SPRYCEL (dasatinib 80 mg) Dailymed
Generic: dasatinib is used for the treatment of Leukemia, Lymphoid Leukemia, Myeloid
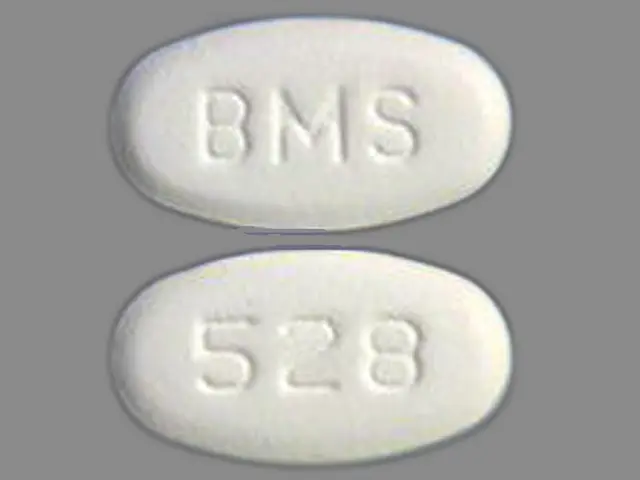
IMPRINT: BMS 528
SHAPE: oval
COLOR: white
All Imprints
dasatinib 70 mg - bms 524 round white
dasatinib 50 mg - bms 528 oval white
dasatinib 20 mg - bms 527 round white
dasatinib 100 mg - bms100 852 oval white

Go PRO for all pill images
1 Indications And Usage
SPRYCEL (dasatinib) is indicated for the treatment of adult patients with
‚ÄĘ newly diagnosed Philadelphia chromosome-positive (Ph+) chronic myeloid leukemia (CML) in chronic phase.‚ÄĘ chronic, accelerated, or myeloid or lymphoid blast phase Ph+ CML with resistance or intolerance to prior therapy including imatinib.‚ÄĘ Philadelphia chromosome-positive acute lymphoblastic leukemia (Ph+ ALL) with resistance or intolerance to prior therapy.
SPRYCEL (dasatinib) is indicated for the treatment of pediatric patients 1 year of age and older with
‚ÄĘ Ph+ CML in chronic phase.‚ÄĘ newly diagnosed Ph+ ALL in combination with chemotherapy.
SPRYCEL is a kinase inhibitor indicated for the treatment of
‚ÄĘ newly diagnosed adults with Philadelphia chromosome-positive (Ph+) chronic myeloid leukemia (CML) in chronic phase.(1 ,14) ‚ÄĘ adults with chronic, accelerated, or myeloid or lymphoid blast phase Ph+ CML with resistance or intolerance to prior therapy including imatinib.(1 ,14) ‚ÄĘ adults with Philadelphia chromosome-positive acute lymphoblastic leukemia (Ph+ ALL) with resistance or intolerance to prior therapy.(1 ,14) ‚ÄĘ pediatric patients 1 year of age and older with Ph+ CML in chronic phase.(1 ,14) ‚ÄĘ pediatric patients 1 year of age and older with newly diagnosed Ph+ ALL in combination with chemotherapy.(1 ,14)
2 Dosage And Administration
 
‚ÄĘ Chronic phase CML in adults: 100 mg once daily.(2) ‚ÄĘ Accelerated phase CML, myeloid or lymphoid blast phase CML, or Ph+ ALL in adults: 140 mg once daily.(2) ‚ÄĘ Chronic phase CML and ALL in pediatrics: starting dose based on body weight.(2) ‚ÄĘ Administer orally, with or without a meal. Do not crush, cut, or chew tablets.(2) 2.1 Dosage of SPRYCEL in Adult Patients
The recommended starting dosage of SPRYCEL for chronic phase CML in adults is 100 mg administered orally once daily. The recommended starting dosage of SPRYCEL for accelerated phase CML, myeloid or lymphoid blast phase CML, or Ph+ ALL in adults is 140 mg administered orally once daily. Tablets should not be crushed, cut, or chewed; they should be swallowed whole. SPRYCEL can be taken with or without a meal, either in the morning or in the evening.
2.2 Dosage of SPRYCEL in Pediatric Patients with CML or Ph+ ALL
The recommended starting dosage for pediatrics is based on body weight as shown in Table 1. The recommended dose should be administered orally once daily with or without food. Recalculate the dose every 3 months based on changes in body weight, or more often if necessary.
Do not crush, cut or chew tablets. Swallow tablets whole. There are additional administration considerations for pediatric patients who have difficulty swallowing tablets whole [see Use in Specific Populations (8.4) and Clinical Pharmacology (12.3)].
Table 1: Dosage of SPRYCEL for Pediatric Patientsa Body Weight (kg)b Daily Dose (mg) a  For pediatric patients with Ph+ ALL, begin SPRYCEL therapy on or before day 15 of induction chemotherapy, when diagnosis is confirmed and continue for 2 years. b  Tablet dosing is not recommended for patients weighing less than 10 kg.
10 to less than 20
40 mg
20 to less than 30
60 mg
30 to less than 45
70 mg
at least 45
100 mg
Refer to Section 2.4 for recommendations on dose escalation in adults with CML and Ph+ ALL, and pediatric patients with CML.
2.3 Dose Modification
Strong CYP3A4 Inducers Avoid the use of concomitant strong CYP3A4 inducers and St. John’s wort. If patients must be coadministered a strong CYP3A4 inducer, consider a SPRYCEL dose increase. If the dose of SPRYCEL is increased, monitor the patient carefully for toxicity [see Drug Interactions (7.1) ].
Strong CYP3A4 Inhibitors Avoid the use of concomitant strong CYP3A4 inhibitors and grapefruit juice. Recommend selecting an alternate concomitant medication with no or minimal enzyme inhibition potential, if possible. If SPRYCEL must be administered with a strong CYP3A4 inhibitor, consider a dose decrease to:
‚ÄĘ 40 mg daily for patients taking SPRYCEL 140 mg daily.
‚ÄĘ 20 mg daily for patients taking SPRYCEL 100 mg daily.
‚ÄĘ 20 mg daily for patients taking SPRYCEL 70 mg daily.
For patients taking SPRYCEL 60 mg or 40 mg daily, consider interrupting SPRYCEL until the inhibitor is discontinued. Allow a washout period of approximately 1 week after the inhibitor is stopped before reinitiating SPRYCEL.
These reduced doses of SPRYCEL are predicted to adjust the area under the curve (AUC) to the range observed without CYP3A4 inhibitors; however, clinical data are not available with these dose adjustments in patients receiving strong CYP3A4 inhibitors. If SPRYCEL is not tolerated after dose reduction, either discontinue the strong CYP3A4 inhibitor or interrupt SPRYCEL until the inhibitor is discontinued. Allow a washout period of approximately 1 week after the inhibitor is stopped before the SPRYCEL dose is increased [see Drug Interactions (7.1)].
2.4 Dose Escalation in Adults with CML and Ph+ ALL, and Pediatric Patients with CML
For adult patients with CML and Ph+ ALL, consider dose escalation to 140 mg once daily (chronic phase CML) or 180 mg once daily (advanced phase CML and Ph+ ALL) in patients who do not achieve a hematologic or cytogenetic response at the recommended starting dosage. For pediatric patients with CML, consider dose escalation to 120 mg once daily (see Table 2 below). Dose escalation is not recommended for pediatric patients with Ph+ ALL, where SPRYCEL is administered in combination with chemotherapy.
Escalate the SPRYCEL dose as shown in Table 2 in pediatric patients with chronic phase CML who do not achieve a hematologic or cytogenetic response at the recommended starting dosage.
Table 2: Dose Escalation for Pediatric CML
Formulation
Dose (maximum dose per day)
Starting Dose
Escalation
Tablets
40 mg
50 mg
60 mg
70 mg
70 mg
90 mg
100 mg
120 mg
2.5 Dose Adjustment for Adverse Reactions
In clinical studies, myelosuppression was managed by dose interruption, dose reduction, or discontinuation of study therapy. Hematopoietic growth factor has been used in patients with resistant myelosuppression. Guidelines for dose modifications for adult and pediatric patients are summarized in Tables 3 and 4, respectively.
