 2B6, 2C9, 2C19, 2D6, and 2E1 systemically.
Nelfinavir, Zidovudine, Lamivudine
Results of a crossover study in healthy subjects showed crofelemer 500 mg administered four times daily (8- times the recommended daily dosage) for five days had no effect on the exposure of zidovudine and nelfinavir when administered as a single dose. A 20% decrease in lamivudine exposure was also observed in the same study but was not considered to be clinically important.
Midazolam
In vitro studies have shown that crofelemer has the potential to inhibit CYP3A4 enzymes at concentrations expected in the gut. The effects of crofelemer on the pharmacokinetics of a single oral dose of 2 mg midazolam, a sensitive CYP3A4 substrate, was evaluated in healthy subjects following crofelemer dosed orally at 500 mg twice daily (4-times the recommended dosage) for six consecutive days. No significant changes in the mean Cmax and AUC were observed for midazolam and its active metabolite, hydroxymidazolam after co- administration with crofelemer compared to that after administration of midazolam alone.
13
13.1
Carcinogenesis
In a 6-month carcinogenicity study in Tg.rasH2 transgenic mice, crofelemer at oral doses up to 125 mg/kg twice a day (250 mg/kg/day) did not produce any drug-related neoplasms in male and female animals. The 250 mg/kg/day dose is about 60 times the recommended clinical dose of 4.2 mg/kg/day (125 mg twice daily).
In a 2-year oral carcinogenicity study in rats, crofelemer at doses up to 100/50 mg/kg twice a day (200/100 mg/kg/day), which is 47.6/23.8 times the recommended clinical dose, did not produce any drug-related neoplastic findings in male and female rats (due to drug-related decreased survival in male rats, 37.5 mg/kg twice daily and 100 mg/kg twice daily doses were reduced to 25 mg/kg twice daily and 50 mg/kg twice daily, respectively, at week 67).
Mutagenesis
Crofelemer was negative in the bacterial reverse mutation assay, chromosomal aberration assay, and rat bone marrow micronucleus assay.
Impairment of Fertility
Crofelemer, at oral doses of up to 738 mg/kg/day (177 times the recommended human daily dose of 125 mg twice daily), had no effects on fertility or reproductive performance of male and female rats.
14
The efficacy of MYTESI was evaluated in a randomized, double-blind, placebo-controlled (one month) and placebo-free (five month), multi-center study. The study enrolled 374 HIV-positive patients on stable anti-retroviral therapy with a history of diarrhea for one month or more. Diarrhea was defined as either persistently loose stools despite regular use of anti-diarrheal medication (e.g., loperamide, diphenoxylate, and bismuth subsalicylate) or one or more watery bowel movements per day without regular anti-diarrheal medicine use.
Patients were excluded if they had a positive gastrointestinal biopsy, gastrointestinal culture, or stool test for multiple bacteria (Salmonella, Shigella, Campylobacter, Yersinia, Mycobacterium), bacterial toxin (Clostridium difficile), ova and parasites (Giardia, Entamoeba, Isospora, Cyclospora, Cryptosporidium, Microsporidium), or viruses (Cytomegalovirus). Patients were also excluded if they had a history of ulcerative colitis, Crohn’s disease, celiac sprue (gluten-enteropathy), chronic pancreatitis, malabsorption, or any other gastrointestinal disease associated with diarrhea.
The study had a two-stage adaptive design. In both stages, patients received placebo for 10 days (screening period) followed by randomization to crofelemer or placebo for 31 days of treatment (double-blind period). Only patients with 1 or more watery bowel movements per day on at least 5 of the last 7 days in the screening period were randomized to the double-blind period. Each stage enrolled patients separately; the dose for the second stage was selected based on an interim analysis of data from the first stage. In the first stage, patients were randomized 1:1:1:1 to one of three crofelemer dosage regimens (125 mg twice daily, or one of two higher dosage regimens) or placebo. In the second stage, patients were randomized 1:1 to MYTESI 125 mg twice daily or placebo. The efficacy analysis was based on results from the double-blind portion of both stages.
Each study stage also had a five month period (placebo-free period) that followed the double-blind period. Patients treated with MYTESI continued the same dose in the placebo-free period. In the first stage, patients that received placebo were re-randomized 1:1:1 to one of the three crofelemer dosage regimens (125 mg twice daily, or one of the two higher dosage regimens) in the placebo-free period. In the second stage, patients that received placebo were treated with MYTESI 125 mg twice daily in the placebo-free period.
The median time since diagnosis of HIV was 12 years. The percentage of patients with a CD4 cell count of less than 404 was 39%. The percentage of patients with a HIV viral load greater than or equal to 1000, 400 to 999, and less than 400 HIV copies/mL was 7%, 3%, and 9%, respectively; the remainder had a viral load that was not detectable. The median time since diarrhea started was 4 years. The median number of daily watery bowel movements was 2.5 per day.
