Darunavir Dailymed
Generic: darunavir is used for the treatment of Acquired Immunodeficiency Syndrome


Go PRO for all pill images
Recent Major Changes Section
Contraindications (4 )Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â 4/2022
1 Indications And Usage
Darunavir tablets, co-administered with ritonavir (darunavir/ritonavir), in combination with other antiretroviral agents, are indicated for the treatment of human immunodeficiency virus (HIV-1) infection in adult and pediatric patients 3 years of age and older [see Use in Specific Populations (8.4) and Clinical Studies (14)].
Darunavir tablets are a human immunodeficiency virus (HIV-1) protease inhibitor indicated for the treatment of HIV-1 infection in adult and pediatric patients 3 years of age and older. Darunavir tablets must be co-administered with ritonavir (darunavir/ritonavir) and with other antiretroviral agents. (1 )
2 Dosage And Administration
- Testing:
- In treatment-experienced patients, treatment history genotypic and/or phenotypic testing is recommended prior to initiation of therapy with darunavir tablets/ritonavir to assess drug susceptibility of the HIV-1 virus (
2.1 ,12.4 )- Monitor serum liver chemistry tests before and during therapy with darunavir tablets/ritonavir. (
2.1 ,2.2 ,5.2 )
- Treatment-naĂŻve adult patients and treatment-experienced adult patients with no darunavir resistance associated substitutions: 800 mg (one 800 mg tablet) taken with ritonavir 100 mg once daily and with food. (
2.3 )- Treatment-experienced adult patients with at least one darunavir resistance associated substitution: 600 mg (one 600 mg tablet) taken with ritonavir 100 mg twice daily and with food. (
2.3 )- Pregnant patients: 600 mg (one 600 mg tablet) taken with ritonavir 100 mg twice daily and with food. (
2.4 )- Pediatric patients (3 to less than 18 years of age and weighing at least 10 kg): dosage of darunavir and ritonavir is based on body weight and should not exceed the adult dose. Darunavir tablets should be taken with ritonavir and with food. (
2.5 )- Darunavir tablets/ritonavir is not recommended for use in patients with severe hepatic impairment. (
2.6 )2.1 Testing Prior to Initiation of darunavir tablets/ritonavir
In treatment-experienced patients, treatment history, genotypic and/or phenotypic testing is recommended to assess drug susceptibility of the HIV-1 virus [see Microbiology (12.4)]. Refer to Dosage and Administration (2.3), (2.4) and (2.5) for dosing recommendations.
Appropriate laboratory testing such as serum liver biochemistries should be conducted prior to initiating therapy with darunavir tablets/ritonavir [see Warnings and Precautions (5.2)].
2.2 Monitoring During Treatment with darunavir tablets/ritonavir
Patients with underlying chronic hepatitis, cirrhosis, or in patients who have pre-treatment elevations of transaminases should be monitored for elevation in serum liver biochemistries, especially during the first several months of darunavir tablets/ritonavir treatment [see Warnings and Precautions (5.2)].
2.3 Recommended Dosage in Adult Patients
Darunavir tablets must be co-administered with ritonavir to exert its therapeutic effect. Failure to correctly co-administer darunavir tablets with ritonavir will result in plasma levels of darunavir that will be insufficient to achieve the desired antiviral effect and will alter some drug interactions.
Patients who have difficulty swallowing darunavir tablets can use the 100 mg per mL darunavir oral suspension.
Treatment-NaĂŻve Adult Patients
The recommended oral dose of darunavir is 800 mg (one 800 mg tablet) taken with ritonavir 100 mg (one 100 mg tablet or capsule or 1.25 mL of a 80 mg per mL ritonavir oral solution) once daily and with food.
Treatment-Experienced Adult Patients
The recommended oral dosage for treatment-experienced adult patients is summarized in Table 1.
Baseline genotypic testing is recommended for dose selection. However, when genotypic testing is not feasible, darunavir 600 mg taken with ritonavir 100 mg twice daily is recommended.
Table 1: Recommended Darunavir/ritonavir Dosage in Treatment-Experienced Adult Patients
Baseline Resistance
Formulation and Recommended Dosing
Darunavir tablets with ritonavir tablets or capsule
With no darunavir resistance associated substitutionsa
One 800 mg darunavir tablet with one 100 mg ritonavir tablet/capsule, taken once daily with food
With at least one darunavir resistance associated substitutionsa, or
with no baseline resistance information
One 600 mg darunavir tablet with one 100 mg ritonavir tablet/capsule, taken twice daily with food
 a V11I, V32I, L33F, I47V, I50V, I54L, I54M, T74P, L76V, I84V and L89V 2.4 Recommended Dosage During Pregnancy
The recommended dosage in pregnant patients is darunavir 600 mg taken with ritonavir 100 mg twice daily with food.
Darunavir 800 mg taken with ritonavir 100 mg once daily should only be considered in certain pregnant patients who are already on a stable darunavir 800 mg with ritonavir 100 mg once daily regimen prior to pregnancy, are virologically suppressed (HIV-1 RNA less than 50 copies per mL), and in whom a change to twice daily darunavir 600 mg with ritonavir 100 mg may compromise tolerability or compliance.
2.5 Recommended Dosage in Pediatric Patients (age 3 to less than 18 years)
Healthcare professionals should pay special attention to accurate dose selection of darunavir tablets, transcription of the medication order, dispensing information and dosing instruction to minimize risk for medication errors, overdose, and underdose.
Prescribers should select the appropriate dose of darunavir/ritonavir for each individual child based on body weight (kg) and should not exceed the recommended dose for adults.
Before prescribing darunavir tablets, children weighing greater than or equal to 15 kg should be assessed for the ability to swallow tablets. If a child is unable to reliably swallow a tablet, the use of darunavir oral suspension should be considered.
The recommended dose of darunavir/ritonavir for pediatric patients (3 to less than 18 years of age and weighing at least 10 kg is based on body weight (see Tables 3 and 5) and should not exceed the recommended adult dose. Darunavir tablets should be taken with ritonavir and with food.
The recommendations for the darunavir/ritonavir dosage regimens were based on pediatric clinical trial data and population pharmacokinetic modeling and simulation [see Use in Specific Populations (8.4) and Clinical Pharmacology (12.3)].
Dosing Recommendations for Treatment-NaĂŻve Pediatric Patients or Antiretroviral Treatment-Experienced Pediatric Patients with No Darunavir Resistance Associated Substitutions
Pediatric Patients Weighing At Least 15 kg
Pediatric patients weighing at least 15 kg can be dosed with darunavir oral tablet(s) using the following table:
Table 3: Recommended Dose for Pediatric Patients Weighing At Least 15 kg Who are Treatment-NaĂŻve or Treatment-Experienced with No Darunavir Resistance Associated Substitutionsa
Body weight (kg)
Formulation: Darunavir tablet(s) and ritonavir capsules or tablets (100 mg)
Dose: once daily with food
Greater than or equal to 15 kg to less than 30 kg
Darunavir 600 mg with ritonavir 100 mg
Greater than or equal to 30 kg to less than 40 kg
Darunavir 675 mg with ritonavir 100 mg
Greater than or equal to 40 kg
Darunavir 800 mg with ritonavir 100 mg
 a darunavir resistance associated substitutions: V11I, V32I, L33F, I47V, I50V, I54M, I54L, T74P, L76V, I84V and L89V
Dosing Recommendations for Treatment-Experienced Pediatric Patients with At Least One Darunavir Resistance Associated Substitutions
Pediatric Patients Weighing At Least 15 kg
Pediatric patients weighing at least 15 kg can be dosed with darunavir oral tablet(s) using the following table:
Table 5: Recommended Dose for Pediatric Patients Weighing At Least 15 kg Who are Treatment-Experienced with At Least One Darunavir Resistance Associated Substitutiona
Body weight (kg)
Formulation: Darunavir tablet(s) and ritonavir tablets, capsules (100 mg) or oral solution (80 mg/mL)
Dose: twice daily with food
Greater than or equal to 15 kg to less than 30 kg
Darunavir 375 mg with ritonavir 0.6 mL (48 mg)
Greater than or equal to 30 kg to less than 40 kg
Darunavir 450 mg with ritonavir 0.75 mL (60 mg)
Greater than or equal to 40 kg
Darunavir 600 mg with ritonavir 100 mg
 a darunavir resistance associated substitutions: V11I, V32I, L33F, I47V, I50V, I54M, I54L, T74P, L76V, I84V and L89V
The use of darunavir tablets/ritonavir in pediatric patients below 3 years of age is not recommended [see Warnings and Precautions (5.10) and Use in Specific Populations (8.4)].
2.6 Not Recommended in Patients with Severe Hepatic Impairment
No dosage adjustment is required in patients with mild or moderate hepatic impairment. No data are available regarding the use of darunavir tablets/ritonavir when co-administered to subjects with severe hepatic impairment; therefore, darunavir tablets/ritonavir is not recommended for use in patients with severe hepatic impairment [see Use in Specific Populations (8.6) and Clinical Pharmacology (12.3)].
3 Dosage Forms And Strengths
- 600 mg: orange, oval-shaped, film-coated tablets debossed with “TV” on one side and “7736” on the other side of the tablet.
- Tablets: 600 mg (
3 )
4 Contraindications
Co-administration of darunavir/ritonavir is contraindicated with drugs that are highly dependent on CYP3A for clearance and for which elevated plasma concentrations are associated with serious and/or life-threatening events (narrow therapeutic index). Examples of these drugs and other contraindicated drugs (which may lead to reduced efficacy of darunavir) are uled below [see Drug Interactions (7.3)]. Due to the need for co-administration of darunavir with ritonavir, please refer to ritonavir prescribing information for a description of ritonavir contraindications.
- Alpha 1-adrenoreceptor antagonist: alfuzosin
- Anti-gout: colchicine, in patients with renal and/or hepatic impairment
- Antimycobacterial: rifampin
- Antipsychotics: lurasidone, pimozide
- Cardiac Disorders: dronedarone, ivabradine, ranolazine
- Ergot derivatives, e.g. dihydroergotamine, ergotamine, methylergonovine
- Herbal product: St. John’s wort (Hypericum perforatum)
- Hepatitis C direct acting antiviral: elbasvir/grazoprevir
- Lipid modifying agents: lomitapide, lovastatin, simvastatin
- Opioid Antagonist: naloxegol
- PDE-5 inhibitor: sildenafil when used for treatment of pulmonary arterial hypertension
- Sedatives/hypnotics: orally administered midazolam, triazolam
- Co-administration of darunavir/ritonavir is contraindicated with drugs that are highly dependent on CYP3A for clearance and for which elevated plasma concentrations are associated with serious and/or life-threatening events (narrow therapeutic index). (
4 )
5 Warnings And Precautions
- Drug-induced hepatitis (e.g., acute hepatitis, cytolytic hepatitis) has been reported with darunavir/ritonavir. Monitor liver function before and during therapy, especially in patients with underlying chronic hepatitis, cirrhosis, or in patients who have pre-treatment elevations of transaminases. Postmarketing cases of liver injury, including some fatalities, have been reported. (
5.2 )- Skin reactions ranging from mild to severe, including Stevens-Johnson Syndrome, toxic epidermal necrolysis, drug rash with eosinophilia and systemic symptoms and acute generalized exanthematous pustulosis, have been reported. Discontinue treatment if severe reaction develops. (
5.3 )- Use with caution in patients with a known sulfonamide allergy. (
5.4 )- Patients may develop new onset diabetes mellitus or hyperglycemia. Initiation or dose adjustments of insulin or oral hypoglycemic agents may be required. (
5.6 )- Patients may develop redistribution/accumulation of body fat or immune reconstitution syndrome. (
5.7 ,5.8 )- Patients with hemophilia may develop increased bleeding events. (
5.9 )- Darunavir/ritonavir is not recommended in pediatric patients below 3 years of age in view of toxicity and mortality observed in juvenile rats dosed with darunavir up to days 23 to 26 of age. (
5.10 )5.1 Importance of Co-administration with Ritonavir
Darunavir must be co-administered with ritonavir and food to achieve the desired antiviral effect. Failure to administer darunavir with ritonavir and food may result in a loss of efficacy of darunavir.
Please refer to ritonavir prescribing information for additional information on precautionary measures.
5.2 Hepatotoxicity
Drug-induced hepatitis (e.g., acute hepatitis, cytolytic hepatitis) has been reported with darunavir/ritonavir. During the clinical development program (N=3,063), hepatitis was reported in 0.5% of patients receiving combination therapy with darunavir/ritonavir. Patients with pre-existing liver dysfunction, including chronic active hepatitis B or C, have an increased risk for liver function abnormalities including severe hepatic adverse events.
Post-marketing cases of liver injury, including some fatalities, have been reported. These have generally occurred in patients with advanced HIV-1 disease taking multiple concomitant medications, having co-morbidities including hepatitis B or C co-infection, and/or developing immune reconstitution syndrome. A causal relationship with darunavir/ritonavir therapy has not been established.
Appropriate laboratory testing should be conducted prior to initiating therapy with darunavir/ritonavir and patients should be monitored during treatment. Increased AST/ALT monitoring should be considered in patients with underlying chronic hepatitis, cirrhosis, or in patients who have pre-treatment elevations of transaminases, especially during the first several months of darunavir/ritonavir treatment.
Evidence of new or worsening liver dysfunction (including clinically significant elevation of liver enzymes and/or symptoms such as fatigue, anorexia, nausea, jaundice, dark urine, liver tenderness, hepatomegaly) in patients on darunavir/ritonavir should prompt consideration of interruption or discontinuation of treatment.
5.3 Severe Skin Reactions
During the clinical development program (n = 3,063), severe skin reactions, accompanied by fever and/or elevations of transaminases in some cases, have been reported in 0.4% of subjects. Stevens-Johnson Syndrome was rarely (less than 0.1%) reported during the clinical development program. During post-marketing experience toxic epidermal necrolysis, drug rash with eosinophilia and systemic symptoms, and acute generalized exanthematous pustulosis have been reported. Discontinue darunavir/ritonavir immediately if signs or symptoms of severe skin reactions develop. These can include but are not limited to severe rash or rash accompanied with fever, general malaise, fatigue, muscle or joint aches, bulers, oral lesions, conjunctivitis, hepatitis and/or eosinophilia.
Rash (all grades, regardless of causality) occurred in 10.3% of subjects treated with darunavir/ritonavir [ see Adverse Reactions (6)]. Rash was mostly mild-to-moderate, often occurring within the first four weeks of treatment and resolving with continued dosing. The discontinuation rate due to rash in subjects using darunavir/ritonavir was 0.5%.
Rash occurred more commonly in treatment-experienced subjects receiving regimens containing darunavir/ritonavir + raltegravir compared to subjects receiving darunavir/ritonavir without raltegravir or raltegravir without darunavir/ritonavir. However, rash that was considered drug related occurred at similar rates for all three groups. These rashes were mild to moderate in severity and did not limit therapy; there were no discontinuations due to rash.
5.4 Sulfa Allergy
Darunavir contains a sulfonamide moiety. Darunavir should be used with caution in patients with a known sulfonamide allergy. In clinical studies with darunavir/ritonavir, the incidence and severity of rash were similar in subjects with or without a history of sulfonamide allergy.
5.5 Risk of Serious Adverse Reactions due to Drug Interactions
Initiation of darunavir/ritonavir, a CYP3A inhibitor, in patients receiving medications metabolized by CYP3A or initiation of medications metabolized by CYP3A in patients already receiving darunavir/ritonavir, may increase plasma concentrations of medications metabolized by CYP3A and reduce plasma concentrations of active metabolite(s) formed by CYP3A.
Initiation of medications that inhibit or induce CYP3A may increase or decrease concentrations of darunavir/ritonavir, respectively. These interactions may lead to:
- Clinically significant adverse reactions, potentially leading to severe, life threatening, or fatal events from greater exposures of concomitant medications.
- Clinically significant adverse reactions from greater exposures of darunavir/ritonavir.
- Loss of therapeutic effect of the concomitant medications from lower exposures of active metabolite(s).
- Loss of therapeutic effect of darunavir/ritonavir and possible development of resistance from lower exposures of darunavir/ritonavir.
See Table 10 for steps to prevent or manage these possible and known significant drug interactions, including dosing recommendations [see Drug Interactions (7)]. Consider the potential for drug interactions prior to and during darunavir/ritonavir therapy; review concomitant medications during darunavir/ritonavir therapy; and monitor for the adverse reactions associated with the concomitant drugs [see Contraindications (4) and Drug Interactions (7)].
5.6 Diabetes Mellitus/Hyperglycemia
New onset diabetes mellitus, exacerbation of pre-existing diabetes mellitus, and hyperglycemia have been reported during postmarketing surveillance in HIV-infected patients receiving protease inhibitor (PI) therapy. Some patients required either initiation or dose adjustments of insulin or oral hypoglycemic agents for treatment of these events. In some cases, diabetic ketoacidosis has occurred. In those patients who discontinued PI therapy, hyperglycemia persisted in some cases. Because these events have been reported voluntarily during clinical practice, estimates of frequency cannot be made and causal relationships between PI therapy and these events have not been established.
5.7 Fat Redistribution
Redistribution/accumulation of body fat, including central obesity, dorsocervical fat enlargement (buffalo hump), peripheral wasting, facial wasting, breast enlargement, and “cushingoid appearance” have been observed in patients receiving antiretroviral therapy. The mechanism and long-term consequences of these events are currently unknown. A causal relationship has not been established.
5.8 Immune Reconstitution Syndrome
Immune reconstitution syndrome has been reported in patients treated with combination antiretroviral therapy, including darunavir. During the initial phase of combination antiretroviral treatment, patients whose immune systems respond may develop an inflammatory response to indolent or residual opportunistic infections (such as Mycobacterium avium infection, cytomegalovirus, Pneumocystis jirovecii pneumonia [PCP], or tuberculosis), which may necessitate further evaluation and treatment.
Autoimmune disorders (such as Graves’ disease, polymyositis, Guillain-Barré syndrome, and autoimmune hepatitis) have also been reported to occur in the setting of immune reconstitution; however, the time to onset is more variable, and can occur many months after initiation of antiretroviral treatment.
5.9 Hemophilia
There have been reports of increased bleeding, including spontaneous skin hematomas and hemarthrosis in patients with hemophilia type A and B treated with PIs. In some patients, additional factor VIII was given. In more than half of the reported cases, treatment with PIs was continued or reintroduced if treatment had been discontinued. A causal relationship between PI therapy and these episodes has not been established.
5.10 Not Recommended in Pediatric Patients Below 3 Years of Age
Darunavir/ritonavir in pediatric patients below 3 years of age is not recommended in view of toxicity and mortality observed in juvenile rats dosed with darunavir (from 20 mg/kg to 1,000 mg/kg) up to days 23 to 26 of age [see Use in Specific Populations (8.1 and 8.4) and Clinical Pharmacology (12.3)].
6 Adverse Reactions
The following adverse reactions are discussed in other sections of labeling:
- Hepatotoxicity [see Warnings and Precautions (5.2)]
- Severe Skin Reactions [see Warnings and Precautions (5.3)]
- Diabetes Mellitus/Hyperglycemia [see Warnings and Precautions (5.6)]
- Fat Redistribution [see Warnings and Precautions (5.7)]
- Immune Reconstitution Syndrome [see Warnings and Precautions (5.8)]
- Hemophilia [see Warnings and Precautions (5.9)]
Due to the need for co-administration of darunavir with ritonavir, please refer to ritonavir prescribing information for ritonavir-associated adverse reactions.