Table 3: Dose Adjustments for Neutropenia and Thrombocytopenia in Adults * ANC: absolute neutrophil count
Chronic Phase CML (starting dose 100 mg once daily)
ANC* <0.5 × 109/LorPlatelets <50 × 109/L
1. Stop SPRYCEL until ANC ‚Č•1.0 √ó 109/L and platelets ‚Č•50 √ó 109/L.2. Resume treatment with SPRYCEL at the original starting dose if recovery occurs in ‚ȧ7¬†days.3. If platelets <25 √ó 109/L or recurrence of ANC <0.5 √ó 109/L for >7 days, repeat Step 1 and resume SPRYCEL at a reduced dose of 80 mg once daily for second episode. For third episode, further reduce dose to 50 mg once daily (for newly diagnosed patients) or discontinue SPRYCEL (for patients resistant or intolerant to prior therapy including imatinib).
Accelerated Phase CML, Blast Phase CML and Ph+ ALL (starting dose 140 mg once daily)
ANC* <0.5 × 109/LorPlatelets <10 × 109/L
1. Check if cytopenia is related to leukemia (marrow aspirate or biopsy).2. If cytopenia is unrelated to leukemia, stop SPRYCEL until ANC ‚Č•1.0 √ó 109/L and platelets ‚Č•20 √ó 109/L and resume at the original starting dose.3. If recurrence of cytopenia, repeat Step 1 and resume SPRYCEL at a reduced dose of 100¬†mg once daily (second episode) or 80 mg once daily (third episode).4. If cytopenia is related to leukemia, consider dose escalation to 180 mg once daily.
Table 4: Dose Adjustments for Neutropenia and Thrombocytopenia in Pediatric Patients with Ph+ CML *ANC: absolute neutrophil count** lower tablet dose not available
  Dose (maximum dose per day)
1. If cytopenia persists for more than 3 weeks, check if cytopenia is related to leukemia (marrow aspirate or biopsy).
2. If cytopenia is unrelated to leukemia, stop SPRYCEL until ANC* ‚Č•1.0 √ó 109/L and platelets ‚Č•75 √ó 109/L and resume at the original starting dose or at a reduced dose.
3. If cytopenia recurs, repeat marrow aspirate/biopsy and resume SPRYCEL at a reduced dose.
  Original Starting Dose
  One-Level Dose Reduction
  Two-Level Dose Reduction
Tablets
  40 mg
  20 mg
  **
  60 mg
  40 mg
  20 mg
  70 mg
  60 mg
  50 mg
  100 mg
  80 mg
  70 mg
For pediatric patients with chronic phase CML, if Grade ‚Č• 3 neutropenia or thrombocytopenia recurs during complete hematologic response (CHR), interrupt SPRYCEL and resume at a reduced dose. Implement temporary dose reductions for intermediate degrees of cytopenia and disease response as needed.
For pediatric patients with Ph+ ALL, if neutropenia and/or thrombocytopenia result in a delay of the next block of treatment by more than 14 days, interrupt SPRYCEL and resume at the same dose level once the next block of treatment is started. If neutropenia and/or thrombocytopenia persist and the next block of treatment is delayed another 7 days, perform a bone marrow assessment to assess cellularity and percentage of blasts. If marrow cellularity is <10%, interrupt treatment with SPRYCEL until ANC >500/őľL (0.5 x 109/L), at which time treatment may be resumed at full dose. If marrow cellularity is >10%, resumption of treatment with SPRYCEL may be considered.
For adults with Ph+ CML and ALL, and pediatric patients with Ph+ CML, if a severe non-hematologic adverse reaction develops with SPRYCEL use, treatment must be withheld until the adverse reaction has resolved or improved. Thereafter, treatment can be resumed as appropriate at a reduced dose depending on the severity and recurrence [see Warnings and Precautions (5)].
For pediatric patients with Ph+ ALL, interrupt treatment for cases of grade ‚Č• 3 non-hematologic adverse reactions with the exception of liver function test abnormalities, and resume at a reduced dose when resolved to grade ‚ȧ1. For elevated direct bilirubin over 5 times the institutional upper limit of normal (ULN), interrupt treatment until improvement to baseline or grade ‚ȧ1. For elevated AST/ALT over 15 times the institutional ULN, interrupt treatment until improvement to baseline or grade <1. For recurrent liver function test abnormalities as above, reduce the dose if this adverse reaction recurs after reinitiation of SPRYCEL. Dose reduction recommendations are described in Table 5.
Table 5: Dose Adjustments for Non-Hematologic Toxicities in Pediatric Patients ** lower tablet dose not available
Dose (maximum dose per day)
1. If a non-hematologic toxicity grade 2 occurs, consider interrupting SPRYCEL if no recovery despite symptomatic therapy; once recovered to grade ‚ȧ1, resume at the original starting dose. Resume SPRYCEL at a reduced dose for recurrent events.
2. If a non-hematologic toxicity grade 3 occurs, stop SPRYCEL until recovery to grade ‚ȧ1 and then resume at a reduced dose.
3. If direct bilirubin is >5 ULN or AST/ALT >15 ULN, interrupt SPRYCEL until recovery to grade ‚ȧ1 and then resume SPRYCEL at the original starting dose. Resume SPRYCEL at a reduced dose for recurrent hepatotoxicity.
Original Starting Dose
One-Level Dose Reduction
Two-Level Dose Reduction
Tablets
40 mg
20 mg
**
60 mg
40 mg
20 mg
70 mg
60 mg
50 mg
100 mg
80 mg
70 mg
2.6 Duration of Treatment
In clinical studies, treatment with SPRYCEL in adults and in pediatric patients with chronic phase CML was continued until disease progression or until no longer tolerated by the patient. The effect of stopping treatment on long-term disease outcome after the achievement of a cytogenetic response (including complete cytogenetic response [CCyR]) or major molecular response (MMR and MR4.5) has not been established.
In clinical studies, treatment with SPRYCEL in pediatric patients with Ph+ ALL was administered for a maximum duration of 2 years [see Dosage and Administration (2.2) and Clinical Studies (14.4)].
SPRYCEL is a hazardous product. Follow applicable special handling and disposal procedures.1
3 Dosage Forms And Strengths
SPRYCEL (dasatinib) Tablets are available as 20-mg, 50-mg, 70-mg, 80-mg, 100-mg, and 140-mg white to off-white, biconvex, film-coated tablets.
Tablets: 20 mg, 50 mg, 70 mg, 80 mg, 100 mg, and 140 mg.(3)
4 Contraindications
None.