Most patients were male (85%). The percentage of patients that were Caucasian was 46%; the percentage of patients that were African-American was 32%. The median age was 45 years with a range of 21 to 68 years.
In the double-blind period of the study, 136 patients received MYTESI 125 mg twice daily, 101 patients received one of the two higher dosage regimens and 138 patients received placebo. The percentages of patients that completed the double-blind period were 92% in the MYTESI 125 mg group and 94% in the placebo arm.
Most patients received concomitant protease inhibitors during the double-blind period (Table 2). The most frequently used anti-retroviral therapies in the MYTESI 125 mg and placebo groups were tenofovir/emtricitabine, ritonavir, and lopinavir/ritonavir.
Table 2: Concomitant Anti-Retroviral Therapy Used in the Double-Blind Period in Patients with HIV MYTESI125 mg twice daily(N = 136)n (%) PlaceboN = 138n (%) Any antiretroviral therapy 135 (99) 134 (97) Any protease inhibitor 87 (64) 97 (70) Tenofovir/Emtricitabine 45 (33) 52 (38) Ritonavir 46 (34) 49 (36) Lopinavir/Ritonavir 30 (22) 40 (29) Efavirenz/Tenofovir/Emtricitabine 30 (22) 21 (15) Tenofovir disoproxil fumarate 18 (13) 14 (10) Atazanavir sulfate 19 (14) 22 (16) Abacavir w/ lamivudine 17 (13) 18 (13) Darunavir 19 (14) 14 (10) Raltegravir 16 (12) 11 (8) Valaciclovir hydrochloride 12 (9) 16 (12) Fosamprenavir 12 (9) 13 (9) Zidovudine w/lamivudine 12 (9) 15 (11) Lamivudine 7 (5) 6 (4) Nevirapine 8 (6) 9 (7) Atazanavir 5 (4) 2 (1)
The primary efficacy endpoint was the proportion of patients with a clinical response, defined as less than or equal to 2 watery bowel movements per week during at least 2 of the 4 weeks of the placebo-controlled phase. Patients who received concomitant anti-diarrheal medications or opiates were counted as clinical non-responders.
A significantly larger proportion of patients in the MYTESI 125 mg twice daily group experienced clinical response compared with patients in the placebo group (18% vs. 8%, 1–sided p < 0.01). In the randomized clinical study, examination of duration of diarrhea, baseline number of daily watery bowel movements, use of protease inhibitors, CD4 cell count and age subgroups did not identify differences in the consistency of the crofelemer treatment effect among these subgroups. There were too few female patients and patients with an HIV viral load > 400 copies/mL to adequately assess differences in effects in these populations. Among race subgroups, there were no differences in the consistency of the crofelemer treatment effect except for the subgroup of African-Americans; crofelemer was less effective in African-Americans than non-African-Americans.
Although the CD4 cell count and HIV viral load did not appear to change over the one month placebo-controlled period, the clinical significance of this finding is unknown because of the short duration of the placebo-controlled period.
Of the 24 clinical responders to MYTESI 125 mg twice daily, 22 entered the placebo-free period; 16 were responding at the end of month 3, and 14 were responding at the end of month 5.
/storage And Handling
MYTESI (crofelemer) 125 mg delayed-release tablets are white, oval tablets printed on one side with 125SLXP.They are available in the following package size: Bottles of 60: NDC 70564-802-60Store at 20°C-25°C (68°F-77°F); excursions permitted between 15°C-30°C (59°F-86°F). See USP Controlled Room Temperature.
17
- Instruct patients that MYTESI tablets may be taken with or without food.
- Instruct patients to swallow MYTESI tablets whole and not to crush or chew the tablets.
Manufactured by Patheon, Inc. for
Napo Pharmaceuticals, Inc., San Francisco, CA 94105
Copyright © Napo Pharmaceuticals, Inc.
US Patent Nos. 7,341,744 and 7,323,195.
NP-367-1 11/2020
70033046
The botanical drug substance of MYTESI is extracted from Croton lechleri (the botanical raw material) that is harvested from the wild in South America.
Package Label.principal Display Panel
PRINCIPAL DISPLAY PANEL
NDC 70564-802-60 Mytesi™ (crofelemer)delayed-release tablets125 mgSwallow tablet whole. Do not crush or chew. 60 enteric coated tablets                Rx only

DISCLAIMER:
"This tool does not provide medical advice, and is for informational and educational purposes only, and is not a substitute for professional medical advice, treatment or diagnosis. Call your doctor to receive medical advice. If you think you may have a medical emergency, please dial 911."
"Do not rely on openFDA to make decisions regarding medical care. While we make every effort to ensure that data is accurate, you should assume all results are unvalidated. We may limit or otherwise restrict your access to the API in line with our Terms of Service."
"This product uses publicly available data from the U.S. National Library of Medicine (NLM), National Institutes of Health, Department of Health and Human Services; NLM is not responsible for the product and does not endorse or recommend this or any other product."
PillSync may earn a commission via links on our site