- The most common clinical adverse drug reactions to darunavir/ritonavir (incidence greater than or equal to 5%) of at least moderate intensity (greater than or equal to Grade 2) were diarrhea, nausea, rash, headache, abdominal pain and vomiting. (
6 )
To report SUSPECTED ADVERSE REACTIONS, contact Teva at 1-888-838-2872 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
Treatment NaĂŻve-Adults: TMC114-C211
The safety assessment is based on all safety data from the Phase 3 trial TMC114-C211 comparing darunavir/ritonavir 800/100 mg once daily versus lopinavir/ritonavir 800/200 mg per day in 689 antiretroviral treatment-naĂŻve HIV-1-infected adult subjects. The total mean exposure for subjects in the darunavir/ritonavir 800/100 mg once daily arm and in the lopinavir/ritonavir 800/200 mg per day arm was 162.5 and 153.5 weeks, respectively.
The majority of the adverse drug reactions (ADRs) reported during treatment with darunavir/ritonavir 800/100 mg once daily were mild in severity. The most common clinical ADRs to darunavir/ritonavir 800/100 mg once daily (greater than or equal to 5%) of at least moderate intensity (greater than or equal to Grade 2) were diarrhea, headache, abdominal pain and rash. 2.3% of subjects in the darunavir/ritonavir arm discontinued treatment due to ADRs.
ADRs to darunavir/ritonavir 800/100 mg once daily of at least moderate intensity (greater than or equal to Grade 2) in antiretroviral treatment-naĂŻve HIV-1-infected adult subjects are presented in Table 6Â and subsequent text below the table.
Table 6: Selected Clinical Adverse Drug Reactions to darunavir/ritonavir 800/100 mg Once Dailya of at Least Moderate Intensity (≥Grade 2) Occurring in ≥2% of Antiretroviral Treatment-Naïve HIV-1-Infected Adult Subjects (Trial TMC114-C211)
System organ class,
preferred term,
%
darunavir/ritonavir
800/100 mg once daily
+ TDF/FTC
N=343
lopinavir/ritonavir
800/200 mg per day
+ TDF/FTC
N=346
Gastrointestinal Disorders
   Abdominal pain
6%
6%
   Diarrhea
9%
16%
   Nausea
4%
4%
  Vomiting
2%
4%
General Disorders and Administration Site Conditions
   Fatigue
<1%
3%
Metabolism and Nutrition Disorders
   Anorexia
2%
<1%
Nervous System Disorders
   Headache
7%
6%
Skin and Subcutaneous Tissue Disorders
   Rash
6%
7%
N=total number of subjects per treatment group; FTC=emtricitabine; TDF=tenofovir disoproxil fumarate a Excluding laboratory abnormalities reported as ADRs.
Less Common Adverse Reactions
Treatment-emergent ADRs of at least moderate intensity (greater than or equal to Grade 2) occurring in less than 2% of antiretroviral treatment-naĂŻve subjects receiving darunavir/ritonavir 800/100 mg once daily are uled below by body system:
Gastrointestinal Disorders: acute pancreatitis, dyspepsia, flatulence
General Disorders and Administration Site Conditions: asthenia
Hepatobiliary Disorders: acute hepatitis (e.g., acute hepatitis, cytolytic hepatitis, hepatotoxicity)
Immune System Disorders: (drug) hypersensitivity, immune reconstitution syndrome
Metabolism and Nutrition Disorders: diabetes mellitus
Musculoskeletal and Connective Tissue Disorders: myalgia, osteonecrosis
Psychiatric Disorders: abnormal dreams
Skin and Subcutaneous Tissue Disorders: angioedema, pruritus, Stevens-Johnson Syndrome, urticaria
Laboratory Abnormalities
Selected Grade 2 to 4 laboratory abnormalities that represent a worsening from baseline observed in antiretroviral treatment-naĂŻve adult subjects treated with darunavir/ritonavir 800/100 mg once daily are presented in Table 7.
Table 7: Grade 2 to 4 Laboratory Abnormalities Observed in Antiretroviral Treatment-NaĂŻve HIV-1-Infected Adult Subjectsa (Trial TMC114-C211)
Laboratory parameter
%
Limit
darunavir/ritonavir
800/100 mg once
daily
+ TDF/FTC
lopinavir/ritonavir
800/200 mg per day
+ TDF/FTC
Biochemistry
Alanine Aminotransferase
   Grade 2
>2.5 to ≤5.0 X ULN
9%
9%
   Grade 3
>5.0 to ≤10.0 X ULN
3%
3%
   Grade 4
>10.0 X ULN
<1%
3%
Aspartate Aminotransferase
   Grade 2
>2.5 to ≤5.0 X ULN
7%
10%
   Grade 3
>5.0 to ≤10.0 X ULN
4%
2%
   Grade 4
>10.0 X ULN
1%
3%
Alkaline Phosphatase
   Grade 2
>2.5 to ≤5.0 X ULN
1%
1%
   Grade 3
>5.0 to ≤10.0 X ULN
0%
<1%
   Grade 4
>10.0 X ULN
0%
0%
Hyperbilirubinemia
   Grade 2
>1.5 to ≤2.5 X ULN
<1%
5%
   Grade 3
>2.5 to ≤5.0 X ULN
<1%
<1%
   Grade 4
>5.0 X ULN
0%
0%
Triglycerides
   Grade 2
5.65-8.48 mmol/L
500-750 mg/dL
3%
10%
   Grade 3
8.49-13.56 mmol/L
751-1,200 mg/dL
2%
5%
   Grade 4
>13.56 mmol/L
>1200 mg/dL
1%
1%
Total Cholesterol
   Grade 2
6.20-7.77 mmol/L
240-300 mg/dL
23%
27%
   Grade 3
>7.77 mmol/L
>300 mg/dL
1%
5%
Low-Density Lipoprotein Cholesterol
   Grade 2
4.13-4.90 mmol/L
160-190 mg/dL
14%
12%
   Grade 3
≥4.91 mmol/L
≥191 mg/dL
9%
6%
Elevated Glucose Levels
   Grade 2
6.95-13.88 mmol/L
126-250 mg/dL
11%
10%
   Grade 3
13.89-27.75 mmol/L
251-500 mg/dL
1%
<1%
   Grade 4
>27.75 mmol/L
>500 mg/dL
0%
0%
Pancreatic Lipase
   Grade 2
>1.5 to ≤3.0 X ULN
3%
2%
   Grade 3
>3.0 to ≤5.0 X ULN
<1%
1%
   Grade 4
>5.0 X ULN
0%
<1%
Pancreatic Amylase
   Grade 2
>1.5 to ≤2.0 X ULN
5%
2%
   Grade 3
>2.0 to ≤5.0 X ULN
5%
4%
   Grade 4
>5.0 X ULN
0%
<1%
N=total number of subjects per treatment group; FTC=emtricitabine; TDF=tenofovir disoproxil fumarate a Grade 4 data not applicable in Division of AIDS grading scale.
Treatment-Experienced Adults: TMC114-C214
The safety assessment is based on all safety data from the Phase 3 trial TMC114-C214 comparing darunavir/ritonavir 600/100 mg twice daily versus lopinavir/ritonavir 400/100 mg twice daily in 595 antiretroviral treatment-experienced HIV-1-infected adult subjects. The total mean exposure for subjects in the darunavir/ritonavir 600/100 mg twice daily arm and in the lopinavir/ritonavir 400/100 mg twice daily arm was 80.7 and 76.4 weeks, respectively.
The majority of the ADRs reported during treatment with darunavir/ritonavir 600/100 mg twice daily were mild in severity. The most common clinical ADRs to darunavir/ritonavir 600/100 mg twice daily (greater than or equal to 5%) of at least moderate intensity (greater than or equal to Grade 2) were diarrhea, nausea, rash, abdominal pain and vomiting. 4.7% of subjects in the darunavir/ritonavir arm discontinued treatment due to ADRs.
ADRs to darunavir/ritonavir 600/100 mg twice daily of at least moderate intensity (greater than or equal to Grade 2) in antiretroviral treatment-experienced HIV-1-infected adult subjects are presented in Table 8Â and subsequent text below the table.
Table 8: Selected Clinical Adverse Drug Reactions to darunavir/ritonavir 600/100 mg Twice Dailya  of at Least Moderate Intensity (≥Grade 2) Occurring in ≥2% of Antiretroviral Treatment-Experienced HIV-1-Infected Adult Subjects (Trial TMC114-C214)
System organ class,
preferred term,
%
darunavir/ritonavir
600/100 mg twice daily
+ OBR
N=298
lopinavir/ritonavir
400/100 mg twice
daily
+ OBR
N=297
Gastrointestinal Disorders
   Abdominal distension
2%
<1%
   Abdominal pain
6%
3%
   Diarrhea
14%
20%
   Dyspepsia
2%
1%
   Nausea
7%
6%
   Vomiting
5%
3%
General Disorders and Administration Site Conditions
   Asthenia
3%
1%
   Fatigue
2%
1%
Metabolism and Nutrition Disorders
   Anorexia
2%
2%
   Diabetes mellitus
2%
<1%
Nervous System Disorders
   Headache
3%
3%
Skin and Subcutaneous Tissue Disorders
   Rash
7%
3%
N=total number of subjects per treatment group; OBR=optimized background regimen a Excluding laboratory abnormalities reported as ADRs.
Less Common Adverse Reactions
Treatment-emergent ADRs of at least moderate intensity (greater than or equal to Grade 2) occurring in less than 2% of antiretroviral treatment-experienced subjects receiving darunavir/ritonavir 600/100 mg twice daily are uled below by body system:
Gastrointestinal Disorders: acute pancreatitis, flatulence
Musculoskeletal and Connective Tissue Disorders: myalgia
Psychiatric Disorders: abnormal dreams
Skin and Subcutaneous Tissue Disorders: pruritus, urticaria
Laboratory Abnormalities
Selected Grade 2 to 4 laboratory abnormalities that represent a worsening from baseline observed in antiretroviral treatment-experienced adult subjects treated with darunavir/ritonavir 600/100 mg twice daily are presented in Table 9.
Table 9: Grade 2 to 4 Laboratory Abnormalities Observed in Antiretroviral Treatment-Experienced HIV-1-Infected Adult Subjectsa (Trial TMC114-C214)
Laboratory parameter,
%
Limit
darunavir/ritonavir
600/100 mg twice daily
+ OBR
lopinavir/ritonavir
400/100 mg twice daily
+ OBR
Biochemistry
Alanine Aminotransferase
   Grade 2
>2.5 to ≤5.0 X ULN
7%
5%
   Grade 3
>5.0 to ≤10.0 X ULN
2%
2%
   Grade 4
>10.0 X ULN
1%
2%
Aspartate Aminotransferase
   Grade 2
>2.5 to ≤5.0 X ULN
6%
6%
   Grade 3
>5.0 to ≤10.0 X ULN
2%
2%
   Grade 4
>10.0 X ULN
<1%
2%
Alkaline Phosphatase
   Grade 2
>2.5 to ≤5.0 X ULN
<1%
0%
   Grade 3
>5.0 to ≤10.0 X ULN
<1%
<1%
   Grade 4
>10.0 X ULN
0%
0%
Hyperbilirubinemia
   Grade 2
>1.5 to ≤2.5 X ULN
<1%
2%
   Grade 3
>2.5 to ≤5.0 X ULN
<1%
<1%
   Grade 4
>5.0 X ULN
<1%
0%
Triglycerides
   Grade 2
5.65-8.48 mmol/L
500-750 mg/dL
10%
11%
   Grade 3
8.49-13.56 mmol/L
751-1,200 mg/dL
7%
10%
   Grade 4
>13.56 mmol/L
>1200 mg/dL
3%
6%
Total Cholesterol
   Grade 2
6.20-7.77 mmol/L
240-300 mg/dL
25%
23%
   Grade 3
>7.77 mmol/L
>300 mg/dL
10%
14%
Low-Density Lipoprotein Cholesterol
   Grade 2
4.13-4.90 mmol/L
160-190 mg/dL
14%
14%
   Grade 3
≥4.91 mmol/L
≥191 mg/dL
8%
9%
Elevated Glucose Levels
   Grade 2
6.95-13.88 mmol/L
126-250 mg/dL
10%
11%
   Grade 3
13.89-27.75 mmol/L
251-500 mg/dL
1%
<1%
   Grade 4
>27.75 mmol/L
>500 mg/dL
<1%
0%
Pancreatic Lipase
   Grade 2
>1.5 to ≤3.0 X ULN
3%
4%
   Grade 3
>3.0 to ≤5.0 X ULN
2%
<1%
   Grade 4
>5.0 X ULN
<1%
0%
Pancreatic Amylase
   Grade 2
>1.5 to ≤2.0 X ULN
6%
7%
   Grade 3
>2.0 to ≤5.0 X ULN
7%
3%
   Grade 4
>5.0 X ULN
0%
0%
N=total number of subjects per treatment group; OBR=optimized background regimen a Grade 4 data not applicable in Division of AIDS grading scale.
Serious ADRs
The following serious ADRs of at least moderate intensity (greater than or equal to Grade 2) occurred in the Phase 2b and Phase 3 trials with darunavir/ritonavir: abdominal pain, acute hepatitis, acute pancreatitis, anorexia, asthenia, diabetes mellitus, diarrhea, fatigue, headache, hepatic enzyme increased, hypercholesterolemia, hyperglycemia, hypertriglyceridemia, immune reconstitution syndrome, low density lipoprotein increased, nausea, pancreatic enzyme increased, rash, Stevens-Johnson Syndrome, and vomiting.
Patients Co-Infected with Hepatitis B and/or Hepatitis C Virus
In subjects co-infected with hepatitis B or C virus receiving darunavir/ritonavir, the incidence of adverse events and clinical chemistry abnormalities was not higher than in subjects receiving darunavir/ritonavir who were not co-infected, except for increased hepatic enzymes [see Warnings and Precautions (5.2)]. The pharmacokinetic exposure in co-infected subjects was comparable to that in subjects without co-infection.
Clinical Trials Experience: Pediatric Patients
Darunavir/ritonavir has been studied in combination with other antiretroviral agents in 3 Phase 2 trials. TMC114-C212, in which 80 antiretroviral treatment-experienced HIV-1-infected pediatric subjects 6 to less than 18 years of age and weighing at least 20 kg were included, TMC114-C228, in which 21 antiretroviral treatment-experienced HIV-1-infected pediatric subjects 3 to less than 6 years of age and weighing at least 10 kg were included, and TMC114-C230 in which 12 antiretroviral treatment-naĂŻve HIV-1 infected pediatric patients aged from 12 to less than 18 years and weighing at least 40 kg were included. The TMC114-C212 and C228 trials evaluated darunavir/ritonavir twice daily dosing and the TMC114-C230 trial evaluated darunavir/ritonavir once daily dosing [see Use in Specific Populations (8.4) and Clinical Studies (14.4)].
Frequency, type, and severity of ADRs in pediatric subjects were comparable to those observed in adults.
TMC114-C212
Clinical ADRs to darunavir/ritonavir (all grades, greater than or equal to 3%), were vomiting (13%), diarrhea (11%), abdominal pain (10%), headache (9%), rash (5%), nausea (4%), and fatigue (3%).
Grade 3 or 4 laboratory abnormalities were ALT increased (Grade 3: 3%; Grade 4: 1%), AST increased (Grade 3: 1%), pancreatic amylase increased (Grade 3: 4%, Grade 4: 1%), pancreatic lipase increased (Grade 3: 1%), total cholesterol increased (Grade 3: 1%), and LDL increased (Grade 3: 3%).
TMC114-C228
Clinical ADRs to darunavir/ritonavir (all grades, greater than or equal to 5%), were diarrhea (24%), vomiting (19%), rash (19%), abdominal pain (5%), and anorexia (5%).
There were no Grade 3 or 4 laboratory abnormalities considered as ADRs in this trial.
TMC114-C230
Clinical ADRs to darunavir/ritonavir (all grades, greater than or equal to 3%), were vomiting (33%), nausea (25%), diarrhea (16.7%), abdominal pain (8.3%), decreased appetite (8.3%), pruritus (8.3%), and rash (8.3%).
There were no Grade 3 or 4 laboratory abnormalities considered as ADRs in this trial.
6.2 Postmarketing Experience
The following adverse reactions have been identified during post approval use of darunavir. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Metabolism and Nutrition Disorders:Â Redistribution of body fat
Musculoskeletal and Connective Tissue Disorders: Rhabdomyolysis (associated with co-administration with HMG-CoA reductase inhibitors and darunavir/ritonavir)
Skin and Subcutaneous Tissue Disorders: Toxic epidermal necrolysis, acute generalized exanthematous pustulosis, drug rash with eosinophilia and systemic symptoms [see Warnings and Precautions (5.3)]
Renal and Urinary Disorders: Crystal nephropathy, crystalluria
7 Drug Interactions
- Co-administration of darunavir/ritonavir with other drugs can alter the concentrations of other drugs and other drugs may alter the concentrations of darunavir. The potential drug-drug interactions must be considered prior to and during therapy. (
4 ,5.5 ,7 ,12.3 ).7.1 Potential for darunavir/ritonavir to Affect Other Drugs
Darunavir co-administered with ritonavir is an inhibitor of CYP3A, CYP2D6, and P-gp. Co-administration of darunavir and ritonavir with drugs that are primarily metabolized by CYP3A and CYP2D6 or are transported by P-gp may result in increased plasma concentrations of such drugs, which could increase or prolong their therapeutic effect and adverse events.
Darunavir co-administered with ritonavir with drugs that have active metabolite(s) formed by CYP3A may result in reduced plasma concentrations of these active metabolite(s), potentially leading to loss of their therapeutic effect (see Table 10).
7.2 Potential for Other Drugs to Affect Darunavir
Darunavir and ritonavir are metabolized by CYP3A. In vitro data indicate that darunavir may be a P-gp substrate. Drugs that induce CYP3A activity would be expected to increase the clearance of darunavir and ritonavir, resulting in lowered plasma concentrations of darunavir and ritonavir. Co-administration of darunavir and ritonavir and other drugs that inhibit CYP3A, or P-gp may decrease the clearance of darunavir and ritonavir and may result in increased plasma concentrations of darunavir and ritonavir (see Table 10).
7.3 Established and Other Potentially Significant Drug Interactions
Table 10 provides dosing recommendations as a result of drug interactions with darunavir/ritonavir. These recommendations are based on either drug interaction studies or predicted interactions due to the expected magnitude of interaction and potential for serious adverse events or loss of efficacy. The table includes examples of potentially significant interactions but is not all inclusive [see Contraindications (4) and Clinical Pharmacology (12.3)], and therefore the label of each drug that is co-administered with darunavir /ritonavir should be consulted for information related to the route of metabolism, interaction pathways, potential risks, and specific actions to be taken with regard to co-administration.
Table 10: Established and Other Potentially Significant Drug Interactions: Alterations in Dose or Regimen May be Recommended Based on Drug Interaction Studies or Predicted Interaction (see Contraindications (4) for a ul of examples of contraindicated drugs) [see Clinical Pharmacology (12.3) for Magnitude of Interaction, Tables 15 and 16]
Concomitant Drug Class
Drug Name Examples
Effect on Concentration of Darunavir Or Concomitant Drug
Clinical Comment
HIV-1-Antiviral Agents: Nucleoside Reverse Transcriptase Inhibitors (NRTIs)
didanosine
↔ darunavir
↔ didanosine
Didanosine should be administered one hour before or two hours after darunavir/ritonavir (which are administered with food).