None.(4)
5 Warnings And Precautions
‚ÄĘ Myelosuppression and Bleeding Events: Severe thrombocytopenia, neutropenia, and anemia may occur. Use caution if used concomitantly with medications that inhibit platelet function or anticoagulants. Monitor complete blood counts regularly. Transfuse and interrupt SPRYCEL when indicated.(2.5 ,5.1 ,5.2) ‚ÄĘ Fluid Retention: Fluid retention, sometimes severe, including pleural effusions. Manage with supportive care measures and/or dose modification.(2.5 ,5.3) ‚ÄĘ Cardiovascular Toxicity: Monitor patients for signs or symptoms and treat appropriately.(5.4) ‚ÄĘ Pulmonary Arterial Hypertension (PAH): SPRYCEL may increase the risk of developing PAH which may be reversible on discontinuation. Consider baseline risk and evaluate patients for signs and symptoms of PAH during treatment. Stop SPRYCEL if PAH is confirmed.(5.5) ‚ÄĘ QT Prolongation: Use SPRYCEL with caution in patients who have or may develop prolongation of the QT interval.(5.6) ‚ÄĘ Severe Dermatologic Reactions: Individual cases of severe mucocutaneous dermatologic reactions have been reported.(5.7) ‚ÄĘ Tumor Lysis Syndrome: Tumor lysis syndrome has been reported. Maintain adequate hydration and correct uric acid levels prior to initiating therapy with SPRYCEL.(5.8) ‚ÄĘ Embryo-Fetal Toxicity: Can cause fetal harm. Advise patients of reproductive potential of potential risk to fetus and to use effective contraception.(5.9 ,8.1 ,8.3) ‚ÄĘ Effects on Growth and Development in Pediatric Patients: epiphyses delayed fusion, osteopenia, growth retardation, and gynecomastia have been reported. Monitor bone growth and development in pediatric patients.(5.10) ‚ÄĘ Hepatotoxicity: Assess liver function before initiation of treatment and monthly thereafter or as clinically indicated. Monitor liver function when combined with chemotherapy known to be associated with liver dysfunction.(5.11) 5.1 Myelosuppression
Treatment with SPRYCEL is associated with severe (NCI CTCAE Grade 3 or 4) thrombocytopenia, neutropenia, and anemia, which occur earlier and more frequently in patients with advanced phase CML or Ph+ ALL than in patients with chronic phase CML [see Adverse Reactions (6.1)].
In patients with chronic phase CML, perform complete blood counts (CBCs) every 2 weeks for 12 weeks, then every 3 months thereafter, or as clinically indicated. In patients with advanced phase CML or Ph+ ALL, perform CBCs weekly for the first 2 months and then monthly thereafter, or as clinically indicated.
In pediatric patients with Ph+ ALL treated with SPRYCEL in combination with chemotherapy, perform CBCs prior to the start of each block of chemotherapy and as clinically indicated. During the consolidation blocks of chemotherapy, perform CBCs every 2 days until recovery.
Myelosuppression is generally reversible and usually managed by withholding SPRYCEL temporarily and/or dose reduction [see Dosage and Administration (2.5) ].
5.2 Bleeding-Related Events
SPRYCEL can cause serious and fatal bleeding. In all CML or Ph+ ALL clinical studies, Grade ‚Č•3 central nervous system (CNS) hemorrhages, including fatalities, occurred in <1% of patients receiving SPRYCEL. The incidence of Grade 3/4 hemorrhage occurred in 5.8% of adult patients and generally required treatment interruptions and transfusions. The incidence of Grade 5 hemorrhage occurred in 0.4% of adult patients. The most frequent site of hemorrhage was gastrointestinal [see Adverse Reactions (6.1)]. Most bleeding events in clinical studies were associated with severe thrombocytopenia. In addition to causing thrombocytopenia in human subjects, dasatinib caused platelet dysfunction in vitro.
Concomitant medications that inhibit platelet function or anticoagulants may increase the risk of hemorrhage.
5.3 Fluid Retention
SPRYCEL may cause fluid retention [see Adverse Reactions (6.1)]. After 5 years of follow-up in the adult randomized newly diagnosed chronic phase CML study (n=258), Grade 3 or 4 fluid retention was reported in 5% of patients, including 3% of patients with Grade 3 or 4 pleural effusion. In adult patients with newly diagnosed or imatinib-resistant or -intolerant chronic phase CML, Grade 3 or 4 fluid retention occurred in 6% of patients treated with SPRYCEL at the recommended dose (n=548). In adult patients with advanced phase CML or Ph+ ALL treated with SPRYCEL at the recommended dose (n=304), Grade 3 or 4 fluid retention was reported in 8% of patients, including Grade 3 or 4 pleural effusion reported in 7% of patients. In pediatric patients with chronic phase CML, cases of Grade 1 or 2 fluid retention were reported in 10.3% of patients.
Evaluate patients who develop symptoms of pleural effusion or other fluid retention, such as new or worsened dyspnea on exertion or at rest, pleuritic chest pain, or dry cough, promptly with a chest x-ray or additional diagnostic imaging as appropriate. Fluid retention events were typically managed by supportive care measures that may include diuretics or short courses of steroids. Severe pleural effusion may require thoracentesis and oxygen therapy. Consider dose reduction or treatment interruption [see Dosage and Administration (2.5) ].
5.4 Cardiovascular Toxicity
SPRYCEL can cause cardiac dysfunction [see Adverse Reactions (6.1)]. After 5 years of follow-up in the randomized newly diagnosed chronic phase CML trial in adults (n=258), the following cardiac adverse reactions occurred: cardiac ischemic events (3.9% dasatinib vs 1.6% imatinib), cardiac-related fluid retention (8.5% dasatinib vs 3.9% imatinib), and conduction system abnormalities, most commonly arrhythmia and palpitations (7.0% dasatinib vs 5.0% imatinib). Two cases (0.8%) of peripheral arterial occlusive disease occurred with imatinib and 2 (0.8%) transient ischemic attacks occurred with dasatinib. Monitor patients for signs or symptoms consistent with cardiac dysfunction and treat appropriately.
5.5 Pulmonary Arterial Hypertension
SPRYCEL may increase the risk of developing pulmonary arterial hypertension (PAH) in adult and pediatric patients which may occur any time after initiation, including after more than 1 year of treatment. Manifestations include dyspnea, fatigue, hypoxia, and fluid retention [see Adverse Reactions (6.1)]. PAH may be reversible on discontinuation of SPRYCEL. Evaluate patients for signs and symptoms of underlying cardiopulmonary disease prior to initiating SPRYCEL and during treatment. If PAH is confirmed, SPRYCEL should be permanently discontinued.
5.6 QT Prolongation
SPRYCEL may increase the risk of prolongation of QTc in patients including those with hypokalemia or hypomagnesemia, patients with congenital long QT syndrome, patients taking antiarrhythmic medicines or other medicinal products that lead to QT prolongation, and cumulative high-dose anthracycline therapy [see Adverse Reactions (6.1)]. Correct hypokalemia or hypomagnesemia prior to and during SPRYCEL administration.
5.7 Severe Dermatologic Reactions
Cases of severe mucocutaneous dermatologic reactions, including Stevens-Johnson syndrome [see Adverse Reactions (6.2)] and erythema multiforme, have been reported in patients treated with SPRYCEL. Discontinue permanently in patients who experience a severe mucocutaneous reaction during treatment if no other etiology can be identified.
5.8 Tumor Lysis Syndrome
Tumor lysis syndrome has been reported in patients with resistance to prior imatinib therapy, primarily in advanced phase disease. Due to potential for tumor lysis syndrome, maintain adequate hydration, correct uric acid levels prior to initiating therapy with SPRYCEL, and monitor electrolyte levels. Patients with advanced stage disease and/or high tumor burden may be at increased risk and should be monitored more frequently [see Adverse Reactions (6.1) ].
5.9 Embryo-Fetal Toxicity
Based on limited human data, SPRYCEL can cause fetal harm when administered to a pregnant woman. Adverse pharmacologic effects of SPRYCEL including hydrops fetalis, fetal leukopenia, and fetal thrombocytopenia have been reported with maternal exposure to SPRYCEL. Advise females of reproductive potential and males with female partners of reproductive potential to use effective contraception during treatment with SPRYCEL and for 30 days after the last dose [see Use in Specific Populations (8.1, 8.3)].
5.10 Effects on Growth and Development in Pediatric Patients
In pediatric trials of SPRYCEL in chronic phase CML after at least 2 years of treatment, adverse reactions associated with bone growth and development were reported in 5 (5.2%) patients, one of which was severe in intensity (Growth Retardation Grade 3). These 5 cases included cases of epiphyses delayed fusion, osteopenia, growth retardation, and gynecomastia [see Adverse Reactions (6.1) and Use in Specific Populations (8.4)]. Of these 5 cases, 1 case of osteopenia and 1 case of gynecomastia resolved during treatment.
Monitor bone growth and development in pediatric patients.
5.11 Hepatotoxicity
SPRYCEL may cause hepatotoxicity as measured by elevations in bilirubin, aspartate aminotransferase (AST), alanine aminotransferase (ALT), and alkaline phosphatase [see Adverse Reactions (6.1)]. Monitor transaminases at baseline and monthly or as clinically indicated during treatment. Reduce dose, withhold, or permanently discontinue SPRYCEL based on severity [see Dosage and Administration (2.5)]. When SPRYCEL is administered in combination with chemotherapy, liver toxicity in the form of transaminase elevation and hyperbilirubinemia has been observed. Monitor hepatic function when SPRYCEL is used in combination with chemotherapy.
6 Adverse Reactions
The following clinically significant adverse reactions are discussed in greater detail in other sections of the labeling:
‚ÄĘ Myelosuppression [see Dosage and Administration (2.5) and Warnings and Precautions (5.1)].‚ÄĘ Bleeding-related events [see Warnings and Precautions (5.2)].‚ÄĘ Fluid retention [see Warnings and Precautions (5.3)].‚ÄĘ Cardiovascular toxicity [see Warnings and Precautions (5.4)].‚ÄĘ Pulmonary arterial hypertension [see Warnings and Precautions (5.5)].‚ÄĘ QT prolongation [see Warnings and Precautions (5.6) ].‚ÄĘ Severe dermatologic reactions [see Warnings and Precautions (5.7)].‚ÄĘ Tumor lysis syndrome [see Warnings and Precautions (5.8)].‚ÄĘ Effects on growth and development in pediatric patients [see Warnings and Precautions (5.10)].‚ÄĘ Hepatotoxicity [see Warnings and Precautions (5.11)].
Most common adverse reactions (‚Č•15%) in patients receiving SPRYCEL as single-agent therapy included myelosuppression, fluid retention events, diarrhea, headache, skin rash, hemorrhage, dyspnea, fatigue, nausea, and musculoskeletal pain.(6)
Most common adverse reactions (‚Č•30%) in pediatric patients receiving SPRYCEL in combination with chemotherapy included mucositis, febrile neutropenia, pyrexia, diarrhea, nausea, vomiting, musculoskeletal pain, abdominal pain, cough, headache, rash, fatigue, constipation, arrhythmia, hypertension, edema, infections (bacterial, viral and fungal), hypotension, decreased appetite, hypersensitivity, dyspnea, epistaxis, peripheral neuropathy, and altered state of consciousness.(6)
To report SUSPECTED ADVERSE REACTIONS, contact Bristol-Myers Squibb at 1-800-721-5072 or FDA at 1-800-FDA-1088 orwww.fda.gov/medwatch .
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The data described below reflect exposure to SPRYCEL administered as single-agent therapy at all doses tested in clinical studies (n=2809), including 324 adult patients with newly diagnosed chronic phase CML, 2388 adult patients with imatinib-resistant or -intolerant chronic or advanced phase CML or Ph+ ALL, and 97 pediatric patients with chronic phase CML. The median duration of therapy in a total of 2712 adult patients was 19.2 months (range 0 to 93.2 months). In a randomized trial in patients with newly diagnosed chronic phase CML, the median duration of therapy was approximately 60 months. The median duration of therapy in 1618 adult patients with chronic phase CML was 29 months (range 0 to 92.9 months).
The median duration of therapy in 1094 adult patients with advanced phase CML or Ph+ ALL was 6.2 months (range 0 to 93.2 months).
In two non-randomized trials in 97 pediatric patients with chronic phase CML (51 patients newly diagnosed and 46 patients resistant or intolerant to previous treatment with imatinib), the median duration of therapy was 51.1 months (range 1.9 to 99.6 months).
In the overall population of 2712 adult patients, 88% of patients experienced adverse reactions at some time and 19% experienced adverse reactions leading to treatment discontinuation.
In the randomized trial in adult patients with newly diagnosed chronic phase CML, drug was discontinued for adverse reactions in 16% of patients with a minimum of 60 months of follow-up. After a minimum of 60 months of follow-up, the cumulative discontinuation rate was 39%. Among the 1618 patients with chronic phase CML, drug-related adverse reactions leading to discontinuation were reported in 329 (20.3%) patients; among the 1094 patients with advanced phase CML or Ph+ ALL, drug-related adverse reactions leading to discontinuation were reported in 191 (17.5%) patients.
Among the 97 pediatric subjects, drug-related adverse reactions leading to discontinuation were reported in 1 patient (1%).
Adverse reactions reported in ‚Č•10% of adult patients, and other adverse reactions of interest, in a randomized trial in patients with newly diagnosed chronic phase CML at a median follow-up of approximately 60 months are presented in Table 6.
Adverse reactions reported in ‚Č•10% of adult patients treated at the recommended dose of 100 mg once daily (n=165), and other adverse reactions of interest, in a randomized dose-optimization trial of patients with chronic phase CML resistant or intolerant to prior imatinib therapy at a median follow-up of approximately 84 months are presented in Table 8.
Adverse reactions reported in ‚Č•10% of pediatric patients at a median follow-up of approximately 51.1 months are presented in Table 11.
Drug-related serious adverse reactions (SARs) were reported for 16.7% of adult patients in the randomized trial of patients with newly diagnosed chronic phase CML. Serious adverse reactions reported in ‚Č•5% of patients included pleural effusion (5%).
Drug-related SARs were reported for 26.1% of patients treated at the recommended dose of 100 mg once daily in the randomized dose-optimization trial of adult patients with chronic phase CML resistant or intolerant to prior imatinib therapy. Serious adverse reactions reported in ‚Č•5% of patients included pleural effusion (10%).
Drug-related SARs were reported for 14.4% of pediatric patients.
Adverse reactions (excluding laboratory abnormalities) that were reported in at least 10% of adult patients are shown in Table 6 for newly diagnosed patients with chronic phase CML and Tables 8 and 10 for CML patients with resistance or intolerance to prior imatinib therapy.
Table 6: Adverse Reactions Reported in ‚Č•10% of Adult Patients with Newly Diagnosed Chronic Phase CML (minimum of 60 months follow-up) ¬† All Grades Grade 3/4 ¬† SPRYCEL (n=258) Imatinib (n=258) SPRYCEL (n=258) Imatinib (n=258) Adverse Reaction Percent (%) of Patients a Includes cardiac failure acute, cardiac failure congestive, cardiomyopathy, diastolic dysfunction, ejection fraction decreased, and left ventricular dysfunction. b Includes erythema, erythema multiforme, rash, rash generalized, rash macular, rash papular, rash pustular, skin exfoliation, and rash vesicular. c Adverse reaction of special interest with <10% frequency. d Includes conjunctival hemorrhage, ear hemorrhage, ecchymosis, epistaxis, eye hemorrhage, gingival bleeding, hematoma, hematuria, hemoptysis, intra-abdominal hematoma, petechiae, scleral hemorrhage, uterine hemorrhage, and vaginal hemorrhage.
Fluid retention
38
45
5
1
   Pleural effusion
28
1
3
0
   Superficial localized edema
14
38
0
<1
   Pulmonary hypertension
5
<1
1
0
   Generalized edema
4
7
0
0
   Pericardial effusion
4
1
1
0
   Congestive heart failure/cardiac dysfunctiona
2
1
<1
<1
   Pulmonary edema
1
0
0
0
Diarrhea
22
23
1
1
Musculoskeletal pain
14
17
0
<1
Rashb
14
18
0
2
Headache
14
11
0
0
Abdominal pain
11
8
0
1
Fatigue
11
12
<1
0
Nausea
10
25
0
0
Myalgia
7
12
0
0
Arthralgia
7
10
0
<1
Hemorrhagec
8
8
1
1
   Gastrointestinal bleeding
2
2
1
0
   Other bleedingd
6
6
0
<1
   CNS bleeding
<1
<1
0
<1
Vomiting
5
12
0
0
Muscle spasms
5
21
0
<1
A comparison of cumulative rates of adverse reactions reported in ‚Č•10% of patients with minimum follow-up of 1 and 5 years in a randomized trial of newly diagnosed patients with chronic phase CML treated with SPRYCEL are shown in Table 7.