HIV-1-Antiviral Agents: HIV-Protease Inhibitors (PIs)
indinavir
(The reference regimen for indinavir was indinavir/ritonavir 800/100 mg twice daily.)
↑ darunavir
↑ indinavir
The appropriate dose of indinavir in combination with darunavir/ritonavir has not been established.
lopinavir/ritonavir
↓ darunavir
↔ lopinavir
Appropriate doses of the combination have not been established. Hence, it is not recommended to co-administer lopinavir/ritonavir and darunavir, with or without ritonavir.
saquinavir
Other HIV protease inhibitors, except atazanavir [see Drug Interactions (7.4)]
↓ darunavir
↔ saquinavir
Appropriate doses of the combination have not been established. Hence, it is not recommended to co-administer saquinavir and darunavir, with or without ritonavir.
As co-administration with darunavir/ritonavir has not been studied, co-administration is not recommended.
HIV-1-Antiviral Agents: CCR5 co-receptor antagonists
maraviroc
↑ maraviroc
When used in combination with darunavir/ritonavir, the dose of maraviroc should be 150 mg twice daily.
Other Agents
Alpha 1-adrenoreceptor antagonist:
alfuzosinÂ
↑ alfuzosin
Co-administration is contraindicated due to potential for serious and/or life-threatening reactions such as hypotension.
Antibacterial:
clarithromycin
↔ darunavir
↑ clarithromycin
No dose adjustment of the combination is required for patients with normal renal function. For co-administration of clarithromycin and darunavir/ritonavir in patients with renal impairment, the following dose adjustments should be considered:
• For subjects with CLcr of 30-60 mL/min, the dose of clarithromycin should be reduced by 50%.
• For subjects with CLcr of <30 mL/min, the dose of clarithromycin should be reduced by 75%.
Anticoagulants: Direct Oral Anticoagulants (DOACs) apixaban
rivaroxaban
dabigatran etexilateedoxaban
Other Anticoagulants
warfarin
↑ apixaban
↑ rivaroxaban
↑ dabigatran↑ edoxaban
↓ warfarin
↔ darunavir
Due to potentially increased bleeding risk, dosing recommendations for co-administration of apixaban with darunavir/ritonavir depend on the apixaban dose. Refer to apixaban dosing instructions for co-administration with P-gp and strong CYP3A inhibitors in apixaban prescribing information.
Co-administration of darunavir/ritonavir and rivaroxaban is not recommended because it may lead to an increased bleeding risk.
Refer to the dabigatran etexilate or edoxaban prescribing information for recommendations regarding co-administration. The specific recommendations are based on indication, renal function, and effect of the co-administered P-gp inhibitors on the concentration of dabigatran or edoxaban. Clinical monitoring is recommended when a DOAC not affected by CYP3A4 but transported by P-gp, including dabigatran etexilate and edoxaban, is co-administered with darunavir/ritonavir.
Warfarin concentrations are decreased when co-administered with darunavir/ritonavir. It is recommended that the international normalized ratio (INR) be monitored when warfarin is combined with darunavir/ritonavir.
Anticonvulsants:
carbamazepine
↔ darunavir
↑ carbamazepine
The dose of either darunavir/ritonavir or carbamazepine does not need to be adjusted when initiating co-administration with darunavir/ritonavir and carbamazepine. Clinical monitoring of carbamazepine concentrations and its dose titration is recommended to achieve the desired clinical response.
clonazepam
phenobarbital, phenytoin
↑ clonazepam
↔ darunavir
↓ phenytoin
↓ phenobarbital
Clinical monitoring of anticonvulsants that are metabolized by CYP3A is recommended.
Phenytoin and phenobarbital levels should be monitored when co-administering with darunavir/ritonavir.
Antidepressants:
Selective Serotonin Reuptake Inhibitors (SSRIs):
paroxetine, sertraline
Tricyclic Antidepressants (TCAs):
amitriptyline, desipramine,
imipramine, nortriptyline
Other:
trazodone
↓ paroxetine
↓ sertraline
↑ amitriptyline
↑ desipramine
↑ imipramine
↑ nortriptyline
↑ trazodone
If either sertraline or paroxetine is initiated in patients receiving darunavir/ritonavir, dose titrating the SSRI based on a clinical assessment of antidepressant response is recommended. Monitor for antidepressant response in patients on a stable dose of sertraline or paroxetine who start treatment with darunavir/ritonavir.
Use a lower dose of the tricyclic antidepressants and trazodone due to potential increased adverse events such as nausea, dizziness, hypotension and syncope.
Antifungals:
itraconazole, isavuconazole,
ketoconazole, posaconazole
voriconazole
↑ darunavir
↑ itraconazole
↑ isavuconazole
↑ ketoconazole
↔ posaconazoleÂ
↓ voriconazole
Monitor for increased darunavir/ritonavir and/or antifungal adverse events with concomitant use of these antifungals. When co-administration is required, the daily dose of ketoconazole or itraconazole should not exceed 200 mg with monitoring for increased antifungal adverse events.
Voriconazole is not recommended for patients receiving darunavir/ritonavir unless an assessment comparing predicted benefit to risk ratio justifies the use of voriconazole.
Anti-gout:
colchicine
↑ colchicine
Co-administration is contraindicated in patients with renal and/or hepatic impairment due to potential for serious and/or life-threatening reactions.
For patients without renal or hepatic impairment:
- Treatment of gout-flares – co-administration of colchicine in patients on darunavir/ritonavir: 0.6 mg (1 tablet) × 1 dose, followed by 0.3 mg (half tablet) 1 hour later. Treatment course to be repeated no earlier than 3 days.
- Prophylaxis of gout-flares – co-administration of colchicine in patients on darunavir/ritonavir:
If the original regimen was 0.6 mg twice a day, the regimen should be adjusted to 0.3 mg once a day.
If the original regimen was 0.6 mg once a day, the regimen should be adjusted to 0.3 mg once every other day.
- Treatment of familial Mediterranean fever – co-administration of colchicine in patients on darunavir/ritonavir:
maximum daily dose of 0.6 mg (may be given as 0.3 mg twice a day).
Antimalarial:
artemether/lumefantrine
↓ artemether
↓ dihydroartemisinin
↑ lumefantrine
↔ darunavir
The combination of darunavir/ritonavir and artemether/lumefantrine can be used without dose adjustments. However, the combination should be used with caution as increased lumefantrine exposure may increase the risk of QT prolongation.
Antimycobacterials:
rifampin rifabutin(The reference regimen for rifabutin was 300 mg once daily.)
rifapentine
↓ darunavir ↑ darunavir
↑ rifabutin
↑ 25-O-desacetylrifabutin
↓ darunavir
Co-administration is contraindicated due to potential for loss of therapeutic effect and development of resistance.
Dose reduction of rifabutin by at least 75% of the usual dose (300 mg once daily) is recommended (i.e., a maximum dose of 150 mg every other day). Increased monitoring for adverse events is warranted in patients receiving this combination and further dose reduction of rifabutin may be necessary.
Co-administration of darunavir/ritonavir with rifapentine is not recommended.
Antineoplastics:
dasatinib, nilotinib
vinblastine, vincristine
↑ antineoplastics
A decrease in the dosage or an adjustment of the dosing interval of dasatinib and nilotinib may be necessary for patients. Please refer to the dasatinib and nilotinib prescribing information for dosing instructions.
For vincristine and vinblastine, consideration should be given to temporarily withholding the ritonavir-containing antiretroviral regimen in patients who develop significant hematologic or gastrointestinal side effects when darunavir/ritonavir is administered concurrently with vincristine or vinblastine. If the antiretroviral regimen must be withheld for a prolonged period, consideration should be given to initiating a revised regimen that does not include a CYP3A or P-gp inhibitor.
Antipsychotics:
lurasidone
pimozide
quetiapine
e.g. perphenazine, risperidone, thioridazine
↑ lurasidone
↑ pimozide
↑ quetiapine
↑ antipsychotics
Co-administration is contraindicated due to potential for serious and/or life-threatening reactions. Co-administration is contraindicated due to potential for serious and/or life-threatening reactions such as cardiac arrhythmias.
Initiation of darunavir with ritonavir in patients taking quetiapine:
Consider alternative antiretroviral therapy to avoid increases in quetiapine exposures. If co-administration is necessary, reduce the quetiapine dose to 1/6 of the current dose and monitor for quetiapine-associated adverse reactions. Refer to the quetiapine prescribing information for recommendations on adverse reaction monitoring.
Initiation of quetiapine in patients taking darunavir with ritonavir:
Refer to the quetiapine prescribing information for initial dosing and titration of quetiapine.
A decrease in the dose of antipsychotics that are metabolized by CYP3A or CYP2D6 may be needed when co-administered with darunavir/ritonavir.
β-Blockers:
e.g. carvedilol, metoprolol, timolol
↑ beta-blockers
Clinical monitoring of patients is recommended. A dose decrease may be needed for these drugs when co-administered with darunavir/ritonavir and a lower dose of the beta blocker should be considered.
Calcium Channel Blockers:
amlodipine, diltiazem, felodipine, nicardipine, nifedipine, verapamil
↑ calcium channel blockers
Clinical monitoring of patients is recommended.
Cardiac Disorders:
ranolazine, ivabradine
dronedarone
Other antiarrhythmics e.g. amiodarone, bepridil, disopyramide, flecainide, lidocaine (systemic), mexiletine, propafenone, quinidine
 digoxinÂ
↑ ranolazine↑ ivabradine
↑ dronedarone
↑ antiarrhythmics
↑ digoxin
Â
Co-administration is contraindicated due to potential for serious and/or life-threatening reactions.
Co-administration is contraindicated due to potential for serious and/or life-threatening reactions such as cardiac arrhythmias.
Therapeutic concentration monitoring, if available, is recommended for antiarrhythmics when co-administered with darunavir/ritonavir.
The lowest dose of digoxin should initially be prescribed. The serum digoxin concentrations should be monitored and used for titration of digoxin dose to obtain the desired clinical effect.
Corticosteroids: dexamethasone (systemic)
Corticosteroids primarily metabolized by CYP3A:e.g. betamethasonebudesonideciclesonidefluticasonemethylprednisolonemometasonetriamcinolone
↓ darunavir
↑ corticosteroids
Co-administration of darunavir/ritonavir with systemic dexamethasone or other systemic corticosteroids that induce CYP3A may result in loss of therapeutic effect and development of resistance to darunavir. Consider alternative corticosteroids.
Co-administration with corticosteroids (all routes of administration) of which exposures are significantly increased by strong CYP3A inhibitors can increase the risk for Cushing’s syndrome and adrenal suppression.
Alternative corticosteroids including beclomethasone, prednisone, and prednisolone (for which PK and/or PD are less affected by strong CYP3A inhibitors relative to other steroids) should be considered, particularly for long term use.Â
Endothelin receptor antagonist:
bosentan
↑ bosentan
Co-administration of bosentan in patients on darunavir/ritonavir:
In patients who have been receiving darunavir/ritonavir for at least 10 days, start bosentan at 62.5 mg once daily or every other day based upon individual tolerability.
Co-administration of darunavir/ritonavir in patients on bosentan:
Discontinue use of bosentan at least 36 hours prior to initiation of darunavir/ritonavir. After at least 10 days following the initiation of darunavir/ritonavir, resume bosentan at 62.5 mg once daily or every other day based upon individual tolerability.
Ergot derivatives: e.g. dihydroergotamine, ergotamine, methylergonovine  ↑ ergot derivatives Co-administration is contraindicated due to potential for serious and/or life-threatening reactions such as acute ergot toxicity characterized by peripheral vasospasm and ischemia of the extremities and other tissues.
Hepatitis C virus (HCV): Direct-Acting Antivirals:elbasvir/grazoprevir
glecaprevir/pibrentasvir
↑ elbasvir/grazoprevir
↑ glecaprevir
↑ pibrentasvir
Co-administration is contraindicated due to potential for the increased risk of alanine transaminase (ALT) elevations.
Co-administration of darunavir/ritonavir with glecaprevir/pibrentasvir is not recommended.
Herbal product:
St. John’s wort (Hypericum perforatum)
↓ darunavir
Co-administration is contraindicated due to potential for reduced plasma concentrations of darunavir, which may result in loss of therapeutic effect and development of resistance.
Hormonal contraceptives: ethinyl estradiol, norethindrone, drospirenone
↓ ethinyl estradiol
↓ norethindrone drospirenone:effects unknown
Â
Effective alternative (non-hormonal) contraceptive method or a barrier method of contraception is recommended [see Use in Specific Populations (8.3)].
For co-administration with drospirenone, clinical monitoring is recommended due to the potential for hyperkalemia. No data are available to make recommendations on co-administration with other hormonal contraceptives.
Immunosuppressants:
e.g. cyclosporine, tacrolimus,
sirolimus
Immunosuppressant/neoplastic:
everolimus
irinotecan
↑ immunosuppressants
Therapeutic concentration monitoring of the immunosuppressive agent is recommended when co-administered with darunavir/ritonavir.
Co-administration of everolimus and darunavir/ritonavir is not recommended.
Discontinue darunavir/ritonavir at least 1 week prior to starting irinotecan therapy. Do not administer darunavir/ritonavir with irinotecan unless there are no therapeutic alternatives.
Inhaled beta agonist:
salmeterol
↑ salmeterol
Co-administration of salmeterol and darunavir/ritonavir is not recommended. The combination may result in increased risk of cardiovascular adverse events associated with salmeterol, including QT prolongation, palpitations and sinus tachycardia.
Lipid Modifying Agents:
HMG-CoA reductase inhibitors:
lovastatin, simvastatin
atorvastatin, pravastatin, rosuvastatin
Other lipid modifying agents: lomitapideÂ
↑ lovastatin
↑ simvastatin
↑ HMG-CoA reductase inhibitors
↑ lomitapide
Â
Co-administration is contraindicated due to potential for serious reactions such as myopathy including rhabdomyolysis.
Co-administration of darunavir/ritonavir with HMG-CoA reductase inhibitors may lead to adverse events such as myopathy. Titrate atorvastatin, pravastatin or rosuvastatin dose carefully and use the lowest necessary dose while monitoring for adverse events. Do not exceed atorvastatin 20 mg/day.
Co-administration is contraindicated due to potential for markedly increased transaminases.Narcotic analgesics metabolized by CYP3A: e.g. fentanyl, oxycodone
↑ fentanyl
↑ oxycodone
 Careful monitoring of therapeutic effects and adverse reactions associated with CYP3A-metabolized narcotic analgesics (including potentially fatal respiratory depression) is recommended with co-administration.  tramadol ↑  tramadol  A dose decrease may be needed for tramadol with concomitant use.
Narcotic analgesics/treatment of opioid dependence:
buprenorphine,
buprenorphine/naloxone
methadone
↔ buprenorphine, naloxone
↑ norbuprenorphine (metabolite)
↓ methadone
No dose adjustment for buprenorphine or buprenorphine/naloxone is required with concurrent administration of darunavir/ritonavir. Clinical monitoring is recommended if darunavir/ritonavir and buprenorphine or buprenorphine/naloxone are co-administered.
No adjustment of methadone dosage is required when initiating co-administration of darunavir/ritonavir. However, clinical monitoring is recommended as the dose of methadone during maintenance therapy may need to be adjusted in some patients.
Opioid Antagonist
naloxegol
↑ naloxegol
Co-administration of darunavir/ritonavir and naloxegol is contraindicated due to potential for precipitating opioid withdrawal symptoms.
PDE-5 inhibitors:
e.g. avanafil, sildenafil, tadalafil,
vardenafil
↑ PDE-5 inhibitors (only the use of sildenafil at doses used for treatment of erectile dysfunction has been studied with darunavir/ritonavir)
Co-administration with darunavir/ritonavir may result in an increase in PDE-5 inhibitor-associated adverse events, including hypotension, syncope, visual disturbances and priapism.
Use of PDE-5 inhibitors for pulmonary arterial hypertension (PAH):
Co-administration with sildenafil used for PAH is contraindicated due to potential for sildenafil associated adverse reactions (which include visual disturbances, hypotension, prolonged erection, and syncope).
The following dose adjustments are recommended for use of tadalafil with darunavir/ritonavir:
- Co-administration of tadalafil in patients on darunavir/ritonavir:
In patients receiving darunavir/ritonavir for at least one week, start tadalafil at 20 mg once daily. Increase to 40 mg once daily based upon individual tolerability.
- Co-administration of darunavir/ritonavir in patients on tadalafil:
Avoid use of tadalafil during the initiation of darunavir/ritonavir. Stop tadalafil at least 24 hours prior to starting darunavir/ritonavir. After at least one week following the initiation of darunavir/ritonavir, resume tadalafil at 20 mg once daily. Increase to 40 mg once daily based upon individual tolerability.
Use of PDE-5 inhibitors for erectile dysfunction: Sildenafil at a single dose not exceeding 25 mg in 48 hours, vardenafil at a single dose not exceeding 2.5 mg dose in 72 hours, or tadalafil at a single dose not exceeding 10 mg dose in 72 hours can be used with increased monitoring for PDE-5 inhibitor-associated adverse events.
Co-administration of darunavir/ritonavir and avanafil is not recommended.
Â
Platelet aggregation inhibitor: ticagrelor
clopidogrel
prasugrelÂ
↑ ticagrelor
↓ clopidogrel active metabolite
↔ prasugrel active metaboliteÂ
Co-administration of darunavir/ritonavir and ticagrelor is not recommended.
Co-administration of darunavir/ritonavir and clopidogrel is not recommended due to potential reduction of the antiplatelet activity of clopidogrel.
No dose adjustment is needed when prasugrel is co-administered with darunavir/ritonavir.
Proton pump inhibitor:
omeprazole
↓ omeprazole
↔ darunavir
When omeprazole is co-administered with darunavir/ritonavir, monitor patients for decreased efficacy of omeprazole. Consider increasing the omeprazole dose in patients whose symptoms are not well controlled; avoid use of more than 40 mg per day of omeprazole.
Sedatives/hypnotics:
orally administered midazolam, triazolam
metabolized by CYP3Ae.g. buspirone, diazepam,estazolam, zolpidem
parenterally administered midazolam
↑ midazolam
↑ triazolam
↑ sedatives/hypnotics
Co-administration is contraindicated due to potential for serious and/or life-threatening reactions such as prolonged or increased sedation or respiratory depression. Triazolam and orally administered midazolam are extensively metabolized by CYP3A. Co-administration of triazolam or orally administered midazolam with darunavir may cause large increases in the concentrations of these benzodiazepines.
Titration is recommended when co-administering darunavir/ritonavir with sedatives/hypnotics metabolized by CYP3A and a lower dose of the sedatives/hypnotics should be considered with monitoring for adverse events.
Co-administration of parenteral midazolam should be done in a setting which ensures close clinical monitoring and appropriate medical management in case of respiratory depression and/or prolonged sedation. Dosage reduction for midazolam should be considered, especially if more than a single dose of midazolam is administered.
Urinary antispasmodics fesoterodine solifenacin
↑ fesoterodine
↑ solifenacin
When fesoterodine is co-administered with darunavir/ritonavir, do not exceed a fesoterodine dose of 4 mg once daily.
When solifenacin is co-administered with darunavir/ritonavir, do not exceed a solifenacin dose of 5 mg once daily.
7.4 Drugs without Clinically Significant Interactions with Darunavir
No dosage adjustments are recommended when darunavir/ritonavir is co-administered with the following medications: atazanavir, dolutegravir, efavirenz, etravirine, nevirapine, nucleoside reverse transcriptase inhibitors (abacavir, emtricitabine, emtricitabine/tenofovir alafenamide, lamivudine, stavudine, tenofovir disoproxil fumarate, zidovudine), pitavastatin, raltegravir, ranitidine, or rilpivirine.