Table 7: Adverse Reactions Reported in ‚Č•10% of Adult Patients with Newly Diagnosed Chronic Phase CML in the SPRYCEL-Treated Arm (n=258) ¬† Minimum of 1 Year Follow-up Minimum of 5 Years Follow-up ¬† All Grades Grade 3/4 All Grades Grade 3/4 Adverse Reaction Percent (%) of Patients a Includes cardiac failure acute, cardiac failure congestive, cardiomyopathy, diastolic dysfunction, ejection fraction decreased, and left ventricular dysfunction. b Includes erythema, erythema multiforme, rash, rash generalized, rash macular, rash papular, rash pustular, skin exfoliation, and rash vesicular.
Fluid retention
19
1
38
5
   Pleural effusion
10
0
28
3
   Superficial localized edema
9
0
14
0
   Pulmonary hypertension
1
0
5
1
   Generalized edema
2
0
4
0
   Pericardial effusion
1
<1
4
1
   Congestive heart failure/cardiac      dysfunctiona
2
<1
2
<1
   Pulmonary edema
<1
0
1
0
Diarrhea
17
<1
22
1
Musculoskeletal pain
11
0
14
0
Rashb
11
0
14
0
Headache
12
0
14
0
Abdominal pain
7
0
11
0
Fatigue
8
<1
11
<1
Nausea
8
0
10
0
At 60 months, there were 26 deaths in dasatinib-treated patients (10.1%) and 26 deaths in imatinib-treated patients (10.1%); 1 death in each group was assessed by the investigator as related to study therapy.
Table 8: Adverse Reactions Reported in ‚Č•10% of Adult Patients with Chronic Phase CML Resistant or Intolerant to Prior Imatinib Therapy (minimum of 84 months follow-up) a Includes drug eruption, erythema, erythema multiforme, erythrosis, exfoliative rash, generalized erythema, genital rash, heat rash, milia, rash, rash erythematous, rash follicular, rash generalized, rash macular, rash maculopapular, rash papular, rash pruritic, rash pustular, skin exfoliation, skin irritation, urticaria vesiculosa, and rash vesicular.
 
100 mg Once Daily
 
Chronic (n=165)
Adverse Reaction
All Grades
Grade 3/4
Percent (%) of Patients
Fluid retention
48
7
   Superficial localized edema
22
0
   Pleural effusion
28
5
   Generalized edema
4
0
   Pericardial effusion
3
1
   Pulmonary hypertension
2
1
Headache
33
1
Diarrhea
28
2
Fatigue
26
4
Dyspnea
24
2
Musculoskeletal pain
22
2
Nausea
18
1
Skin rasha
18
2
Myalgia
13
0
Arthralgia
13
1
Infection (including bacterial, viral, fungal,    and non-specified)
13
1
Abdominal pain
12
1
Hemorrhage
12
1
   Gastrointestinal bleeding
2
1
Pruritus
12
1
Pain
11
1
Constipation
10
1
Cumulative rates of selected adverse reactions that were reported over time in patients treated with the 100 mg once daily recommended starting dose in a randomized dose-optimization trial of imatinib-resistant or -intolerant patients with chronic phase CML are shown in Table 9.
Table 9: Selected Adverse Reactions Reported in Adult Dose Optimization Trial (Imatinib-Intolerant or -Resistant Chronic Phase CML)a Minimum of 2 Years Follow-up Minimum of 5 Years Follow-up Minimum of 7 Years Follow-up Adverse Reaction All Grades Grade 3/4 All Grades Grade 3/4 All Grades Grade 3/4 Percent (%) of Patients a  Randomized dose-optimization trial results reported in the recommended starting dose of 100 mg once daily (n=165) population.
Diarrhea
27
2
28
2
28
2
Fluid retention
34
4
42
6
48
7
   Superficial edema
18
0
21
0
22
0
   Pleural effusion
18
2
24
4
28
5
   Generalized edema
3
0
4
0
4
0
   Pericardial effusion
2
1
2
1
3
1
   Pulmonary hypertension
0
0
0
0
2
1
Hemorrhage
11
1
11
1
12
1
   Gastrointestinal bleeding
2
1
2
1
2
1
Table 10: Adverse Reactions Reported in ‚Č•10% of Adult Patients with Advanced Phase CML Resistant or Intolerant to Prior Imatinib Therapy a Includes ventricular dysfunction, cardiac failure, cardiac failure congestive, cardiomyopathy, congestive cardiomyopathy, diastolic dysfunction, ejection fraction decreased, and ventricular failure. b Includes drug eruption, erythema, erythema multiforme, erythrosis, exfoliative rash, generalized erythema, genital rash, heat rash, milia, rash, rash erythematous, rash follicular, rash generalized, rash macular, rash maculopapular, rash papular, rash pruritic, rash pustular, skin exfoliation, skin irritation, urticaria vesiculosa, and rash vesicular.
140 mg Once Daily
Accelerated (n=157)
Myeloid Blast (n=74)
Lymphoid Blast (n=33)
Adverse Reaction
All Grades
Grade 3/4
All Grades
Grade 3/4
All Grades
Grade 3/4
Percent (%) of Patients
Fluid retention
35
8
34
7
21
6
   Superficial localized edema
18
1
14
0
3
0
   Pleural effusion
21
7
20
7
21
6
   Generalized edema
1
0
3
0
0
0
   Pericardial effusion
3
1
0
0
0
0
   Congestive heart failure/cardiac      dysfunctiona
0
0
4
0
0
0
   Pulmonary edema
1
0
4
3
0
0
Headache
27
1
18
1
15
3
Diarrhea
31
3
20
5
18
0
Fatigue
19
2
20
1
9
3
Dyspnea
20
3
15
3
3
3
Musculoskeletal pain
11
0
8
1
0
0
Nausea
19
1
23
1
21
3
Skin rashb
15
0
16
1
21
0
Arthralgia
10
0
5
1
0
0
Infection (including bacterial, viral, fungal,    and non-specified)
10
6
14
7
9
0
Hemorrhage
26
8
19
9
24
9
   Gastrointestinal bleeding
8
6
9
7
9
3
   CNS bleeding
1
1
0
0
3
3
Vomiting
11
1
12
0
15
0
Pyrexia
11
2
18
3
6
0
Febrile neutropenia
4
4
12
12
12
12
Table 11: Adverse Reactions Reported in ‚Č•10% of Dasatinib-Treated Pediatric Patients with Chronic Phase CML (n=97) All Grades Grade 3/4 Adverse Reaction Percent (%) of Patients
Headache
28
3
Nausea
20
0
Diarrhea
21
0
Skin rash
19
0
Vomiting
13
0
Pain in extremity
19
1
Abdominal pain
16
0
Fatigue
10
0
Arthralgia
10
1
Adverse reactions associated with bone growth and development were reported in 5 (5.2%) of pediatric patients with chronic phase CML [see Warnings and Precautions (5.10)].
Laboratory Abnormalities
Myelosuppression was commonly reported in all patient populations. The frequency of Grade 3 or 4 neutropenia, thrombocytopenia, and anemia was higher in patients with advanced phase CML than in chronic phase CML (Tables 12 and 13). Myelosuppression was reported in patients with normal baseline laboratory values as well as in patients with pre-existing laboratory abnormalities.
In patients who experienced severe myelosuppression, recovery generally occurred following dose interruption or reduction; permanent discontinuation of treatment occurred in 2% of adult patients with newly diagnosed chronic phase CML and 5% of adult patients with resistance or intolerance to prior imatinib therapy [see Warnings and Precautions (5.1)].
Grade 3 or 4 elevations of transaminases or bilirubin and Grade 3 or 4 hypocalcemia, hypokalemia, and hypophosphatemia were reported in patients with all phases of CML but were reported with an increased frequency in patients with myeloid or lymphoid blast phase CML. Elevations in transaminases or bilirubin were usually managed with dose reduction or interruption. Patients developing Grade 3 or 4 hypocalcemia during SPRYCEL therapy often had recovery with oral calcium supplementation.
Laboratory abnormalities reported in adult patients with newly diagnosed chronic phase CML are shown in Table 12. There were no discontinuations of SPRYCEL therapy in this patient population due to biochemical laboratory parameters.
Table 12: CTC Grade 3/4 Laboratory Abnormalities in Adult Patients with Newly Diagnosed Chronic Phase CML (minimum of 60 months follow-up) SPRYCEL (n=258) Imatinib (n=258) ¬† Percent (%) of Patients CTC grades: neutropenia (Grade 3 ‚Č•0.5‚Äď<1.0 √ó 109/L, Grade 4 <0.5 √ó 109/L); thrombocytopenia (Grade 3 ‚Č•25‚Äď<50 √ó 109/L, Grade 4 <25 √ó 109/L); anemia (hemoglobin Grade 3 ‚Č•65‚Äď<80 g/L, Grade 4 <65 g/L); elevated creatinine (Grade 3 >3‚Äď6 √ó upper limit of normal range (ULN), Grade 4 >6 √ó ULN); elevated bilirubin (Grade 3 >3‚Äď10 √ó ULN, Grade 4 >10 √ó ULN); elevated SGOT or SGPT (Grade 3 >5‚Äď20 √ó ULN, Grade 4 >20 √ó ULN); hypocalcemia (Grade 3 <7.0‚Äď6.0 mg/dL, Grade 4 <6.0 mg/dL); hypophosphatemia (Grade 3 <2.0‚Äď1.0 mg/dL, Grade 4 <1.0 mg/dL); hypokalemia (Grade 3 <3.0‚Äď2.5 mmol/L, Grade 4 <2.5¬†mmol/L).
Hematology Parameters
 
 
   Neutropenia
29
24
   Thrombocytopenia
22
14
   Anemia
13
9
Biochemistry Parameters
 
 
   Hypophosphatemia
7
31
   Hypokalemia
0
3
   Hypocalcemia
4
3
   Elevated SGPT (ALT)
<1
2
   Elevated SGOT (AST)
<1
1
   Elevated Bilirubin
1
0
   Elevated Creatinine
1
1
Laboratory abnormalities reported in patients with CML resistant or intolerant to imatinib who received the recommended starting doses of SPRYCEL are shown by disease phase in Table 13.
Table 13: CTC Grade 3/4 Laboratory Abnormalities in Clinical Studies of CML in Adults: Resistance or Intolerance to Prior Imatinib Therapy CTC grades: neutropenia (Grade 3 ‚Č•0.5‚Äď<1.0 √ó 109/L, Grade 4 <0.5 √ó 109/L); thrombocytopenia (Grade 3 ‚Č•25‚Äď<50 √ó 109/L, Grade 4 <25 √ó 109/L); anemia (hemoglobin Grade 3 ‚Č•65‚Äď<80 g/L, Grade 4 <65 g/L); elevated creatinine (Grade 3 >3‚Äď6 √ó upper limit of normal range (ULN), Grade 4 >6 √ó ULN); elevated bilirubin (Grade 3 >3‚Äď10 √ó ULN, Grade 4 >10 √ó ULN); elevated SGOT or SGPT (Grade 3 >5‚Äď20 √ó ULN, Grade 4 >20 √ó ULN); hypocalcemia (Grade 3 <7.0‚Äď6.0 mg/dL, Grade 4 <6.0 mg/dL); hypophosphatemia (Grade 3 <2.0‚Äď1.0 mg/dL, Grade 4 <1.0 mg/dL); hypokalemia (Grade 3 <3.0‚Äď2.5 mmol/L, Grade 4 <2.5 mmol/L).*¬†¬†Hematology parameters for 100 mg once-daily dosing in chronic phase CML reflects 60-month minimum follow-up.
Chronic Phase CML100 mg Once Daily
Advanced Phase CML140 mg Once Daily
Accelerated Phase
Myeloid Blast Phase
Lymphoid Blast Phase
(n=165)
(n=157)
(n=74)
(n=33)
 
Percent (%) of Patients
Hematology Parameters*
 
 
 
 
   Neutropenia
36
58
77
79
   Thrombocytopenia
24
63
78
85
   Anemia
13
47
74
52
Biochemistry Parameters
 
 
 
 
   Hypophosphatemia
10
13
12
18
   Hypokalemia
2
7
11
15
   Hypocalcemia
<1
4
9
12
   Elevated SGPT (ALT)
0
2
5
3
   Elevated SGOT (AST)
<1
0
4
3
   Elevated Bilirubin
<1
1
3
6
   Elevated Creatinine
0
2
8
0
Among adult patients with chronic phase CML with resistance or intolerance to prior imatinib therapy, cumulative Grade 3 or 4 cytopenias were similar at 2 and 5 years including: neutropenia (36% vs 36%), thrombocytopenia (23% vs 24%), and anemia (13% vs 13%).
In the pediatric studies in CML, the rates of laboratory abnormalities were consistent with the known profile for laboratory parameters in adults.
A total of 135 adult patients with Ph+¬†ALL were treated with SPRYCEL in clinical studies. The median duration of treatment was 3 months (range 0.03‚Äď31 months). The safety profile of patients with Ph+¬†ALL was similar to those with lymphoid blast phase CML. The most frequently reported adverse reactions included fluid retention events, such as pleural effusion (24%) and superficial edema (19%), and gastrointestinal disorders, such as diarrhea (31%), nausea (24%), and vomiting (16%). Hemorrhage (19%), pyrexia (17%), rash (16%), and dyspnea (16%) were also frequently reported. Serious adverse reactions reported in ‚Č•5% of patients included pleural effusion (11%), gastrointestinal bleeding (7%), febrile neutropenia (6%), and infection (5%).
The safety of SPRYCEL administered continuously in combination with multiagent chemotherapy was determined in a multicohort study of 81 pediatric patients with newly diagnosed Ph+ ALL. [seeClinical Studies (14.4)]. The median duration of therapy was 24 months (range 2 to 27 months).
Fatal adverse reactions occurred in 3 patients (4%), all of which were due to infections. Eight (10%) patients experienced adverse reactions leading to treatment discontinuation, including fungal sepsis, hepatotoxicity in the setting of graft versus host disease, thrombocytopenia, CMV infection, pneumonia, nausea, enteritis and drug hypersensitivity.
The most common serious adverse reactions (incidence ‚Č•10%) were pyrexia, febrile neutropenia, mucositis, diarrhea, sepsis, hypotension, infections (bacterial, viral and fungal), hypersensitivity, vomiting, renal insufficiency, abdominal pain, and musculoskeletal pain.
The incidence of common adverse reactions (incidence ‚Č•20%) on study are shown in Table 14:
Table 14: Adverse Reactions Reported in ‚Č•20% of Pediatric Patients with Ph+ ALL Treated with SPRYCEL in Combination with Chemotherapy CA180372 (N=81)
Percent (%) of Patients
Adverse Reaction
All Grades
Grade 3/4
Mucositis
93
60
Febrile neutropenia
86
86
Pyrexia
85
17
Diarrhea
84
31
Nausea
84
11
Vomiting
83
17
Musculoskeletal pain
83
25
Abdominal pain
78
17
Cough
78
1
Headache
77
15
Rash
68
7
Fatigue
59
3
Constipation
57
1
Arrhythmia
47
12
Hypertension
47
10
Edema
47
6
Viral infection
40
12
Hypotension
40
26
Decreased appetite
38
22
Hypersensitivity
36
20
Upper respiratory tract infection
36
10
Dyspnea
35
10
Epistaxis
31
6
Peripheral neuropathy
31
7
Sepsis (excluding fungal)
n/a
31
Altered state of consciousness
30
4
Fungal infection
30
11
Pneumonia (excluding fungal)
28
25
Pruritus
28
-
Clostridial infection (excluding sepsis)
25
14
Urinary Tract Infection
24
14
Bacteremia (excluding fungal)
22
20
Erythema
22
6
Chills
21
-
Pleural effusion
21
9
Sinusitis
21
10
Dehydration
20
9
Renal insufficiency
20
9
Visual impairment
20
-
The incidence of common adverse reactions attributed by the investigator to SPRYCEL (reported at a frequency of ‚Č•10%, all grades and grade 3/4, respectively) on study (N=81), included febrile neutropenia (23%, 23%), nausea (21%, 4%), vomiting (19%, 4%), mucositis (17%, 6%), musculoskeletal pain (17%, 2%), abdominal pain (16%, 5%), diarrhea (16%, 7%), rash (15%, 0%), fatigue (12%, 0%), pyrexia (12%, 6%), and headache (12%, 5%).