8 Use In Specific Populations
- Pregnancy: Total darunavir exposures were generally lower during pregnancy compared to postpartum period. The reduction in darunavir exposures during pregnancy were greater for once daily dosing compared to the twice daily dosing regimen. (
8.1 ,12.3 )- Lactation: Women infected with HIV should be instructed not to breastfeed due to the potential for HIV transmission. (
8.2 )- Pediatrics: Not recommended for patients less than 3 years of age. (
8.4 )8.1 Pregnancy
Pregnancy Exposure Registry
There is a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to darunavir during pregnancy. Healthcare providers are encouraged to register patients by calling the Antiretroviral Pregnancy Registry (APR) 1-800-258-4263.
Risk Summary
Prospective pregnancy data from the APR are not sufficient to adequately assess the risk of birth defects or miscarriage. Available limited data from the APR show no statistically significant difference in the overall risk of major birth defects for darunavir compared with the background rate for major birth defects of 2.7% in a U.S. reference population of the Metropolitan Atlanta Congenital Defects Program (MACDP) [see Data].
The rate of miscarriage is not reported in the APR. The estimated background rate of miscarriage in clinically recognized pregnancies in the U.S. general population is 15-20%. The background risk of major birth defects and miscarriage for the indicated population is unknown.
Studies in animals did not show evidence of developmental toxicity. Exposures (based on AUC) in rats were 3-fold higher, whereas in mice and rabbits, exposures were lower (less than 1-fold) than human exposures at the recommended daily dose [see Data].
Clinical Considerations
The recommended dosage in pregnant patients is darunavir 600 mg taken with ritonavir 100 mg twice daily with food.
Darunavir 800 mg taken with ritonavir 100 mg once daily should only be considered in certain pregnant patients who are already on a stable darunavir 800 mg with ritonavir 100 mg once daily regimen prior to pregnancy, are virologically suppressed (HIV-1 RNA less than 50 copies per mL), and in whom a change to twice daily darunavir 600 mg with ritonavir 100 mg may compromise tolerability or compliance [see Dosage and Administration (2.4) and Clinical Pharmacology (12.3)].
Data
Human Data
Darunavir/ritonavir (600/100 mg twice daily or 800/100 mg once daily) in combination with a background regimen was evaluated in a clinical trial of 36 pregnant women during the second and third trimesters, and postpartum. Eighteen subjects were enrolled in each BID and QD treatment arms. Twenty-nine subjects completed the trial through the postpartum period (6-12 weeks after delivery) and 7 subjects discontinued before trial completion, 5 subjects in the BID arm and 2 subjects in the QD arm.
The pharmacokinetic data demonstrate that exposure to darunavir and ritonavir as part of an antiretroviral regimen was lower during pregnancy compared with postpartum (6-12 weeks). Exposure reductions during pregnancy were greater for the once daily regimen as compared to the twice daily regimen [see Clinical Pharmacology (12.3)].
Virologic response was preserved. In the BID arm, the proportion of subjects with HIV-1 RNA <50 copies/mL were 39% (7/18) at baseline, 61% (11/18) through the third trimester visit, and 61% (11/18) through the 6-12 week postpartum visit. Virologic outcomes during the third trimester visit showed HIV-1 RNA ≥50 copies/mL for 11% (2/18) of subjects and were missing for 5 subjects (1 subject discontinued prematurely due to virologic failure). In the QD arm, the proportion of subjects with HIV-1 RNA <50 copies/mL were 61% (11/18) at baseline, 83% (15/18) through the third trimester visit, and 78% (14/18) through the 6-12 week postpartum visit. Virologic outcomes during the third trimester visit showed HIV-1 RNA ≥50 copies/mL for none of the subjects and were missing for 3 subjects (1 subject discontinued prematurely due to virologic failure).
Darunavir/ritonavir was well tolerated during pregnancy and postpartum. There were no new clinically relevant safety findings compared with the known safety profile of darunavir/ritonavir in HIV-1-infected adults.
Among the 31 infants with HIV test results available data, born to the 31 HIV-infected pregnant women who completed trial through delivery or postpartum period, all 31 infants had test results that were negative for HIV-1 at the time of delivery and/or through 16 weeks postpartum. All 31 infants received antiretroviral prophylactic treatment containing zidovudine.
Based on prospective reports to the APR of over 980 exposures to darunavir-containing regimens during pregnancy resulting in live births (including over 660 exposed in the first trimester and over 320 exposed in the second/third trimester), the prevalence of birth defects in live births was 3.6% (95% CI: 2.3% to 5.3.%) with first trimester exposure to darunavir-containing regimens and 2.5% (95% CI: 1.1% to 4.8%) with second/third trimester exposure to darunavir-containing regimens.
Animal Data
Reproduction studies conducted with darunavir showed no embryotoxicity or teratogenicity in mice (doses up to 1,000 mg/kg from gestation day (GD) 6-15 with darunavir alone) and rats (doses up to 1,000 mg/kg from GD 7-19 in the presence or absence of ritonavir) as well as in rabbits (doses up to 1,000 mg/kg/day from GD 8-20 with darunavir alone). In these studies, darunavir exposures (based on AUC) were higher in rats (3-fold), whereas in mice and rabbits, exposures were lower (less than 1-fold) compared to those obtained in humans at the recommended clinical dose of darunavir boosted with ritonavir.
8.2 Lactation
Risk Summary
The Centers for Disease Control and Prevention recommend that HIV-infected mothers not breastfeed their infants to avoid risking postnatal transmission of HIV.
There are no data on the presence of darunavir in human milk, the effects on the breastfed infant, or the effects on milk production. Darunavir is present in the milk of lactating rats [see Data]. Because of the potential for (1) HIV transmission (in HIV-negative infants), (2) developing viral resistance (in HIV-positive infants) and (3) serious adverse reactions in a breastfed infant, instruct mothers not to breastfeed if they are receiving darunavir [see Use in Specific Populations (8.4)].
Data
Animal Data
Studies in rats (with darunavir alone or with ritonavir) have demonstrated that darunavir is secreted in the milk. In the rat pre- and postnatal development study, a reduction in pup body weight gain was observed due to exposure of pups to drug substances via milk. The maximal maternal plasma exposures achieved with darunavir (up to 1,000 mg/kg with ritonavir) were approximately 50% of those obtained in humans at the recommended clinical dose with ritonavir.
8.3 Females and Males of Reproductive Potential
Contraception
Use of darunavir may reduce the efficacy of combined hormonal contraceptives and the progestin only pill. Advise patients to use an effective alternative (non-hormonal) contraceptive method or add a barrier method of contraception. For co-administration with drospirenone, clinical monitoring is recommended due to the potential for hyperkalemia [see Drug Interactions (7.3)].
8.4 Pediatric Use
Darunavir/ritonavir is not recommended in pediatric patients below 3 years of age because of toxicity and mortality observed in juvenile rats dosed with darunavir (from 20 mg/kg to 1,000 mg/kg) up to days 23 to 26 of age [see Warnings and Precautions (5.10), Use in Specific Populations (8.1) and Clinical Pharmacology (12.3)].
The safety, pharmacokinetic profile, and virologic and immunologic responses of darunavir/ritonavir administered twice daily were evaluated in treatment-experienced HIV-1-infected pediatric subjects 3 to less than 18 years of age and weighing at least 10 kg. These subjects were evaluated in clinical trials TMC114-C212 (80 subjects, 6 to less than 18 years of age) and TMC114-228 (21 subjects, 3 to less than 6 years of age) [see Adverse Reactions (6.1), Clinical Pharmacology (12.3) and Clinical Studies (14.4)]. Frequency, type, and severity of adverse drug reactions in pediatric subjects were comparable to those observed in adults [see Adverse Reactions (6.1)]. Refer to Dosage and Administration (2.5) for twice-daily dosing recommendations for pediatric subjects 3 to less than 18 years of age and weighing at least 10 kg.
In clinical trial TMC114-C230, the safety, pharmacokinetic profile and virologic and immunologic responses of darunavir/ritonavir administered once daily were evaluated in treatment-naĂŻve HIV-1 infected pediatric subjects 12 to less than 18 years of age (12 subjects) [see Adverse Reactions (6.1), Clinical Pharmacology (12.3) and Clinical Studies (14.4)]. Frequency, type, and severity of adverse drug reactions in pediatric subjects were comparable to those observed in adults [see Adverse Reactions (6.1)]. Once daily dosing recommendations for pediatric patients 3 to less than 12 years of age were derived using population pharmacokinetic modeling and simulation. Although a darunavir/ritonavir once daily dosing pediatric trial was not conducted in children less than 12 years of age, there is sufficient clinical safety data to support the predicted darunavir exposures for the dosing recommendations in this age group [see Clinical Pharmacology (12.3)]. Please see Dosage and Administration (2.5) for once-daily dosing recommendations for pediatric subjects 3 to less than 18 years of age and weighing at least 10 kg.
Juvenile Animal Data
In a juvenile toxicity study where rats were directly dosed with darunavir (up to 1,000 mg/kg), deaths occurred from post-natal day 5 at plasma exposure levels ranging from 0.1 to 1.0 of the human exposure levels. In a 4-week rat toxicology study, when dosing was initiated on post-natal day 23 (the human equivalent of 2 to 3 years of age), no deaths were observed with a plasma exposure (in combination with ritonavir) 2 times the human plasma exposure levels.
8.5 Geriatric Use
Clinical studies of darunavir did not include sufficient numbers of patients aged 65 years and over to determine whether they respond differently from younger patients. In general, caution should be exercised in the administration and monitoring of darunavir in elderly patients, reflecting the greater frequency of decreased hepatic function, and of concomitant disease or other drug therapy [see Clinical Pharmacology (12.3)].
8.6 Hepatic Impairment
No dosage adjustment of darunavir/ritonavir is necessary for patients with either mild or moderate hepatic impairment. No pharmacokinetic or safety data are available regarding the use of darunavir/ritonavir in subjects with severe hepatic impairment. Therefore, darunavir/ritonavir is not recommended for use in patients with severe hepatic impairment [see Dosage and Administration (2.6) and Clinical Pharmacology (12.3)].
8.7 Renal Impairment
Population pharmacokinetic analysis showed that the pharmacokinetics of darunavir were not significantly affected in HIV-infected subjects with moderate renal impairment (CrCL between 30-60 mL/min, n=20). No pharmacokinetic data are available in HIV-1-infected patients with severe renal impairment or end stage renal disease; however, because the renal clearance of darunavir is limited, a decrease in total body clearance is not expected in patients with renal impairment. As darunavir and ritonavir are highly bound to plasma proteins, it is unlikely that they will be significantly removed by hemodialysis or peritoneal dialysis [see Clinical Pharmacology (12.3)].
10 Overdosage
Human experience of acute overdose with darunavir/ritonavir is limited. No specific antidote is available for overdose with darunavir. Treatment of overdose with darunavir consists of general supportive measures including monitoring of vital signs and observation of the clinical status of the patient. Since darunavir is highly protein bound, dialysis is unlikely to be beneficial in significant removal of the active substance.
11 Description
Darunavir is an inhibitor of the human immunodeficiency virus (HIV-1) protease.
Darunavir, (present as darunavir hydrate) has the following chemical name: [(1S,2R)-3-[[(4-aminophenyl) sulfonyl](2-methylpropyl)amino]-2-hydroxy-1-(phenylmethyl)propyl]-carbamic acid(3R,3aS,6aR)-hexahydrofuro[2,3-b]furan-3-yl ester hydrate. Darunavir hydrate has the following structural formula:
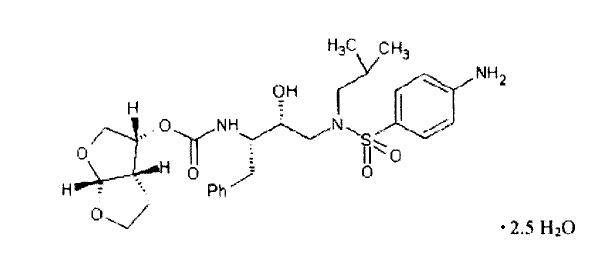
C27H37N3O7S•2.5H2O        M.W. 592.67
Darunavir hydrate is a white to off-white crystalline powder and is soluble in methanol, freely soluble in acetone and dimethylsulphoxide.
Darunavir 600 mg tablets are available as orange, oval-shaped, film-coated tablets for oral administration. Each 600 mg tablet contains darunavir 600 mg (present as darunavir hydrate).
Each tablet also contains the inactive ingredients colloidal silicon dioxide, crospovidone, FD&C red #40 aluminum lake, FD&C yellow #6 aluminum lake, magnesium stearate, microcrystalline cellulose, polyethylene glycol, polyvinyl alcohol, talc, and titanium dioxide.
The strength of darunavir tablets is expressed in terms of the free form of darunavir.
12 Clinical Pharmacology
12.1 Mechanism of Action
Darunavir is an HIV-1 antiviral drug [see Microbiology (12.4)].
12.2 Pharmacodynamics
Cardiac Electrophysiology
In a thorough QT/QTc study in 40 healthy subjects, darunavir/ritonavir doses of 1.33 times the maximum recommended dose did not affect the QT/QTc interval.
12.3 Pharmacokinetics
Pharmacokinetics in Adults
General
Darunavir is primarily metabolized by CYP3A. Ritonavir inhibits CYP3A, thereby increasing the plasma concentrations of darunavir. When a single dose of darunavir 600 mg was given orally in combination with 100 mg ritonavir twice daily, there was an approximate 14-fold increase in the systemic exposure of darunavir. Therefore, darunavir should only be used in combination with 100 mg of ritonavir to achieve sufficient exposures of darunavir.
The pharmacokinetics of darunavir, co-administered with low dose ritonavir (100 mg), has been evaluated in healthy adult volunteers and in HIV-1-infected subjects. Table 11 displays the population pharmacokinetic estimates of darunavir after oral administration of darunavir/ritonavir 600/100 mg twice daily (based on sparse sampling in 285 patients in trial TMC114-C214, 278 patients in trial TMC114-C229 and 119 patients [integrated data] from trials TMC114-C202 and TMC114-C213) and darunavir/ritonavir 800/100 mg once daily (based on sparse sampling in 335 patients in trial TMC114-C211 and 280 patients in trial TMC114-C229) to HIV-1-infected patients.
Table 11: Population Pharmacokinetic Estimates of Darunavir at darunavir/ritonavir 800/100 mg Once Daily (Trial TMC114-C211, 48-Week Analysis and Trial TMC114-C229, 48-Week Analysis) and darunavir/ritonavir 600/100 mg Twice Daily (Trial TMC114-C214, 48-Week Analysis, Trial TMC114-C229, 48-Week Analysis and Integrated Data from Trials TMC114-C213 and TMC114-C202, Primary 24-Week Analysis)
darunavir/ritonavir
800/100 mg once daily
darunavir/ritonavir
600/100 mg twice daily
Parameter
TMC114-C211
N=335
TMC114-C229
N=280
TMC114-C214
N=285
TMC114-C229 N=278
TMC114-C213 + TMC114-C202 (integrated data)
N=119
AUC24h (ng•h/mL)a
  Mean ± Standard Deviation
93,026 ± 27,050
93,334 ± 28,626
116,796 ± 33,594
114,302 ± 32,681
124,698 ± 32,286
  Median (Range)
87,854 (45,000-219,240)
87,788 (45,456-236,920)
111,632 (64,874-355,360)
109,401 (48,934-323,820)
123,336 (67,714-212,980)
C0h (ng/mL)
  Mean ± Standard Deviation
2,282 ± 1,168
2,160 ± 1,201
3,490 ± 1,401
3,386 ± 1,372
3,578 ± 1,151
  Median (Range)
2,041 (368-7,242)
1,896 (184-7,881)
3,307 (1,517-13,198)
3,197 (250-11,865)
3,539 (1,255-7,368)
N=number of subjects with data a AUC24h is calculated as AUC12h*2.
Absorption and Bioavailability
Darunavir, co-administered with 100 mg ritonavir twice daily, was absorbed following oral administration with a Tmax of approximately 2.5-4 hours. The absolute oral bioavailability of a single 600 mg dose of darunavir alone and after co-administration with 100 mg ritonavir twice daily was 37% and 82%, respectively. In vivo data suggest that darunavir/ritonavir is an inhibitor of the P-glycoprotein (P-gp) transporters.
Effects of Food on Oral Absorption
When darunavir tablets were administered with food, the Cmax and AUC of darunavir, co-administered with ritonavir, is approximately 40% higher relative to the fasting state. Within the range of meals studied, darunavir exposure is similar. The total caloric content of the various meals evaluated ranged from 240 Kcal (12 gms fat) to 928 Kcal (56 gms fat).
Distribution
Darunavir is approximately 95% bound to plasma proteins. Darunavir binds primarily to plasma alpha 1-acid glycoprotein (AAG).
Metabolism
In vitro experiments with human liver microsomes (HLMs) indicate that darunavir primarily undergoes oxidative metabolism. Darunavir is extensively metabolized by CYP enzymes, primarily by CYP3A. A mass balance study in healthy volunteers showed that after a single dose administration of 400 mg 14C-darunavir, co-administered with 100 mg ritonavir, the majority of the radioactivity in the plasma was due to darunavir. At least 3 oxidative metabolites of darunavir have been identified in humans; all showed activity that was at least 90% less than the activity of darunavir against wild-type HIV-1.
Elimination
A mass balance study in healthy volunteers showed that after single dose administration of 400 mg 14C-darunavir, co-administered with 100 mg ritonavir, approximately 79.5% and 13.9% of the administered dose of 14C-darunavir was recovered in the feces and urine, respectively. Unchanged darunavir accounted for approximately 41.2% and 7.7% of the administered dose in feces and urine, respectively. The terminal elimination half-life of darunavir was approximately 15 hours when co-administered with ritonavir. After intravenous administration, the clearance of darunavir, administered alone and co-administered with 100 mg twice daily ritonavir, was 32.8 L/h and 5.9 L/h, respectively.
Special Populations
Hepatic Impairment
Darunavir is primarily metabolized by the liver. The steady-state pharmacokinetic parameters of darunavir were similar after multiple dose co-administration of darunavir/ritonavir 600/100 mg twice daily to subjects with normal hepatic function (n=16), mild hepatic impairment (Child-Pugh Class A, n=8), and moderate hepatic impairment (Child-Pugh Class B, n=8). The effect of severe hepatic impairment on the pharmacokinetics of darunavir has not been evaluated [see Dosage and Administration (2.6) and Use in Specific Populations (8.6)].
Hepatitis B or Hepatitis C Virus Co-infection
The 48-week analysis of the data from Studies TMC114-C211 and TMC114-C214 in HIV-1-infected subjects indicated that hepatitis B and/or hepatitis C virus co-infection status had no apparent effect on the exposure of darunavir.
Renal Impairment
Results from a mass balance study with 14C-darunavir/ritonavir showed that approximately 7.7% of the administered dose of darunavir is excreted in the urine as unchanged drug. As darunavir and ritonavir are highly bound to plasma proteins, it is unlikely that they will be significantly removed by hemodialysis or peritoneal dialysis. Population pharmacokinetic analysis showed that the pharmacokinetics of darunavir were not significantly affected in HIV-1-infected subjects with moderate renal impairment (CrCL between 30-60 mL/min, n=20). There are no pharmacokinetic data available in HIV-1-infected patients with severe renal impairment or end stage renal disease [see Use in Specific Populations (8.7)].
Gender
Population pharmacokinetic analysis showed higher mean darunavir exposure in HIV-1-infected females compared to males. This difference is not clinically relevant.
Race
Population pharmacokinetic analysis of darunavir in HIV-1-infected subjects indicated that race had no apparent effect on the exposure to darunavir.
Geriatric Patients
Population pharmacokinetic analysis in HIV-1-infected subjects showed that darunavir pharmacokinetics are not considerably different in the age range (18 to 75 years) evaluated in HIV-1-infected subjects (n=12, age greater than or equal to 65) [see Use in Specific Populations (8.5)].
Pediatric Patients
Darunavir/ritonavir administered twice daily
The pharmacokinetics of darunavir in combination with ritonavir in 93 antiretroviral treatment-experienced HIV-1-infected pediatric subjects 3 to less than 18 years of age and weighing at least 10 kg showed that the administered weight-based dosages resulted in similar darunavir exposure when compared to the darunavir exposure achieved in treatment-experienced adults receiving darunavir/ritonavir 600/100 mg twice daily [see Dosage and Administration (2.5)].
Darunavir/ritonavir administered once daily
The pharmacokinetics of darunavir in combination with ritonavir in 12 antiretroviral treatment-naĂŻve HIV-1-infected pediatric subjects 12 to less than 18 years of age and weighing at least 40 kg receiving darunavir/ritonavir 800/100 mg once daily resulted in similar darunavir exposures when compared to the darunavir exposure achieved in treatment-naĂŻve adults receiving darunavir/ritonavir 800/100 mg once daily [see Dosage and Administration (2.5)].
Based on population pharmacokinetic modeling and simulation, the proposed darunavir/ritonavir once daily dosing regimens for pediatric patients 3 to less than 12 years of age is predicted to result in similar darunavir exposures when compared to the darunavir exposures achieved in treatment-naĂŻve adults receiving darunavir/ritonavir 800/100 mg once daily [see Dosage and Administration (2.5)].
The population pharmacokinetic parameters in pediatric subjects with darunavir/ritonavir administered once or twice daily are summarized in the table below:
Table 12: Population Pharmacokinetic Estimates of Darunavir Exposure (Trials TMC114-C230, TMC114-C212 and TMC114-C228) Following Administration of Doses in Tables 2 and 3
darunavir/ritonavir once daily
darunavir/ritonavir
twice daily
Parameter
TMC114-C230a
N=12
TMC114-C212
N=74
TMC114-C228c
10 to less than 15 kgb
N=10
15 to less than 20 kgd
N=13
AUC24h (ng•h/mL)e
  Mean ± Standard Deviation
84,390 ± 23,587
126,377 ± 34,356
137,896 ± 51,420
157,760 ± 54,080
  Median (Range)
86,741
(35,527-123,325)
127,340
(67,054-230,720)
124,044
(89,688-261,090)
132,698
(112,310-294,840)
C0h (ng/mL)
  Mean ± Standard Deviation
2,141 ± 865
3,948 ± 1,363
4,510 ± 2,031
4,848 ± 2,143
  Median (Range)
2,234
(542-3,776)
3,888
(1,836-7,821)
4,126
(2,456-9,361)
3,927
(3,046-10,292)
N=number of subjects with data. a Summary statistics for population pharmacokinetic parameter estimates for DRV after administration of DRV/rtv at 800/100 mg once daily in treatment-naïve HIV-1 infected subjects from 12 to <18 years of age – Week-48 Analyses. b Calculated from individual pharmacokinetic parameters estimated for Week 2 and Week 4, based on the Week 48 analysis that   evaluated a darunavir dose of 20 mg/kg twice daily with ritonavir 3 mg/kg twice daily. c Subjects may have contributed pharmacokinetic data to both the 10 kg to less than 15 kg weight group and the 15 kg to less than 20 kg weight group. d The 15 kg to less than 20 kg weight group received 380 mg (3.8 mL) darunavir oral suspension twice daily with 48 mg (0.6 mL) ritonavir oral solution twice daily in TMC114-C228. Calculated from individual pharmacokinetic parameters estimated for Week 2 post-dose adjustment visit; Week 24 and Week 48 based on the – Week 48 analysis that evaluated a darunavir dose of 380 mg twice daily. e AUC24h is calculated as AUC12h*2.
Pregnancy and Postpartum
The exposure to total darunavir and ritonavir after intake of darunavir/ritonavir 600/100 mg twice daily and darunavir/ritonavir 800/100 mg once daily as part of an antiretroviral regimen was generally lower during pregnancy compared with postpartum (see Table 13, Table 14 and Figure 1).
Table 13: Pharmacokinetic Results of Total Darunavir After Administration of darunavir/ritonavir at 600/100 mg Twice Daily as Part of an Antiretroviral Regimen, During the 2nd Trimester of Pregnancy, the 3rd Trimester of Pregnancy and Postpartum
Pharmacokinetics of total darunavir
(mean ± standard deviation)
2nd Trimester of pregnancy
(n=12)a
3rd Trimester of pregnancy
(n=12)
Postpartum
(6-12 Weeks)
(n=12)
Cmax, ng/mL
4,668 ± 1,097
5,328 ± 1,631
6,659 ± 2,364
AUC24h, ng.h/mLb
78,740 ± 19,194
91,760 ± 34,720
113,780 ± 52,680
Cmin, ng/mL
1,922 ± 825
2,661 ± 1,269
2,851 ± 2,216
a n=11 for AUC24h b AUC24h is calculated as AUC12h*2.
Table 14: Pharmacokinetic Results of Total Darunavir After Administration of darunavir/ritonavir at 800/100 mg Once Daily as Part of an Antiretroviral Regimen, During the 2nd Trimester of Pregnancy, the 3rd Trimester of Pregnancy and Postpartum
Pharmacokinetics of total darunavir
(mean ± standard deviation)
2nd Trimester of pregnancy
(n=17)
3rd Trimester of pregnancy
(n=15)
Postpartum
(6-12 Weeks)
(n=16)
Cmax, ng/mL
4,964 ± 1,505
5,132 ± 1,198
7,310 ± 1,704
AUC24h, ng.h/mL
62,289 ± 16,234
61,112 ± 13,790
92,116 ± 29,241
Cmin, ng/mL
1,248 ± 542
1,075 ± 594
1,473 ± 1,141
Due to an increase in the unbound fraction of darunavir during pregnancy compared to postpartum, unbound darunavir exposures were less reduced during pregnancy as compared to postpartum. Exposure reductions during pregnancy were greater for the once daily regimen as compared to the twice daily regimen (see Figure 1).
Figure 1: Pharmacokinetic Results (Within-Subject Comparison) of Total and Unbound Darunavir After Administration of darunavir/ritonavir at 600/100 mg Twice Daily or 800/100 mg Once Daily as Part of an Antiretroviral Regimen, During the 2nd and 3rd Trimester of Pregnancy Compared to Postpartum
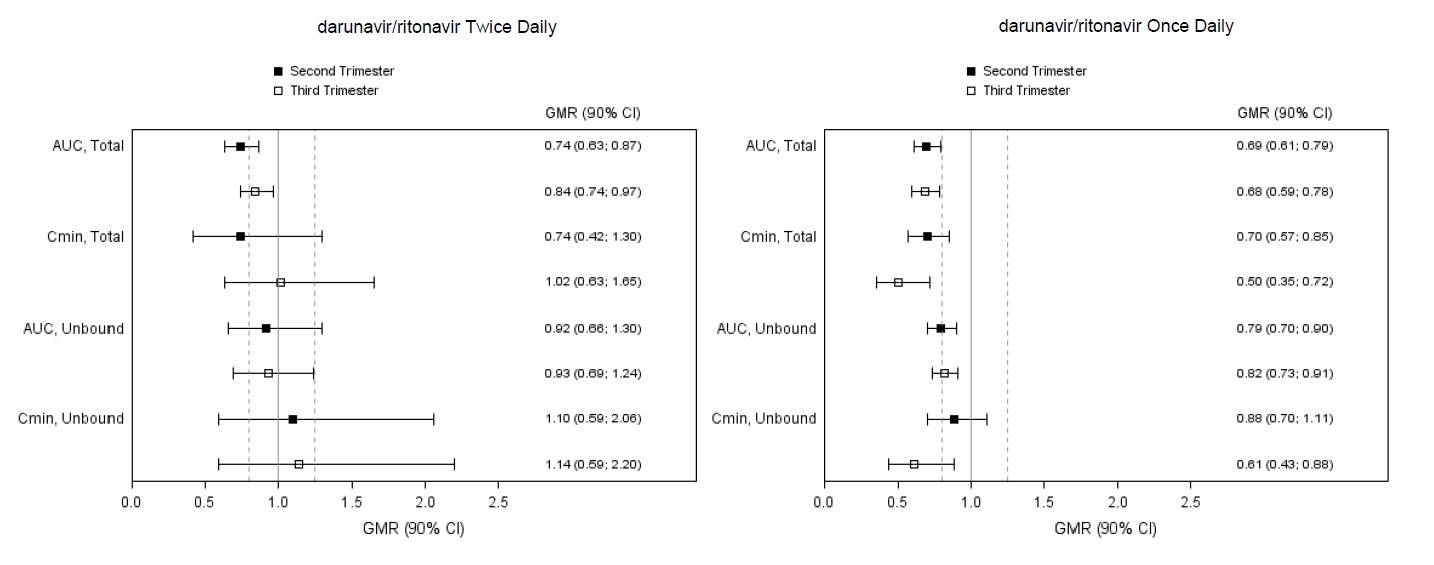
Legend: 90% CI: 90% confidence interval; GMR: geometric mean ratio. Solid vertical line: ratio of 1.0; dotted vertical lines: reference lines of 0.8 and 1.25.
Drug Interactions
[See also Contraindications (4), Warnings and Precautions (5.5) and Drug Interactions (7).]
Darunavir co-administered with ritonavir is an inhibitor of CYP3A, CYP2D6, and P-gp. Co-administration of darunavir and ritonavir with drugs primarily metabolized by CYP3A and CYP2D6, or are transported by P-gp, may result in increased plasma concentrations of such drugs, which could increase or prolong their therapeutic effect and adverse events.
Darunavir and ritonavir are metabolized by CYP3A. In vitro data indicate that darunavir may be a P-gp substrate. Drugs that induce CYP3A activity would be expected to increase the clearance of darunavir and ritonavir, resulting in lowered plasma concentrations of darunavir and ritonavir. Co-administration of darunavir and ritonavir and other drugs that inhibit CYP3A or P-gp may decrease the clearance of darunavir and ritonavir and may result in increased plasma concentrations of darunavir and ritonavir.
Drug interaction studies were performed with darunavir and other drugs likely to be co-administered and some drugs commonly used as probes for pharmacokinetic interactions. The effects of co-administration of darunavir on the AUC, Cmax, and Cmin values are summarized in Table 15 (effect of other drugs on darunavir) and Table 16 (effect of darunavir on other drugs). For information regarding clinical recommendations, see Drug Interactions (7).
Several interaction studies have been performed with a dose other than the recommended dose of the co-administered drug or darunavir; however, the results are applicable to the recommended dose of the co-administered drug and/or darunavir.
Table 15: Drug Interactions: Pharmacokinetic Parameters for Darunavir in the Presence of Co-Administered Drugs
Co-administered
drug
Dose/Schedule
LS Mean ratio (90% CI) of darunavir
Pharmacokinetic parameters with/without co-administered drug
no effect =1.00
Co-administered Drug
Darunavir/
ritonavir
N
PK
Cmax
AUC
Cmin
Co-administration with other HIV protease inhibitors
Atazanavir
300 mg q.d.a
400/100 mg b.i.d.b
13
↔
1.02
(0.96-1.09)
1.03
(0.94-1.12)
1.01
(0.88-1.16)
Indinavir
800 mg b.i.d.
400/100 mg b.i.d.
9
↑
1.11
(0.98-1.26)
1.24
(1.09-1.42)
1.44
(1.13-1.82)
Lopinavir/ritonavir
400/100 mg b.i.d.
1200/100 mg b.i.d.c
14
↓
0.79
(0.67-0.92)
0.62
(0.53-0.73)
0.49
(0.39-0.63)
533/133.3 mg b.i.d.
1200 mg b.i.d.c
15
↓
0.79
(0.64-0.97)
0.59
(0.50-0.70)
0.45
(0.38-0.52)
Saquinavir hard gel capsule
1000 mg b.i.d.
400/100 mg b.i.d.
14
↓
0.83
(0.75-0.92)
0.74
(0.63-0.86)
0.58
(0.47-0.72)
Co-administration with other HIV antiretrovirals
Didanosine
400 mg q.d.
600/100 mg b.i.d.
17
↔
0.93
(0.86-1.00)
1.01
(0.95-1.07)
1.07
(0.95-1.21)
Efavirenz
600 mg q.d.
300/100 mg b.i.d.
12
↓
0.85
(0.72-1.00)
0.87
(0.75-1.01)
0.69
(0.54-0.87)
Etravirine
200 mg b.i.d.
600/100 mg b.i.d.
15
↔
1.11
(1.01-1.22)
1.15
(1.05-1.26)
1.02
(0.90-1.17)
Nevirapine
200 mg b.i.d.
400/100 mg b.i.d.
8
↑
1.40d
(1.14-1.73)
1.24d
(0.97-1.57)
1.02d
(0.79-1.32)
Rilpivirine
150 mg q.d.
800/100 mg q.d.
15
↔
0.90
(0.81-1.00)
0.89
(0.81-0.99)
0.89
(0.68-1.16)
Tenofovir
disoproxil
fumarate
300 mg q.d.
300/100 mg b.i.d.
12
↑
1.16
(0.94-1.42)
1.21
(0.95-1.54)
1.24
(0.90-1.69)
Co-administration with other drugs
Artemether/lumefantrine
80/480 mg (6 doses at 0, 8, 24, 36, 48, and 60 hours)
600/100 mg b.i.d.
14
↔
1.00
(0.93-1.07)
0.96
(0.90-1.03)
0.87
(0.77-0.98)
Carbamazepine
200 mg b.i.d.
600/100 mg b.i.d.
16
↔
1.04
(0.93-1.16)
0.99
(0.90-1.08)
0.85
(0.73-1.00)
Clarithromycin
500 mg b.i.d.
400/100 mg b.i.d.
17
↔
0.83
(0.72-0.96)
0.87
(0.75-1.01)
1.01
(0.81-1.26)
Ketoconazole
200 mg b.i.d.
400/100 mg b.i.d.
14
↑
1.21
(1.04-1.40)
1.42
(1.23-1.65)
1.73
(1.39-2.14)
Omeprazole
20 mg q.d.
400/100 mg b.i.d.
16
↔
1.02
(0.95-1.09)
1.04
(0.96-1.13)
1.08
(0.93-1.25)
Paroxetine
20 mg q.d.
400/100 mg b.i.d.
16
↔
0.97
(0.92-1.02)
1.02
(0.95-1.10)
1.07
(0.96-1.19)
Pitavastatin
4 mg q.d.
800/100 mg q.d.
27
↔
1.06 (1.00-1.12)
1.03 (0.95-1.12)
NA
Ranitidine
150 mg b.i.d.
400/100 mg b.i.d.
16
↔
0.96
(0.89-1.05)
0.95
(0.90-1.01)
0.94
(0.90-0.99)
Rifabutin
150 mg q.o.d.e
600/100 mg b.i.d.
11
↑
1.42
(1.21-1.67)
1.57
(1.28-1.93)
1.75
(1.28-2.37)
Sertraline
50 mg q.d.
400/100 mg b.i.d.
13
↔
1.01
(0.89-1.14)
0.98
(0.84-1.14)
0.94
(0.76-1.16)
N = number of subjects with data a  q.d. = once daily b b.i.d. = twice daily c The pharmacokinetic parameters of darunavir in this study were compared with the pharmacokinetic parameters following administration of darunavir/ritonavir 600/100 mg twice daily. d Ratio based on between-study comparison. e q.o.d. = every other day
Table 16: Drug Interactions: Pharmacokinetic Parameters for Co-Administered Drugs in the Presence of Darunavir/ritonavir
Co-administered
drug
Dose/Schedule
N
PK
LS Mean ratio (90% CI) of
co-administered drug pharmacokinetic parameters
with/without darunavir
no effect=1.00
Co-administered drug
Darunavir/
ritonavir
Cmax
AUC
Cmin
Co-administration with other HIV protease inhibitors
Atazanavir
300 mg q.d.a/100 mg ritonavir q.d. when administered alone
400/100 mg b.i.d.b
13
↔
0.89
(0.78-1.01)
1.08
(0.94-1.24)
1.52
(0.99-2.34)
300 mg q.d. when administered with darunavir/ritonavir
Indinavir
800 mg b.i.d./100 mg ritonavir b.i.d. when administered alone
400/100 mg b.i.d.
9
↑
1.08
(0.95-1.22)
1.23
(1.06-1.42)
2.25
(1.63-3.10)
800 mg b.i.d. when administered with darunavir/ritonavir
Lopinavir/ritonavir
400/100 mg b.i.d.c
1,200/100 mg b.i.d.
14
↔
0.98
(0.78-1.22)
1.09
(0.86-1.37)
1.23
(0.90-1.69)
533/133.3 mg b.i.d.c
1,200 mg b.i.d.
15
↔
1.11
(0.96-1.30)
1.09
(0.96-1.24)
1.13
(0.90-1.42)
Saquinavir hard gel capsule
1,000 mg b.i.d./100 mg ritonavir b.i.d. when administered alone
400/100 mg b.i.d.
12
↔
0.94
(0.78-1.13)
0.94
(0.76-1.17)
0.82
(0.52-1.30)
1,000 mg b.i.d. when administered with darunavir/ritonavir
Co-administration with other HIV antiretrovirals
Didanosine
400 mg q.d.
600/100 mg b.i.d.
17
↔
0.84
(0.59-1.20)
0.91
(0.75-1.10)
-
Dolutegravir
30 mg q.d.
600/100 mg b.i.d.
15
↓
0.89
(0.83-0.97)
0.78
(0.72-0.85)
0.62d
(0.56-0.69)
Dolutegravir
50 mg q.d.
600/100 mg b.i.d. with 200 mg b.i.d. etravirine
9
↓
0.88
(0.78-1.00)
0.75
(0.69-0.81)
0.63d
(0.52-0.76)
Efavirenz
600 mg q.d.
300/100 mg b.i.d.
12
↑
1.15
(0.97-1.35)
1.21
(1.08-1.36)
1.17
(1.01-1.36)
Etravirine
100 mg b.i.d.
600/100 mg b.i.d.
14
↓
0.68
(0.57-0.82)
0.63
(0.54-0.73)
0.51
(0.44-0.61)
Nevirapine
200 mg b.i.d.
400/100 mg b.i.d.
8
↑
1.18
(1.02-1.37)
1.27
(1.12-1.44)
1.47
(1.20-1.82)
Rilpivirine
150 mg q.d.
800/100 mg q.d.
14
↑
1.79
(1.56-2.06)
2.30
(1.98-2.67)
2.78
(2.39-3.24)
Tenofovir
disoproxil
fumarate
300 mg q.d.
300/100 mg b.i.d.
12
↑
1.24
(1.08-1.42)
1.22
(1.10-1.35)
1.37
(1.19-1.57)
Maraviroc
150 mg b.i.d.
600/100 mg b.i.d.