CTCAE grade 3/4 laboratory abnormalities in pediatric patients with Ph+ ALL treated with SPRYCEL in combination with chemotherapy are shown in Table 15.
Table 15: CTCAE Grade 3/4 Laboratory Abnormalities in ‚Č•10% of Pediatric Patients with Ph+ ALL Treated with SPRYCEL in Combination with Chemotherapy CA180372 (N=81) Percent (%) of Patients
Hematology Parameters
     Neutropenia
96
     Thrombocytopenia
88
     Anemia
82
Biochemistry Parameters
     Elevated SGPT (ALT)
47
     Hypokalemia
40
     Elevated SGOT (AST)
26
     Hypocalcemia
19
     Hyponatremia
19
     Elevated Bilirubin
11
     Hypophosphatemia
11
Toxicity grading is per CTCAE version 4.
Additional Pooled Data from Clinical Trials
The following additional adverse reactions were reported in adult and pediatric patients (n=2809) in SPRYCEL CML clinical studies and adult patients in Ph+ ALL clinical studies at a frequency of ‚Č•10%, 1%‚Äst<10%, 0.1%‚Äst<1%, or <¬†0.1%. These adverse reactions are included based on clinical relevance.
Gastrointestinal Disorders: 1%‚Äď<10% ‚Äď mucosal inflammation (including mucositis/stomatitis), dyspepsia, abdominal distension, constipation, gastritis, colitis (including neutropenic colitis), oral soft tissue disorder; 0.1%‚Äď<1% ‚Äď ascites, dysphagia, anal fissure, upper gastrointestinal ulcer, esophagitis, pancreatitis, gastroesophageal reflux disease; <0.1% ‚Äď protein losing gastroenteropathy, ileus, acute pancreatitis, anal fistula.
General Disorders and Administration-Site Conditions: ¬†‚Č•10% ‚Äď peripheral edema, face edema; 1%‚Äď<10% ‚Äď asthenia, chest pain, chills; 0.1%‚Äď<1% ‚Äď malaise, other superficial edema, peripheral swelling; <0.1% ‚Äď gait disturbance.
Skin and Subcutaneous Tissue Disorders: 1%‚Äď<10% ‚Äď alopecia, acne, dry skin, hyperhidrosis, urticaria, dermatitis (including eczema); 0.1%‚Äď<1% ‚Äď pigmentation disorder, skin ulcer, bullous conditions, photosensitivity, nail disorder, neutrophilic dermatosis, panniculitis, palmar-plantar erythrodysesthesia syndrome, hair disorder; <0.1% ‚Äď leukocytoclastic vasculitis, skin fibrosis.
Respiratory, Thoracic, and Mediastinal Disorders: 1%‚Äď<10% ‚Äď lung infiltration, pneumonitis, cough; 0.1%‚Äst<1% ¬†‚Äď ¬†asthma, bronchospasm, dysphonia, pulmonary arterial hypertension; <0.1% ‚Äď acute respiratory distress syndrome, pulmonary embolism.
Nervous System Disorders: 1%‚Äď<10% ‚Äď neuropathy (including peripheral neuropathy), dizziness, dysgeusia, somnolence; 0.1%‚Äď<1% ‚Äď amnesia, tremor, syncope, balance disorder; <0.1% ‚Äď convulsion, cerebrovascular accident, transient ischemic attack, optic neuritis, VIIth nerve paralysis, dementia, ataxia.
Blood and Lymphatic System Disorders: 0.1%‚Äď<1% ‚Äď lymphadenopathy, lymphopenia; <0.1% ‚Äď aplasia pure red cell.
Musculoskeletal and Connective Tissue Disorders: 1%‚Äď<10% ‚Äď muscular weakness, musculoskeletal stiffness; 0.1%‚Äď<1% ‚Äď rhabdomyolysis, tendonitis, muscle inflammation, osteonecrosis, arthritis; <0.1% ‚Äď epiphyses delayed fusion (reported at 1%‚Äď<10% in the pediatric studies), growth retardation (reported at 1%‚Äď<10% in the pediatric studies).
Investigations: 1%‚Äď<10% ‚Äď weight increased, weight decreased; 0.1%‚Äď<1% ‚Äď blood creatine phosphokinase increased, gamma-glutamyltransferase increased.
Infections and Infestations: 1%‚Äď<10% ‚Äď pneumonia (including bacterial, viral, and fungal), upper respiratory tract infection/inflammation, herpes virus infection, enterocolitis infection, sepsis (including fatal outcomes [0.2%]).
Metabolism and Nutrition Disorders: 1%‚Äď<10% ‚Äď appetite disturbances, hyperuricemia; 0.1%‚Äď<1% ‚Äď hypoalbuminemia, tumor lysis syndrome, dehydration, hypercholesterolemia; <0.1% ‚Äď diabetes mellitus.
Cardiac Disorders: 1%‚Äď<10% ‚Äď arrhythmia (including tachycardia), palpitations; 0.1%‚Äď<1% ‚Äď angina pectoris, cardiomegaly, pericarditis, ventricular arrhythmia (including ventricular tachycardia), electrocardiogram T-wave abnormal, troponin increased; <0.1% ‚Äď cor pulmonale, myocarditis, acute coronary syndrome, cardiac arrest, electrocardiogram PR prolongation, coronary artery disease, pleuropericarditis.
Eye Disorders: 1%‚Äď<10% ‚Äď visual disorder (including visual disturbance, vision blurred, and visual acuity reduced), dry eye; 0.1%‚Äď<1% ‚Äď conjunctivitis, visual impairment, lacrimation increased, <0.1% ‚Äď photophobia.
Vascular Disorders: 1%‚Äď<10% ‚Äď flushing, hypertension; 0.1%‚Äď<1% ‚Äď hypotension, thrombophlebitis, thrombosis; <0.1% ‚Äď livedo reticularis, deep vein thrombosis, embolism.
Psychiatric Disorders: 1%‚Äď<10% ‚Äď insomnia, depression; 0.1%‚Äď<1% ‚Äď anxiety, affect lability, confusional state, libido decreased.
Pregnancy, Puerperium, and Perinatal Conditions: <0.1% ‚Äď abortion.
Reproductive System and Breast Disorders: 0.1%‚Äď<1% ‚Äď gynecomastia, menstrual disorder.
Injury, Poisoning, and Procedural Complications: 1%‚Äď<10% ‚Äď contusion.
Ear and Labyrinth Disorders: 1%‚Äď<10% ‚Äď tinnitus; 0.1%‚Äď<1% ‚Äď vertigo, hearing loss.
Hepatobiliary Disorders: 0.1%‚Äď<1% ‚Äď cholestasis, cholecystitis, hepatitis.
Renal and Urinary Disorders: 0.1%‚Äď<1% ‚Äď urinary frequency, renal failure, proteinuria; <0.1% ‚Äď renal impairment.
Immune System Disorders: 0.1%‚Äď<1% ‚Äď hypersensitivity (including erythema nodosum).
Endocrine Disorders: 0.1%‚Äď<1% ‚Äď hypothyroidism; <0.1% ‚Ästhyperthyroidism, thyroiditis.