12
↑
2.29
(1.46-3.59)
4.05
(2.94-5.59)
8.00
(6.35-10.1)
600/100 mg b.i.d. with 200 mg b.i.d. etravirine
10
↑
1.77
(1.20-2.60)
3.10
(2.57-3.74)
5.27
(4.51-6.15)
Co-administration with other drugs
Atorvastatin
40 mg q.d. when administered alone
10 mg q.d. when administered with darunavir/ritonavir
300/100 mg b.i.d.
15
↑
0.56
(0.48-0.67)
0.85
(0.76-0.97)
1.81
(1.37-2.40)
Artemether
80 mg
single dose
600/100 mg b.i.d.
15
↓
0.85
(0.68-1.05)
0.91
(0.78-1.06)
-
Dihydroartemisinin
15
↑
1.06
(0.82-1.39)
1.12
(0.96-1.30)
-
Artemether
artemether/lumefantrine 80/480 mg (6 doses at 0, 8, 24, 36, 48, and 60 hours)
600/100 mg b.i.d.
15
↓
0.82
(0.61-1.11)
0.84
(0.69-1.02)
0.97
(0.90-1.05)
Dihydroartemisinin
15
↓
0.82
(0.66-1.01)
0.82
(0.74-0.91)
1.00
(0.82-1.22)
Lumefantrine
15
↑
1.65
(1.49-1.83)
2.75
(2.46-3.08)
2.26
(1.92-2.67)
Buprenorphine/Naloxone
8/2 mg to 16 mg/4 mg q.d.
600/100 mg b.i.d.
17
↔
0.92e
(0.79-1.08)
0.89e
(0.78-1.02)
0.98e
(0.82-1.16)
Norbuprenorphine
17
↑
1.36
(1.06-1.74)
1.46
(1.15-1.85)
1.71
(1.29-2.27)
Carbamazepine
200 mg b.i.d.
600/100 mg b.i.d.
16
↑
1.43
(1.34-1.53)
1.45
(1.35-1.57)
1.54
(1.41-1.68)
Carbamazepine epoxide
16
↓
0.46
(0.43-0.49)
0.46
(0.44-0.49)
0.48
(0.45-0.51)
Clarithromycin
500 mg b.i.d.
400/100 mg b.i.d.
17
↑
1.26
(1.03-1.54)
1.57
(1.35-1.84)
2.74
(2.30-3.26)
 Dabigatran etexilate  150 mg  800/100 mg single dose 800/100 mg q.d.f
14
13Â
 ↑
↑
 1.64 (1.21-2.23) 1.22 (0.89-1.67)
1.72 (1.33-2.23) 1.18
(0.90-1.53)Â
 - -
Dextromethorphan
30 mg
600/100 mg b.i.d.
12
↑
2.27
(1.59-3.26)
2.70
(1.80-4.05)
-
Dextrorphan
↓
0.87
(0.77-0.98)
0.96
(0.90-1.03)
-
Digoxin
0.4 mg
600/100 mg b.i.d.
8
↑
1.15
(0.89-1.48)
1.36
(0.81-2.27)
-
Ethinyl estradiol
(EE)
Ortho-Novum 1/35
(35 mcg EE/1 mg NE)
600/100 mg b.i.d.
11
↓
0.68
(0.61-0.74)
0.56
(0.50-0.63)
0.38
(0.27-0.54)
Norethindrone (NE)
11
↓
0.90
(0.83-0.97)
0.86
(0.75-0.98)
0.70
(0.51-0.97)
Ketoconazole
200 mg b.i.d.
400/100 mg b.i.d.
15
↑
2.11
(1.81-2.44)
3.12
(2.65-3.68)
9.68
(6.44-14.55)
R-Methadone
55-150 mg q.d.
600/100 mg b.i.d.
16
↓
0.76
(0.71-0.81)
0.84
(0.78-0.91)
0.85
(0.77-0.94)
Omeprazole
40 mg single dose
600/100 mg b.i.d.
12
↓
0.66
(0.48-0.90)
0.58
(0.50-0.66)
-
5-hydroxy
omeprazole
↓
0.93
(0.71-1.21)
0.84
(0.77-0.92)
-
Paroxetine
20 mg q.d.
400/100 mg b.i.d.
16
↓
0.64
(0.59-0.71)
0.61
(0.56-0.66)
0.63
(0.55-0.73)
Pitavastatin
4 mg q.d.
800/100 mg q.d.
27
↓
0.96
(0.84-1.09)
0.74
(0.69-0.80)
NA
Pravastatin
40 mg single dose
600/100 mg b.i.d.
14
↑
1.63
(0.95-2.82)
1.81
(1.23-2.66)
-
Rifabutin
150 mg q.o.d.g
when administered with darunavir/ritonavir
600/100 mg b.i.d.h
11
↑
0.72
(0.55-0.93)
0.93
(0.80-1.09)
1.64
(1.48-1.81)
25-O-desacetyl-rifabutin
300 mg q.d. when administered alone
11
↑
4.77
(4.04-5.63)
9.81
(8.09-11.9)
27.1
(22.2-33.2)
Sertraline
50 mg q.d.
400/100 mg b.i.d.
13
↓
0.56
(0.49-0.63)
0.51
(0.46-0.58)
0.51
(0.45-0.57)
Sildenafil
100 mg (single dose) administered alone
25 mg (single dose) when administered with darunavir/ritonavir
400/100 mg b.i.d.
16
↑
0.62
(0.55-0.70)
0.97
(0.86-1.09)
-
S-warfarin
10 mg single dose
600/100 mg b.i.d.
12
↓
0.92
(0.86-0.97)
0.79
(0.73-0.85)
-
7-OH-S-warfarin
12
↑
1.42
(1.24-1.63)
1.23
(0.97-1.57)
-
N = number of subjects with data; - = no information available a q.d. = once daily b b.i.d. = twice daily c The pharmacokinetic parameters of lopinavir in this study were compared with the pharmacokinetic parameters following administration of lopinavir/ritonavir 400/100 mg twice daily. d Noted as Cτ or C24 in the dolutegravir U.S. prescribing information e Ratio is for buprenorphine; mean Cmax and AUC24 for naloxone were comparable when buprenorphine/naloxone was administered with or without darunavir/ritonavir f 800/100 mg q.d. for 14 days before co-administered with dabigatran etexilate. g q.o.d. = every other day h In comparison to rifabutin 300 mg once daily.
12.4 Microbiology
Mechanism of Action
Darunavir is an inhibitor of the HIV-1 protease. It selectively inhibits the cleavage of HIV-1 encoded Gag-Pol polyproteins in infected cells, thereby preventing the formation of mature virus particles.
Antiviral Activity
Darunavir exhibits activity against laboratory strains and clinical isolates of HIV-1 and laboratory strains of HIV-2 in acutely infected T-cell lines, human peripheral blood mononuclear cells and human monocytes/macrophages with median EC50 values ranging from 1.2 to 8.5 nM (0.7 to 5.0 ng/mL). Darunavir demonstrates antiviral activity in cell culture against a broad panel of HIV-1 group M (A, B, C, D, E, F, G), and group O primary isolates with EC50 values ranging from less than 0.1 to 4.3 nM. The EC50 value of darunavir increases by a median factor of 5.4 in the presence of human serum. Darunavir did not show antagonism when studied in combination with the PIs amprenavir, atazanavir, indinavir, lopinavir, nelfinavir, ritonavir, saquinavir, or tipranavir, the N(t)RTIs abacavir, didanosine, emtricitabine, lamivudine, stavudine, tenofovir, zalcitabine, or zidovudine, the NNRTIs delavirdine, rilpivirine, efavirenz, etravirine, or nevirapine, and the fusion inhibitor enfuvirtide.
Resistance
Cell Culture: HIV-1 isolates with a decreased susceptibility to darunavir have been selected in cell culture and obtained from subjects treated with darunavir/ritonavir. Darunavir-resistant virus derived in cell culture from wild-type HIV-1 had 21- to 88-fold decreased susceptibility to darunavir and developed 2 to 4 of the following amino acid substitutions S37D, R41E/T, K55Q, H69Q, K70E, T74S, V77I, or I85V in the protease. Selection in cell culture of darunavir resistant HIV-1 from nine HIV-1 strains harboring multiple PI resistance-associated mutations resulted in the overall emergence of 22 mutations in the protease gene, coding for amino acid substitutions L10F, V11I, I13V, I15V, G16E, L23I, V32I, L33F, S37N, M46I, I47V, I50V, F53L, L63P, A71V, G73S, L76V, V82I, I84V, T91A/S, and Q92R, of which L10F, V32I, L33F, S37N, M46I, I47V, I50V, L63P, A71V, and I84V were the most prevalent. These darunavir-resistant viruses had at least eight protease substitutions and exhibited 50- to 641-fold decreases in darunavir susceptibility with final EC50 values ranging from 125 nM to 3461 nM.
Clinical trials of darunavir/ritonavir in treatment-experienced subjects: In a pooled analysis of the 600/100 mg darunavir/ritonavir twice daily arms of trials TMC114-C213, TMC114-C202, TMC114-C215, and the control arms of etravirine trials TMC125-C206 and TMC125-C216, the amino acid substitutions V32I and I54L or M developed most frequently on darunavir/ritonavir in 41% and 25%, respectively, of the treatment-experienced subjects who experienced virologic failure, either by rebound or by never being suppressed (less than 50 copies/mL). Other substitutions that developed frequently in darunavir/ritonavir virologic failure isolates occurred at amino acid positions V11I, I15V, L33F, I47V, I50V, and L89V. These amino acid substitutions were associated with decreased susceptibility to darunavir; 90% of the virologic failure isolates had a greater than 7-fold decrease in susceptibility to darunavir at failure. The median darunavir phenotype (fold change from reference) of the virologic failure isolates was 4.3-fold at baseline and 85-fold at failure. Amino acid substitutions were also observed in the protease cleavage sites in the Gag polyprotein of some darunavir/ritonavir virologic failure isolates. In trial TMC114-C212 of treatment-experienced pediatric subjects, the amino acid substitutions V32I, I54L and L89M developed most frequently in virologic failures on darunavir/ritonavir.
In the 96-week as-treated analysis of the Phase 3 trial TMC114-C214, the percent of virologic failures (never suppressed, rebounders and discontinued before achieving suppression) was 21% (62/298) in the group of subjects receiving darunavir/ritonavir 600/100 mg twice daily compared to 32% (96/297) of subjects receiving lopinavir/ritonavir 400/100 mg twice daily. Examination of subjects who failed on darunavir/ritonavir 600/100 mg twice daily and had post-baseline genotypes and phenotypes showed that 7 subjects (7/43; 16%) developed PI substitutions on darunavir/ritonavir treatment resulting in decreased susceptibility to darunavir. Six of the 7 had baseline PI resistance-associated substitutions and baseline darunavir phenotypes greater than 7. The most common emerging PI substitutions in these virologic failures were V32I, L33F, M46I or L, I47V, I54L, T74P and L76V. These amino acid substitutions were associated with 59- to 839-fold decreased susceptibility to darunavir at failure. Examination of individual subjects who failed in the comparator arm on lopinavir/ritonavir and had post-baseline genotypes and phenotypes showed that 31 subjects (31/75; 41%) developed substitutions on lopinavir treatment resulting in decreased susceptibility to lopinavir (greater than 10-fold) and the most common substitutions emerging on treatment were L10I or F, M46I or L, I47V or A, I54V and L76V. Of the 31 lopinavir/ritonavir virologic failure subjects, 14 had reduced susceptibility (greater than 10-fold) to lopinavir at baseline.
In the 48-week analysis of the Phase 3 trial TMC114-C229, the number of virologic failures (including those who discontinued before suppression after Week 4) was 26% (75/294) in the group of subjects receiving darunavir/ritonavir 800/100 mg once daily compared to 19% (56/296) of subjects receiving darunavir/ritonavir 600/100 mg twice daily. Examination of isolates from subjects who failed on darunavir/ritonavir 800/100 mg once daily and had post-baseline genotypes showed that 8 subjects (8/60; 13%) had isolates that developed IAS-USA defined PI resistance-associated substitutions compared to 5 subjects (5/39; 13%) on darunavir/ritonavir 600/100 mg twice daily. Isolates from 2 subjects developed PI resistance associated substitutions associated with decreased susceptibility to darunavir; 1 subject isolate in the darunavir/ritonavir 800/100 mg once daily arm, developed substitutions V32I, M46I, L76V and I84V associated with a 24-fold decreased susceptibility to darunavir, and 1 subject isolate in the darunavir/ritonavir 600/100 mg twice daily arm developed substitutions L33F and I50V associated with a 40-fold decreased susceptibility to darunavir. In the darunavir/ritonavir 800/100 mg once daily and darunavir/ritonavir 600/100 mg twice daily groups, isolates from 7 (7/60; 12%) and 4 (4/42; 10%) virologic failures, respectively, developed decreased susceptibility to an NRTI included in the treatment regimen.
Clinical trials of darunavir/ritonavir in treatment-naĂŻve subjects: In the 192-week as-treated analysis censoring those who discontinued before Week 4 of the Phase 3 trial TMC114-C211, the percentage of virologic failures (never suppressed, rebounders and discontinued before achieving suppression) was 22% (64/288) in the group of subjects receiving darunavir/ritonavir 800/100 mg once daily compared to 29% (76/263) of subjects receiving lopinavir/ritonavir 800/200 mg per day. In the darunavir/ritonavir arm, emergent PI resistance-associated substitutions were identified in 11 of the virologic failures with post-baseline genotypic data (n=43). However, none of the darunavir virologic failures had a decrease in darunavir susceptibility (greater than 7-fold change) at failure. In the comparator lopinavir/ritonavir arm, emergent PI resistance-associated substitutions were identified in 17 of the virologic failures with post-baseline genotypic data (n=53), but none of the lopinavir/ritonavir virologic failures had decreased susceptibility to lopinavir (greater than 10-fold change) at failure. The reverse transcriptase M184V substitution and/or resistance to emtricitabine, which was included in the fixed background regimen, was identified in 4 virologic failures from the darunavir/ritonavir arm and 7 virologic failures in the lopinavir/ritonavir arm.
Cross-resistance
Cross-resistance among PIs has been observed. Darunavir has a less than 10-fold decreased susceptibility in cell culture against 90% of 3,309 clinical isolates resistant to amprenavir, atazanavir, indinavir, lopinavir, nelfinavir, ritonavir, saquinavir and/or tipranavir showing that viruses resistant to these PIs remain susceptible to darunavir.
Darunavir-resistant viruses were not susceptible to amprenavir, atazanavir, indinavir, lopinavir, nelfinavir, ritonavir or saquinavir in cell culture. However, six of nine darunavir-resistant viruses selected in cell culture from PI-resistant viruses showed a fold change in EC50 values less than 3 for tipranavir, indicative of limited cross-resistance between darunavir and tipranavir. In trials TMC114-C213, TMC114-C202, and TMC114-C215, 34% (64/187) of subjects in the darunavir/ritonavir arm whose baseline isolates had decreased susceptibility to tipranavir (tipranavir fold change greater than 3) achieved less than 50 copies/mL serum HIV-1 RNA levels at Week 96. Of the viruses isolated from subjects experiencing virologic failure on darunavir/ritonavir 600/100 mg twice daily (greater than 7-fold change), 41% were still susceptible to tipranavir and 10% were susceptible to saquinavir while less than 2% were susceptible to the other protease inhibitors (amprenavir, atazanavir, indinavir, lopinavir or nelfinavir).
In trial TMC114-C214, the 7 darunavir/ritonavir virologic failures with reduced susceptibility to darunavir at failure were also resistant to the approved PIs (fos)amprenavir, atazanavir, lopinavir, indinavir, and nelfinavir at failure. Six of these 7 were resistant to saquinavir and 5 were resistant to tipranavir. Four of these virologic failures were already PI-resistant at baseline.
Cross-resistance between darunavir and nucleoside/nucleotide reverse transcriptase inhibitors, non-nucleoside reverse transcriptase inhibitors, fusion inhibitors, CCR5 co-receptor antagonists, or integrase inhibitors is unlikely because the viral targets are different.
Baseline Genotype/Phenotype and Virologic Outcome Analyses
Genotypic and/or phenotypic analysis of baseline virus may aid in determining darunavir susceptibility before initiation of darunavir/ritonavir 600/100 mg twice daily therapy. The effect of baseline genotype and phenotype on virologic response at 96 weeks was analyzed in as-treated analyses using pooled data from the Phase 2b trials (Trials TMC114-C213, TMC114-C202, and TMC114-C215) (n=439). The findings were confirmed with additional genotypic and phenotypic data from the control arms of etravirine trials TMC125-C206 and TMC125-C216 at Week 24 (n = 591).
Diminished virologic responses were observed in subjects with 5 or more baseline IAS-defined primary protease inhibitor resistance-associated substitutions (D30N, V32I, L33F, M46I/L, I47A/V, G48V, I50L/V, I54L/M, L76V, V82A/F/L/S/T, I84V, N88S, L90M) (see Table 17).
Table 17: Response to darunavir/ritonavir 600/100 mg Twice Daily by Baseline Number of IAS-Defined Primary PI Resistance-Associated Substitutions: As-treated Analysis of Trials TMC114-C213, TMC114-C202, and TMC114-C215
# IAS-defined primary PI
substitutions
Proportion of subjects with <50 copies/mL at Week 96
N=439
Overall
de novo ENF
Re-used/No ENF
All
44% (192/439)
54% (61/112)
40% (131/327)
0-4
50% (162/322)
58% (49/85)
48% (113/237)
5
22% (16/74)
47% (9/19)
13% (7/55)
≥ 6
9% (3/32)
17% (1/6)
8% (2/26)
ENF=enfuvirtide
IAS Primary PI Substitutions (2008): D30N, V32I, L33F, M46I/L, I47A/V, G48V, I50L/V, I54L/M, L76V, V82A/F/L/S/T, I84V, N88S, L90M.
The presence at baseline of two or more of the substitutions V11I, V32I, L33F, I47V, I50V, I54L or M, T74P, L76V, I84V or L89V was associated with a decreased virologic response to darunavir/ritonavir. In subjects not taking enfuvirtide de novo, the proportion of subjects achieving viral load less than 50 plasma HIV-1 RNA copies/mL at 96 weeks was 59%, 29%, and 12% when the baseline genotype had 0-1, 2 and greater than or equal to 3 of these substitutions, respectively.
Baseline darunavir phenotype (shift in susceptibility relative to reference) was shown to be a predictive factor of virologic outcome. Response rates assessed by baseline darunavir phenotype are shown in Table 18. These baseline phenotype groups are based on the select patient populations in the trials TMC114-C213, TMC114-C202, and TMC114-C215, and are not meant to represent definitive clinical susceptibility breakpoints for darunavir/ritonavir. The data are provided to give clinicians information on the likelihood of virologic success based on pre-treatment susceptibility to darunavir.
Table 18: Response (HIV-1 RNA <50 copies/mL at Week 96) to darunavir/ritonavir 600/100 mg Twice Daily by Baseline Darunavir Phenotype and by Use of Enfuvirtide: As-treated Analysis of Trials TMC114-C213, TMC114-C202, and TMC114-C215
Baseline DRV phenotype
Proportion of subjects with <50 copies/mL at Week 96
N=417
All
de novo ENF
Re-used/
No ENF
Overall
175/417 (42%)
61/112 (54%)
131/327 (40%)
0-7
148/270 (55%)
44/65 (68%)
104/205 (51%)
> 7-20
16/53 (30%)
7/17 (41%)
9/36 (25%)
> 20
11/94 (12%)
6/23 (26%)
5/71 (7%)
ENF=enfuvirtide
13 Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis and Mutagenesis
Darunavir was evaluated for carcinogenic potential by oral gavage administration to mice and rats up to 104 weeks. Daily doses of 150, 450 and 1,000 mg/kg were administered to mice and doses of 50, 150 and 500 mg/kg was administered to rats. A dose-related increase in the incidence of hepatocellular adenomas and carcinomas were observed in males and females of both species as well as an increase in thyroid follicular cell adenomas in male rats. The observed hepatocellular findings in rodents are considered to be of limited relevance to humans. Repeated administration of darunavir to rats caused hepatic microsomal enzyme induction and increased thyroid hormone elimination, which predispose rats, but not humans, to thyroid neoplasms. At the highest tested doses, the systemic exposures to darunavir (based on AUC) were between 0.4- and 0.7-fold (mice) and 0.7- and 1-fold (rats), relative to those observed in humans at the recommended therapeutic doses (600/100 mg twice daily or 800/100 mg once daily).
Darunavir was not mutagenic or genotoxic in a battery of in vitro and in vivo assays including bacterial reserve mutation (Ames), chromosomal aberration in human lymphocytes and in vivo micronucleus test in mice.
Impairment of Fertility
No effects on fertility or early embryonic development were observed with darunavir in rats.
14 Clinical Studies
14.1 Description of Adult Clinical Trials
The evidence of efficacy of darunavir/ritonavir is based on the analyses of 192-week data from a randomized, controlled open-label Phase 3 trial in treatment-naĂŻve (TMC114-C211) HIV-1-infected adult subjects and 96-week data from a randomized, controlled, open-label Phase 3 trial in antiretroviral treatment-experienced (TMC114-C214) HIV-1-infected adult subjects. In addition, 96-week data are included from 2 randomized, controlled Phase 2b trials, TMC114-C213 and TMC114-C202, in antiretroviral treatment-experienced HIV-1-infected adult subjects.
14.2 Treatment-Nave Adult Subjects
TMC114-C211
TMC114-C211 is a randomized, controlled, open-label Phase 3 trial comparing darunavir/ritonavir 800/100 mg once daily versus lopinavir/ritonavir 800/200 mg per day (given as a twice daily or as a once daily regimen) in antiretroviral treatment-naĂŻve HIV-1-infected adult subjects. Both arms used a fixed background regimen consisting of tenofovir disoproxil fumarate 300 mg once daily (TDF) and emtricitabine 200 mg once daily (FTC).
HIV-1-infected subjects who were eligible for this trial had plasma HIV-1 RNA greater than or equal to 5,000 copies/mL. Randomization was stratified by screening plasma viral load (HIV-1 RNA less than 100,000 copies/mL or greater than or equal to 100,000 copies/mL) and screening CD4+ cell count (less than 200 cells/mm3 or greater than or equal to 200 cells/mm3). Virologic response was defined as a confirmed plasma HIV-1 RNA viral load less than 50 copies/mL. Analyses included 689 subjects in trial TMC114-C211 who had completed 192 weeks of treatment or discontinued earlier.
Demographics and baseline characteristics were balanced between the darunavir/ritonavir arm and the lopinavir/ritonavir arm (see Table 19). Table 19Â compares the demographic and baseline characteristics between subjects in the darunavir/ritonavir 800/100 mg once daily arm and subjects in the lopinavir/ritonavir 800/200 mg per day arm in trial TMC114-C211.
Table 19: Demographic and Baseline Characteristics of Subjects in Trial TMC114-C211
darunavir/ritonavir
800/100 mg once daily
+ TDF/FTC
N=343
lopinavir/ritonavir
800/200 mg per day
+ TDF/FTC
N=346
Demographic characteristics
Median age (years) (range, years)
34
(18-70)
33
(19- 68)
Sex
  Male
70%
70%
  Female
30%
30%
Race
  White
40%
45%
  Black
23%
21%
  Hispanic
23%
22%
  Asian
13%
11%
Baseline characteristics
Mean baseline plasma HIV-1 RNA (log10 copies/mL)
4.86
4.84
Median baseline CD4+ cell count
(cells/mm3)
(range, cells/mm3)
228
(4-750)
218
(2-714)
Percentage of patients with baseline viral load ≥100,000 copies/mL
34%
35%
Percentage of patients with baseline CD4+ cell count <200 cells/mm3
41%
43%
FTC=emtricitabine; TDF=tenofovir disoproxil fumarate
Week 192 outcomes for subjects on darunavir/ritonavir 800/100 mg once daily from trial TMC114-C211 are shown in Table 20.
Table 20: Virologic Outcome of Randomized Treatment of Trial TMC114-C211 at 192 Weeks
darunavir/ritonavir
800/100 mg once daily
+ TDF/FTC
N=343
lopinavir/ritonavir
800/200 mg per day
+ TDF/FTC
N=346
Virologic success HIV-1 RNA <50 copies/mL
70%a
61%
Virologic failureb
12%
15%
No virologic data at Week 192 windowc
Reasons
Discontinued trial due to adverse event or deathd
5%
13%
Discontinued trial for other reasonse
13%
12%
Missing data during windowc but on trial
<1%
0%
N = total number of subjects with data; FTC=emtricitabine; TDF=tenofovir disoproxil fumarate a 95% CI: 1.9; 16.1 b Includes patients who discontinued prior to Week 192 for lack or loss of efficacy and patients who are ≥50 copies in the 192-week window and patients who had a change in their background regimen that was not permitted by the protocol. c Window 186-198 Weeks. d Includes patients who discontinued due to adverse event or death at any time point from Day 1 through the time window if this resulted in no virologic data on treatment during the specified window. e Other includes: withdrew consent, loss to follow-up, etc., if the viral load at the time of discontinuation was <50 copies/mL.
In trial TMC114-C211 at 192 weeks of treatment, the median increase from baseline in CD4+ cell counts was 258 cells/mm3 in the darunavir/ritonavir 800/100 mg once daily arm and 263 cells/mm3 in the lopinavir/ritonavir 800/200 mg per day arm. Of the darunavir/ritonavir subjects with a confirmed virologic response of <50 copies/mL at Week 48, 81% remained undetectable at Week 192 versus 68% with lopinavir/ritonavir. In the 192 week analysis, statistical superiority of the darunavir/ritonavir regimen over the lopinavir/ritonavir regimen was demonstrated for both ITT and OP populations.
14.3 Treatment-Experienced Adult Subjects
TMC114-C229
TMC114-C229 is a randomized, open-label trial comparing darunavir/ritonavir 800/100 mg once daily to darunavir/ritonavir 600/100 mg twice daily in treatment-experienced HIV-1-infected patients with screening genotype resistance test showing no darunavir resistance associated substitutions (i.e. V11I, V32I, L33F, I47V, I50V, I54L, I54M, T74P, L76V, I84V, L89V) and a screening viral load of greater than 1,000 HIV-1 RNA copies/mL. Both arms used an optimized background regimen consisting of greater than or equal to 2 NRTIs selected by the investigator.
HIV-1-infected subjects who were eligible for this trial were on a highly active antiretroviral therapy regimen (HAART) for at least 12 weeks. Virologic response was defined as a confirmed plasma HIV-1 RNA viral load less than 50 copies/mL. Analyses included 590 subjects who had completed 48 weeks of treatment or discontinued earlier.
Table 21Â compares the demographic and baseline characteristics between subjects in the darunavir/ritonavir 800/100 mg once daily arm and subjects in the darunavir/ritonavir 600/100 mg twice daily arm in trial TMC114-C229. No imbalances between the 2 arms were noted.
Table 21: Demographic and Baseline Characteristics of Subjects in Trial TMC114-C229
darunavir/ritonavir
800/100 mg once daily
+ OBR
N=294
darunavir/ritonavir
600/100 mg twice daily
+ OBR
N=296
Demographic characteristics
Median age (years)
(range, years)
40
(18-70)
40
(18-77)
Sex
  Male
61%
67%
  Female
39%
33%
Race
  White
35%
37%
  Black
28%
24%
  Hispanic
16%
20%
  Asian
16%
14%
Baseline characteristics
Mean baseline plasma HIV-1 RNA (log10 copies/mL)
4.19
4.13
Median baseline CD4+ cell count
(cells/mm3)
(range, cells/mm3)
219
(24-1,306)
236
(44-864)
Percentage of patients with baseline viral load ≥100,000 copies/mL
13%
11%
Percentage of patients with baseline CD4+ cell count <200 cells/mm3
43%
39%
Median darunavir fold change
(range)a
0.50
(0.1-1.8)
0.50
(0.1-1.9)
Median number of resistance-associatedb:
PI mutations
3
4
NNRTI mutations
2
1
NRTI mutations
1
1
Percentage of subjects susceptible to all available PIs at baseline
88%
86%
Percentage of subjects with number of baseline primary protease inhibitor mutationsb:
0
1
2
≥3
84%
8%
5%
3%
84%
9%
4%
2%
Median number of ARVs previously usedc:
NRTIs
NNRTIs
PIs (excluding low-dose ritonavir)
3
1
1
3
1
1
OBR=optimized background regimen a Based on phenotype (Antivirogram®). b Johnson VA, Brun-Vézinet F, Clotet B, et al. Update of the drug resistance mutations in HIV-1: December 2008. Top HIV Med 2008; 16(5): 138-145. c Only counting ARVs, excluding low-dose ritonavir.
Week 48 outcomes for subjects on darunavir/ritonavir 800/100 mg once daily from trial TMC114-C229 are shown in Table 22.
Table 22: Virologic Outcome of Randomized Treatment of Trial TMC114-C229 at 48 Weeks
darunavir/ritonavir
800/100 mg once daily
+ OBR
N=294
darunavir/ritonavir
600/100 mg twice daily
+ OBR
N=296
Virologic success
HIV-1 RNA <50 copies/mL
69%
69%
Virologic failurea
26%
23%
No virologic data at Week 48 windowb
Reasons
Discontinued trial due to
adverse event or deathc
3%
4%
Discontinued trial for other
reasonsd
2%
3%
Missing data during windowb but on trial
0%
<1%
N = total number of subjects with data; OBR=optimized background regimen a Includes patients who discontinued prior to Week 48 for lack or loss of efficacy, patients who are ≥50 copies in the 48-week window, patients who had a change in their background regimen that was not permitted in the protocol (provided the switch occurred before the earliest onset of an AE leading to permanent stop of trial medication) and patients who discontinued for reasons other than AEs/death and lack or loss of efficacy (provided their last available viral load was detectable (HIV RNA ≥50 copies/mL). b Window 42-54 Weeks c Patients who discontinued due to adverse event or death at any time point from Day 1 through the time window if this resulted in no virologic data on treatment during the specified window. d Other includes: withdrew consent, loss to follow-up, etc., if the viral load at the time of discontinuation was <50 copies/mL.
The mean increase from baseline in CD4+ cell counts was comparable for both treatment arms (108 cells/mm3 and 112 cells/mm3 in the darunavir/ritonavir 800/100 mg once daily arm and the darunavir/ritonavir 600/100 mg twice daily arm, respectively).
TMC114-C214
TMC114-C214 is a randomized, controlled, open-label Phase 3 trial comparing darunavir/ritonavir 600/100 mg twice daily versus lopinavir/ritonavir 400/100 mg twice daily in antiretroviral treatment-experienced, lopinavir/ritonavir-naĂŻve HIV-1-infected adult subjects. Both arms used an optimized background regimen consisting of at least 2 antiretrovirals (NRTIs with or without NNRTIs).
HIV-1-infected subjects who were eligible for this trial had plasma HIV-1 RNA greater than 1,000 copies/mL and were on a highly active antiretroviral therapy regimen (HAART) for at least 12 weeks. Virologic response was defined as a confirmed plasma HIV-1 RNA viral load less than 400 copies/mL. Analyses included 595 subjects in trial TMC114-C214 who had completed 96 weeks of treatment or discontinued earlier.
Demographics and baseline characteristics were balanced between the darunavir/ritonavir arm and the lopinavir/ritonavir arm (see Table 23). Table 23 compares the demographic and baseline characteristics between subjects in the darunavir/ritonavir 600/100 mg twice daily arm and subjects in the lopinavir/ritonavir 400/100 mg twice daily arm in trial TMC114-C214.
Table 23: Demographic and Baseline Characteristics of Subjects in Trial TMC114-C214
darunavir/ritonavir
600/100 mg twice daily
+ OBR
N=298
lopinavir/ritonavir
400/100 mg twice daily
+ OBR
N=297
Demographic characteristics
Median age (years)
40
41
(range, years)
(18-68)
(22-76)
Sex
  Male
77%
81%
  Female
23%
19%
Race
  White
54%
57%
  Black
18%
17%
  Hispanic
15%
15%
  Asian
9%
9%
Baseline characteristics
Mean baseline plasma HIV-1 RNA (log10 copies/mL)
4.33
4.28
Median baseline CD4+ cell count
235
230
(cells/mm3)
(3-831)
(2-1,096)
(range, cells/mm3)
Percentage of patients with baseline viral load ≥100,000 copies/mL
19%
17%
Percentage of patients with baseline CD4+ cell count <200 cells/mm3
40%
40%
Median darunavir fold change
0.60
0.60
(range)
(0.10-37.40)
(0.1-43.8)
Median lopinavir fold change
0.70
0.80
(range)
(0.40-74.40)
(0.30-74.50)
Median number of resistance-associateda:
PI mutations
4
4
NNRTI mutations
1
1
NRTI mutations
2
2
Percentage of subjects with number of baseline primary protease inhibitor mutationsa:
≤1
78%
80%
2
8%
9%
≥3
13%
11%
Median number of ARVs previously usedb:
NRTIs
4
4
NNRTIs
1
1
PIs (excluding low-dose ritonavir)
1
1
Percentage of subjects resistantc to all availabled PIs at baseline, excluding darunavir
2%
3%
OBR=optimized background regimen a Johnson VA, Brun-Vezinet F, Clotet B, et al. Update of the drug resistance mutations in HIV-1: Fall 2006. Top HIV Med 2006; 14(3): 125-130. b Only counting ARVs, excluding low-dose ritonavir. c Based on phenotype (Antivirogram®). d Commercially available PIs at the time of trial enrollment.
Week 96 outcomes for subjects on darunavir/ritonavir 600/100 mg twice daily from trial TMC114-C214 are shown in Table 24.
Table 24: Virologic Outcome of Randomized Treatment of Trial TMC114-C214 at 96 Weeks
darunavir/ritonavir
600/100 mg twice daily
+ OBR
N=298
lopinavir/ritonavir
400/100 mg twice daily
+ OBR
N=297
Virologic success
HIV-1 RNA
<50 copies/mL
58%
52%
Virologic failurea
26%
33%
No virologic data at Week 96 windowb
Reasons
Discontinued trial
due to adverse
event or deathc
7%
8%
Discontinued trial for
other reasonsd
8%
7%
Missing data during
windowb but on trial
1%
<1%
N = total number of subjects with data; OBR=optimized background regimen a Includes patients who discontinued prior to Week 96 for lack or loss of efficacy and patients who are ≥50 copies in the 96-week window and patients who had a change in their OBR that was not permitted by the protocol. b Window 90-102 Weeks. c Includes patients who discontinued due to adverse event or death at any time point from Day 1 through the time window if this resulted in no virologic data on treatment during the specified window. d Other includes: withdrew consent, loss to follow-up, etc., if the viral load at the time of discontinuation was <50 copies/mL.
In trial TMC114-C214 at 96 weeks of treatment, the median increase from baseline in CD4+ cell counts was 81 cells/mm3 in the darunavir/ritonavir 600/100 mg twice daily arm and 93 cells/mm3 in the lopinavir/ritonavir 400/100 mg twice daily arm.
TMC114-C213 and TMC114-C202
TMC114-C213 and TMC114-C202 are randomized, controlled, Phase 2b trials in adult subjects with a high level of PI resistance consisting of 2 parts: an initial partially-blinded, dose-finding part and a second long-term part in which all subjects randomized to darunavir/ritonavir received the recommended dose of 600/100 mg twice daily.
HIV-1-infected subjects who were eligible for these trials had plasma HIV-1 RNA greater than 1,000 copies/mL, had prior treatment with PI(s), NNRTI(s) and NRTI(s), had at least one primary PI mutation (D30N, M46I/L, G48V, I50L/V, V82A/F/S/T, I84V, L90M) at screening, and were on a stable PI-containing regimen at screening for at least 8 weeks. Randomization was stratified by the number of PI mutations, screening viral load, and the use of enfuvirtide.
The virologic response rate was evaluated in subjects receiving darunavir/ritonavir plus an OBR versus a control group receiving an investigator-selected PI(s) regimen plus an OBR. Prior to randomization, PI(s) and OBR were selected by the investigator based on genotypic resistance testing and prior ARV history. The OBR consisted of at least 2 NRTIs with or without enfuvirtide. Selected PI(s) in the control arm included: lopinavir in 36%, (fos)amprenavir in 34%, saquinavir in 35% and atazanavir in 17%; 98% of control subjects received a ritonavir boosted PI regimen out of which 23% of control subjects used dual-boosted PIs. Approximately 47% of all subjects used enfuvirtide, and 35% of the use was in subjects who were ENF-naĂŻve. Virologic response was defined as a decrease in plasma HIV-1 RNA viral load of at least 1 log10 versus baseline.
In the pooled analysis for TMC114-C213 and TMC114-C202, demographics and baseline characteristics were balanced between the darunavir/ritonavir arm and the comparator PI arm (see Table 25). Table 25 compares the demographic and baseline characteristics between subjects in the darunavir/ritonavir 600/100 mg twice daily arm and subjects in the comparator PI arm in the pooled analysis of trials TMC114-C213 and TMC114-C202.
Table 25: Demographic and Baseline Characteristics of Subjects in the Trials TMC114-C213 and TMC114-C202 (Pooled Analysis)
darunavir/ritonavir
600/100 mg twice daily
+ OBR
N=131
Comparator PI(s)
+ OBR
N=124
Demographic characteristics
Median age (years)
43
44
(range, years)
(27-73)
(25-65)
Sex
  Male
89%
88%
  Female
11%
12%
Race
  White
81%
73%
  Black
10%
15%
  Hispanic
7%
8%
Baseline characteristics
Mean baseline plasma HIV-1 RNA (log10 copies/mL)
4.61
4.49
Median baseline CD4+ cell count
153
163
(cells/mm3)
(3-776)
(3-1,274)
(range, cells/mm3)
Percentage of patients with baseline viral load >100,000 copies/mL
24%
29%
Percentage of patients with baseline CD4+ cell count <200 cells/mm3
67%
58%
Median darunavir fold change
4.3
3.3
Median number of resistance-associateda:
PI mutations
12
12
NNRTI mutations
1
1
NRTI mutations
5
5
Percentage of subjects with number of baseline primary protease inhibitor mutationsa:
≤1
8%
9%
2
22%
21%
≥3
70%
70%
Median number of ARVs previously usedb:
NRTIs
6
6
NNRTIs
1
1
PIs (excluding low-dose ritonavir)
5
5
Percentage of subjects resistantb to all availablec PIs at baseline, excluding tipranavir and darunavir
63%
61%
Percentage of subjects with prior use of enfuvirtide
20%
17%
OBR=optimized background regimen a Johnson VA, Brun-Vezinet F, Clotet B, et al. Update of the drug resistance mutations in HIV-1: Fall 2006. Top HIV Med 2006; 14(3): 125-130. b Based on phenotype (Antivirogram®). c Commercially available PIs at the time of trial enrollment.
Week 96 outcomes for subjects on the recommended dose darunavir/ritonavir 600/100 mg twice daily from the pooled trials TMC114-C213 and TMC114-C202 are shown in Table 26.
Table 26: Outcomes of Randomized Treatment Through Week 96 of the Trials TMC114-C213 and TMC114-C202 (Pooled Analysis)
Randomized trials TMC114-C213 and TMC114-C202
darunavir/ritonavir
600/100 mg twice daily
+ OBR
N=131
Comparator PI(s)
+ OBR
N=124
Virologic responders confirmed at least 1 log10 HIV-1 RNA below baseline through Week 96 (<50 copies/mL at Week 96)
57%
(39%)
10%
(9%)
Virologic failures
29%
80%
Lack of initial responsea
8%
53%
Rebounderb
17%
19%
Never suppressedc
4%
8%
Death or discontinuation due to adverse events
9%
3%
Discontinuation due to other reasons
5%
7%
OBR=optimized background regimen a Subjects who did not achieve at least a confirmed 0.5 log10 HIV-1 RNA drop from baseline at Week 12. b Subjects with an initial response (confirmed 1 log10 drop in viral load), but without a confirmed 1 log10 drop in viral load at Week 96. c Subjects who never reached a confirmed 1 log10 drop in viral load before Week 96.
In the pooled trials TMC114-C213 and TMC114-C202 through 48 weeks of treatment, the proportion of subjects with HIV-1 RNA less than 400 copies/mL in the arm receiving darunavir/ritonavir 600/100 mg twice daily compared to the comparator PI arm was 55.0% and 14.5%, respectively. In addition, the mean changes in plasma HIV-1 RNA from baseline were –1.69 log10 copies/mL in the arm receiving darunavir/ritonavir 600/100 mg twice daily and –0.37 log10 copies/mL for the comparator PI arm. The mean increase from baseline in CD4+ cell counts was higher in the arm receiving darunavir/ritonavir 600/100 mg twice daily (103 cells/mm3) than in the comparator PI arm (17 cells/mm3).
14.4 Pediatric Patients
The pharmacokinetic profile, safety and antiviral activity of darunavir/ritonavir were evaluated in 3 randomized, open-label, multicenter studies.
TMC114-C212
Treatment-experienced pediatric subjects between the ages of 6 and less than 18 years and weighing at least 20 kg were stratified according to their weight (greater than or equal to 20 kg to less than 30 kg, greater than or equal to 30 kg to less than 40 kg, greater than or equal to 40 kg) and received darunavir tablets with either ritonavir capsules or oral solution plus background therapy consisting of at least two non-protease inhibitor antiretroviral drugs. Eighty patients were randomized and received at least one dose of darunavir/ritonavir. Pediatric subjects who were at risk of discontinuing therapy due to intolerance of ritonavir oral solution (e.g., taste aversion) were allowed to switch to the capsule formulation. Of the 44 pediatric subjects taking ritonavir oral solution, 23 subjects switched to the 100 mg capsule formulation and exceeded the weight-based ritonavir dose without changes in observed safety.
The 80 randomized pediatric subjects had a median age of 14 (range 6 to less than 18 years), and were 71% male, 54% Caucasian, 30% Black, 9% Hispanic and 8% other. The mean baseline plasma HIV-1 RNA was 4.64 log10 copies/mL, and the median baseline CD4+ cell count was 330 cells/mm3 (range: 6 to 1,505 cells/mm3). Overall, 38% of pediatric subjects had baseline plasma HIV-1 RNA ≥100,000 copies/mL. Most pediatric subjects (79%) had previous use of at least one NNRTI and 96% of pediatric subjects had previously used at least one PI.
Seventy-seven pediatric subjects (96%) completed the 24-week period. Of the patients who discontinued, one patient discontinued treatment due to an adverse event. An additional 2 patients discontinued for other reasons, one patient due to compliance and another patient due to relocation.
The proportion of pediatric subjects with HIV-1 RNA less than 400 copies/mL and less than 50 copies/mL was 64% and 50%, respectively. The mean increase in CD4+ cell count from baseline was 117 cells/mm3.
TMC114-C228
Treatment-experienced pediatric subjects between the ages of 3 and less than 6 years and weighing greater than or equal to 10 kg to less than 20 kg received darunavir oral suspension with ritonavir oral solution plus background therapy consisting of at least two active non-protease inhibitor antiretroviral drugs. Twenty-one subjects received at least one dose of darunavir/ritonavir.
The 21 subjects had a median age of 4.4 years (range 3 to less than 6 years), and were 48% male, 57% Black, 29%, Caucasian and 14% other. The mean baseline plasma HIV-1 was 4.34 log10 copies/mL, the median baseline CD4+ cell count was 927 x 106 cells/L (range: 209 to 2,429 x 106 cells/L) and the median baseline CD4+ percentage was 27.7% (range: 15.6% to 51.1%). Overall, 24% of subjects had a baseline plasma HIV-1 RNA greater than or equal to 100,000 copies/mL. All subjects had used greater than or equal to 2 NRTIs, 62% of subjects had used greater than or equal to 1 NNRTI and 76% had previously used at least one HIV PI.
Twenty subjects (95%) completed the 48 week period. One subject prematurely discontinued treatment due to vomiting assessed as related to ritonavir.
The proportion of subjects with HIV-1 RNA less than 50 copies/mL at Week 48 was 71%. The mean increase in CD4+ percentage from baseline was 4%. The mean change in CD4+ cell count from baseline was 187 x 106 cells/L.
TMC114-C230
Treatment-naĂŻve pediatric subjects between the ages of 12 and less than 18 years and weighing at least 40 kg received the adult recommended dose of darunavir/ritonavir 800/100 mg once daily plus background therapy consisting of at least two non-protease inhibitor antiretroviral drugs.
The 12 randomized pediatric subjects had a median age of 14.4 years (range 12.6 to 17.3 years), and were 33.3% male, 58.3% Caucasian and 41.7% Black. The mean baseline plasma HIV-1 RNA was 4.72 log10 copies/mL, and the median baseline CD4+ cell count was 282 cells/mm3 (range: 204 to 515 cells/mm3). Overall, 41.7% of pediatric subjects had baseline plasma HIV-1 RNA ≥100,000 copies/mL.
All subjects completed the 48 week treatment period.
The proportion of subjects with HIV-1 RNA less than 50 copies/mL and less than 400 copies/mL was 83.3% and 91.7%, respectively. The mean increase in CD4+ cell count from baseline was 221 x 106 cells/L.
16 How Supplied/storage And Handling
Darunavir tablets, 600 mg are supplied as orange, oval-shaped, film-coated tablets debossed with “TV” on one side and “7736” on the other side of the tablet. Tablets are available in bottles of 60 (NDC 0480-7736-06).
Storage
Store at 20° to 25°C (68° to 77°F); excursions permitted to 15° to 30°C (59° to 86°F) [see USP Controlled Room Temperature].
Keep darunavir tablets out of reach of children.
17 Patient Counseling Information
Advise the patient to read the FDA-approved patient labeling (Patient Information).
Instructions for Use
Advise patients to take darunavir and ritonavir with food every day on a regular dosing schedule, as missed doses can result in development of resistance. Darunavir must always be used with ritonavir in combination with other antiretroviral drugs. Advise patients not to alter the dose of either darunavir or ritonavir, discontinue ritonavir, or discontinue therapy with darunavir without consulting their physician [see Dosage and Administration (2)].
Hepatotoxicity
Inform patients that drug-induced hepatitis (e.g., acute hepatitis, cytolytic hepatitis) has been reported with darunavir co-administered with 100 mg of ritonavir. Advise patients about the signs and symptoms of liver problems [see Warnings and Precautions (5.2)].
Severe Skin Reactions
Inform patients that skin reactions ranging from mild to severe, including Stevens-Johnson Syndrome, drug rash with eosinophilia and systemic symptoms, and toxic epidermal necrolysis, have been reported with darunavir co-administered with 100 mg of ritonavir. Advise patients to discontinue darunavir/ritonavir immediately if signs or symptoms of severe skin reactions develop. These can include but are not limited to severe rash or rash accompanied with fever, general malaise, fatigue, muscle or joint aches, bulers, oral lesions, conjunctivitis, hepatitis and/or eosinophilia [see Warnings and Precautions (5.3)].
Drug Interactions
Darunavir/ritonavir may interact with many drugs; therefore, advise patients to report to their healthcare provider the use of any other prescription or nonprescription medication or herbal products, including St. John’s wort [see Contraindications (4), Warnings and Precautions (5.4, 5.5) and Drug Interactions (7)].
Contraception
Instruct patients receiving combined hormonal contraception or the progestin only pill to use an effective alternative (non-hormonal) contraceptive method or add a barrier method during therapy with darunavir/ritonavir because hormonal levels may decrease [see Drug Interactions (7.3) and Use in Specific Populations (8.3)].
Fat Redistribution
Inform patients that redistribution or accumulation of body fat may occur in patients receiving antiretroviral therapy, including darunavir/ritonavir, and that the cause and long-term health effects of these conditions are not known at this time [see Warnings and Precautions (5.7)].
Immune Reconstitution Syndrome
Advise patients to inform their healthcare provider immediately of any symptoms of infection, as in some patients with advanced HIV infection (AIDS), signs and symptoms of inflammation from previous infections may occur soon after anti-HIV treatment is started [see Warnings and Precautions (5.8)].
Pregnancy Registry
Inform patients that there is an antiretroviral pregnancy registry to monitor fetal outcomes of pregnant women exposed to darunavir [see Use in Specific Populations (8.1)].
Lactation
Instruct women with HIV-1 infection not to breastfeed because HIV-1 can be passed to the baby in breast milk [see Use in Specific Populations (8.2)].
Brands uled are the trademarks of their respective owners.
Manufactured In Croatia By: Pliva Hrvatska d.o.o. Zagreb, Croatia
Manufactured For: Teva Pharmaceuticals Parsippany, NJ 07054
Rev. B 4/2023
Spl Patient Package Insert Section
 PATIENT INFORMATION Darunavir (dar ue' na vir) Tablets Read this Patient Information before you start taking darunavir tablets and each time you get a refill. There may be new information. This information does not take the place of talking to your healthcare provider about your medical condition or your treatment. Also read the Patient Information leaflet for ritonavir. What is the most important information I should know about darunavir tablets?
- Ask your healthcare provider or pharmacist about medicines that should not be taken with darunavir tablets. For more information, see “Who should not take darunavir tablets?” and “What should I tell my healthcare provider before taking darunavir tablets?”
- Darunavir tablets may cause liver problems. Some people taking darunavir tablets in combination with ritonavir have developed liver problems, which may be life-threatening. Your healthcare provider should do blood tests before and during your darunavir tablets and ritonavir combination treatment. If you have chronic hepatitis B or C infection, your healthcare provider should check your blood tests more often because you have an increased chance of developing liver problems. Tell your healthcare provider if you have any of the below signs and symptoms of liver problems.
- dark (tea colored) urine                                ○ vomiting                           Â
- yellowing of your skin or whites of your eyes               ○ pain or tenderness on your right side below your ribs
- pale colored stools (bowel movements)Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â â—‹Â loss of appetite
- nausea                                              ○ tiredness
- Darunavir tablets may cause severe or life-threatening skin reactions or rash. Sometimes these skin reactions and skin rashes can become severe and require treatment in a hospital. Tell your healthcare provider right away if you develop a rash. Stop taking darunavir tablets and ritonavir combination treatment and tell your healthcare provider right away if you have any skin changes with symptoms below:
- fever                                              ○ bulers or skin lesions
- tiredness                                           ○ mouth sores or ulcers
- muscle or joint pain                                  ○ red or inflamed eyes, like “pink eye” (conjunctivitis)
Rash occurred more often in people taking darunavir tablets and raltegravir together than with either drug separately, but was generally mild.
See “What are the possible side effects of darunavir tablets?” for more information about side effects.What are darunavir tablets?
Darunavir tablets are a prescription HIV-1 (Human Immunodeficiency Virus-type 1) medicine used with ritonavir and other antiretroviral medicines to treat HIV-1 infection in adults and children 3 years of age and older. HIV is the virus that causes AIDS (Acquired Immune Deficiency Syndrome).
Darunavir tablets should not be used in children under 3 years of age.
When used with other antiretroviral medicines to treat HIV-1 infection, darunavir tablets may help:
- reduce the amount of HIV-1 in your blood. This is called “viral load”.
- increase the number of CD4+ (T) cells in your blood that help fight off other infections.
Reducing the amount of HIV-1 and increasing the CD4+ (T) cells in your blood may improve your immune system. This may reduce your risk of death or getting infections that can happen when your immune system is weak (opportunistic infections).
Darunavir tablets do not cure HIV-1 infection or AIDS. You must keep taking HIV-1 medicines to control HIV-1 infection and decrease HIV-related illnesses.
Avoid doing things that can spread HIV-1 infection to others:
Ask your healthcare provider if you have any questions on how to prevent passing HIV to other people.
- Do not share or re-use needles or other injection equipment.
- Do not share personal lis that can have blood or body fluids on them, like toothbrushes and razor blades.
- Do not have any kind of sex without protection. Always practice safe sex by using a latex or polyurethane condom to lower the chance of sexual contact with semen, vaginal secretions, or blood.
Who should not take darunavir tablets?
Do not take darunavir tablets with any medicine that contains:
Serious problems can happen if you or your child take any of these medicines with darunavir tablets. This is not a complete ul of medicines. Therefore, tell your healthcare provider about all medicines you take.
- alfuzosin
- colchicine, if you have liver or kidney problems
- dronedarone
- elbasvir and grazoprevir
- ergot-containing medicines:
- dihydroergotamine
- ergotamine tartrate
- methylergonovine
- ivabradine
- lomitapide
- lovastatin
- lurasidone
- midazolam, when taken by mouth
- naloxegol
- pimozide
- ranolazine
- rifampin
- sildenafil, when used for the treatment of pulmonary arterial hypertension (PAH)
- simvastatin
- St. John’s wort (Hypericum perforatum)
- triazolam
What should I tell my healthcare provider before taking darunavir tablets?
Before taking darunavir tablets, tell your healthcare provider if you:
- have liver problems, including hepatitis B or hepatitis C
- are allergic to sulfa medicines
- have high blood sugar (diabetes)
- have hemophilia
- have any other medical conditions
- are pregnant or plan to become pregnant. Tell your healthcare provider if you become pregnant while taking darunavir tablets.
- Pregnancy Registry: There is a pregnancy registry for women who take antiretroviral medicines during pregnancy. The purpose of this registry is to collect information about the health of you and your baby. Talk to your healthcare provider about how you can take part in this registry.
- are breastfeeding or plan to breastfeed. Do not breastfeed if you take darunavir tablets.
- You should not breastfeed if you have HIV-1 because of the risk of passing HIV-1 to your baby.
- It is not known if darunavir can pass into your breast milk.
- Talk to your healthcare provider about the best way to feed your baby.
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, topical creams, vitamins, and herbal supplements. Some medicines interact with darunavir tablets. Keep a ul of your medicines to show your healthcare provider and pharmacist.
- You can ask your healthcare provider or pharmacist for a ul of medicines that interact with darunavir tablets.
- Do not start taking a new medicine without telling your healthcare provider. Your healthcare provider can tell you if it is safe to take darunavir tablets with other medicines.
How should I take darunavir tablets?
- Take darunavir tablets exactly as your healthcare provider tells you.
- You must take ritonavir at the same time as darunavir tablets.
- Do not change your dose or stop treatment with darunavir tablets without talking to your healthcare provider.
- Take darunavir tablets and ritonavir with food.
- If you have difficulty swallowing darunavir tablets, darunavir oral suspension is also available. Your healthcare provider will help decide whether darunavir tablets or oral suspension is right for you.
- If your child is taking darunavir tablets, your child's healthcare provider will decide the right dose based on your child's weight. Your child's healthcare provider will tell you how many darunavir tablets and how much ritonavir (capsules, tablets or solution) your child should take. Your child should take darunavir tablets with ritonavir with food. If your child does not tolerate ritonavir oral solution, ask your child's healthcare provider for advice.
- It is important that you do not miss or skip doses of darunavir tablets during treatment.
- If you take too many darunavir tablets, call your healthcare provider or go to the nearest hospital emergency room right away.
What are the possible side effects of darunavir tablets?
Darunavir tablets may cause serious side effects, including:
- See “What is the most important information I should know about darunavir tablets?”
- Diabetes and high blood sugar (hyperglycemia). Some people who take protease inhibitors including darunavir tablets can get high blood sugar, develop diabetes, or your diabetes can get worse. Tell your healthcare provider if you notice an increase in thirst or urinate often while taking darunavir tablets.
- Changes in body fat can happen in people who take HIV-1 medicines. The changes may include an increased amount of fat in the upper back and neck (“buffalo hump”), breast, and around the middle of your body (trunk). Loss of fat from the legs, arms, and face may also happen. The exact cause and long-term health effects of these conditions are not known.
- Changes in your immune system (Immune Reconstitution Syndrome) can happen when you start taking HIV-1 medicines. Your immune system may get stronger and begin to fight infections that have been hidden in your body for a long time. Tell your healthcare provider right away if you start having new symptoms after starting your HIV-1 medicine.
- Increased bleeding for hemophiliacs. Some people with hemophilia have increased bleeding with protease inhibitors including darunavir tablets.
The most common side effects of darunavir tablets include:
- diarrhea                               •  headache
- nausea                                •  stomach-area (abdominal) pain
- rash                                  •  vomiting
Tell your healthcare provider if you have any side effect that bothers you or that does not go away.
These are not all of the possible side effects of darunavir tablets.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.How should I store darunavir tablets? Keep darunavir tablets and all medicines out of the reach of children.
- Store darunavir tablets at room temperature 68° to 77°F (20° to 25°C).
General information about the safe and effective use of darunavir tablets.
Medicines are sometimes prescribed for purposes other than those uled in a Patient Information leaflet. Do not use darunavir tablets for a condition for which they were not prescribed. Do not give darunavir tablets to other people even if they have the same condition you have. They may harm them.
This leaflet summarizes the most important information about darunavir tablets. If you would like more information, talk to your healthcare provider. You can ask your healthcare provider or pharmacist for information about darunavir tablets that is written for health professionals. For more information, call 1-888-838-2872.What are the ingredients in darunavir tablets?
Active ingredient: darunavir
Inactive ingredients: colloidal silicon dioxide, crospovidone, FD&C red #40 aluminum lake, FD&C yellow #6 aluminum lake, magnesium stearate, microcrystalline cellulose, polyethylene glycol, polyvinyl alcohol, talc, and titanium dioxide.
Manufactured In Croatia By: Pliva Hrvatska d.o.o., Zagreb, Croatia Manufactured For: Teva Pharmaceuticals, Parsippany, NJ 07054
This Patient Information has been approved by the U.S. Food and Drug Administration.                     Rev. A 11/2022
Package Label.principal Display Panel
NDCÂ 0480-7736-06
Darunavir Tablets
600 mg
Rx only
60 Tablets

DISCLAIMER:
"This tool does not provide medical advice, and is for informational and educational purposes only, and is not a substitute for professional medical advice, treatment or diagnosis. Call your doctor to receive medical advice. If you think you may have a medical emergency, please dial 911."
"Do not rely on openFDA to make decisions regarding medical care. While we make every effort to ensure that data is accurate, you should assume all results are unvalidated. We may limit or otherwise restrict your access to the API in line with our Terms of Service."
"This product uses publicly available data from the U.S. National Library of Medicine (NLM), National Institutes of Health, Department of Health and Human Services; NLM is not responsible for the product and does not endorse or recommend this or any other product."
PillSync may earn a commission via links on our site