6.2 Postmarketing Experience
The following additional adverse reactions have been identified during post approval use of SPRYCEL. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Infections: hepatitis B virus reactivation
Cardiac disorders: atrial fibrillation/atrial flutter
Respiratory, thoracic, and mediastinal disorders: interstitial lung disease, chylothorax
Skin and subcutaneous tissue disorders: Stevens-Johnson syndrome
Renal and urinary disorders: nephrotic syndrome
Blood and lymphatic system disorders: thrombotic microangiopathy
Hepatobiliary disorders: hepatotoxicity
7 Drug Interactions
‚ÄĘ Strong CYP3A4 Inhibitors: Dose reduction may be necessary.(2.3 ,7.1) ‚ÄĘ Strong CYP3A4 Inducers: Dose increase may be necessary.(2.3 ,7.1) ‚ÄĘ Antacids: Avoid simultaneous administration.(7.1) ‚ÄĘ H2 Antagonists and Proton Pump Inhibitors: Avoid coadministration.(7.1) 7.1 Effect of Other Drugs on Dasatinib
Strong CYP3A4 Inhibitors The coadministration with strong CYP3A inhibitors may increase dasatinib concentrations [see Clinical Pharmacology (12.3)]. Increased dasatinib concentrations may increase the risk of toxicity. Avoid concomitant use of strong CYP3A4 inhibitors. If concomitant administration of a strong CYP3A4 inhibitor cannot be avoided, consider a SPRYCEL dose reduction [see Dosage and Administration (2.5) ].
Strong CYP3A4 Inducers The coadministration of SPRYCEL with strong CYP3A inducers may decrease dasatinib concentrations [see Clinical Pharmacology (12.3)]. Decreased dasatinib concentrations may reduce efficacy. Consider alternative drugs with less enzyme induction potential. If concomitant administration of a strong CYP3A4 inducer cannot be avoided, consider a SPRYCEL dose increase.
Gastric Acid Reducing Agents The coadministration of SPRYCEL with a gastric acid reducing agent may decrease the concentrations of dasatinib. Decreased dasatinib concentrations may reduce efficacy.
Do not administer H2 antagonists or proton pump inhibitors with SPRYCEL. Consider the use of antacids in place of H2 antagonists or proton pump inhibitors. Administer the antacid at least 2 hours prior to or 2 hours after the dose of SPRYCEL. Avoid simultaneous administration of SPRYCEL with antacids.
8 Use In Specific Populations
‚ÄĘ Lactation: Advise women not to breastfeed.(8.2) 8.1 Pregnancy
Based on limited human data, SPRYCEL can cause fetal harm when administered to a pregnant woman. Adverse pharmacologic effects including hydrops fetalis, fetal leukopenia, and fetal thrombocytopenia have been reported with maternal exposure to SPRYCEL. Animal reproduction studies in rats have demonstrated extensive mortality during organogenesis, the fetal period, and in neonates. Skeletal malformations were observed in a limited number of surviving rat and rabbit conceptuses. These findings occurred at dasatinib plasma concentrations below those in humans receiving therapeutic doses of dasatinib [see Data]. Advise a pregnant woman of the potential risk to a fetus.
The estimated background risk in the U.S. general population of major birth defects is 2% to 4% and of miscarriage is 15% to 20% of clinically recognized pregnancies.
Fetal/Neonatal Adverse Reactions
Transplacental transfer of dasatinib has been reported. Dasatinib has been measured in fetal plasma and amniotic fluid at concentrations comparable to those in maternal plasma. Hydrops fetalis, fetal leukopenia, and fetal thrombocytopenia have been reported with maternal exposure to dasatinib. These adverse pharmacologic effects on the fetus are similar to adverse reactions observed in adult patients and may result in fetal harm or neonatal death [see Warnings and Precautions (5.1, 5.3)].
Human Data
Based on human experience, dasatinib is suspected to cause congenital malformations, including neural tube defects, and harmful pharmacological effects on the fetus when administered during pregnancy.
Animal Data
In nonclinical studies at plasma concentrations below those observed in humans receiving therapeutic doses of dasatinib, embryo-fetal toxicities were observed in rats and rabbits. Fetal death was observed in rats. In both rats and rabbits, the lowest doses of dasatinib tested (rat: 2.5 mg/kg/day [15 mg/m2/day] and rabbit: 0.5 mg/kg/day [6 mg/m2/day]) resulted in embryo-fetal toxicities. These doses produced maternal AUCs of 105 ng‚ÄĘh/mL and 44 ng‚ÄĘh/mL (0.1-fold the human AUC) in rats and rabbits, respectively. Embryo-fetal toxicities included skeletal malformations at multiple sites (scapula, humerus, femur, radius, ribs, and clavicle), reduced ossification (sternum; thoracic, lumbar, and sacral vertebrae; forepaw phalanges; pelvis; and hyoid body), edema, and microhepatia. In a pre- and postnatal development study in rats, administration of dasatinib from gestation day (GD) 16 through lactation day (LD) 20, GD 21 through LD 20, or LD 4 through LD 20 resulted in extensive pup mortality at maternal exposures that were below the exposures in patients treated with dasatinib at the recommended labeling dose.
8.2 Lactation
No data are available regarding the presence of dasatinib in human milk, the effects of the drug on the breastfed child, or the effects of the drug on milk production. However, dasatinib is present in the milk of lactating rats. Because of the potential for serious adverse reactions in nursing children from SPRYCEL, breastfeeding is not recommended during treatment with SPRYCEL and for 2 weeks after the last dose.
8.3 Females and Males of Reproductive Potential
SPRYCEL can cause fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)].
Advise females of reproductive potential and males with female partners of reproductive potential to use effective contraception during treatment with SPRYCEL and for 30 days after the last dose.
Based on animal data, dasatinib may result in damage to female and male reproductive tissues [see Nonclinical Toxicology (13.1)].
8.4 Pediatric Use
Ph+ CML in Chronic Phase
The safety and effectiveness of SPRYCEL monotherapy have been demonstrated in pediatric patients with newly diagnosed chronic phase CML [see Clinical Studies (14.3)]. There are no data in children under 1 year of age. Adverse reactions associated with bone growth and development were reported in 5 (5.2%) of patients [see Warnings and Precautions (5.10)].
Ph+ ALL
The safety and effectiveness of SPRYCEL in combination with chemotherapy have been demonstrated in pediatric patients one year and over with newly diagnosed Ph+ ALL. Use of SPRYCEL in pediatric patients is supported by evidence from one pediatric study. There are no data in children under 1 year of age. One case of grade 1 osteopenia was reported.
The safety profile of SPRYCEL in pediatric subjects was comparable to that reported in studies in adult subjects [see Adverse Reactions (6.1) and Clinical Studies (14.3, 14.4)].
Monitor bone growth and development in pediatric patients [see Warnings and Precautions (5.10)].
Pediatric Patients with Difficulty Swallowing Tablets
Five patients with Ph+ ALL 2 to 10 years of age received at least one dose of SPRYCEL tablet dispersed in juice on Study CA180372. The exposure for dispersed tablets was 36% lower as compared to intact tablets in pediatric patients [see Clinical Pharmacology (12.3)]. Due to the limited available clinical data, it is unclear whether dispersing SPRYCEL tablets significantly alters the safety and/or efficacy of SPRYCEL.
8.5 Geriatric Use
Of the 2712 patients in clinical studies of SPRYCEL, 617 (23%) were 65 years of age and older, and 123 (5%) were 75 years of age and older. No differences in confirmed Complete Cytogenetic Response (cCCyR) and MMR were observed between older and younger patients. While the safety profile of SPRYCEL in the geriatric population was similar to that in the younger population, patients aged 65 years and older are more likely to experience the commonly reported adverse reactions of fatigue, pleural effusion, diarrhea, dyspnea, cough, lower gastrointestinal hemorrhage, and appetite disturbance, and more likely to experience the less frequently reported adverse reactions of abdominal distention, dizziness, pericardial effusion, congestive heart failure, hypertension, pulmonary edema, and weight decrease, and should be monitored closely.
10 Overdosage
Experience with overdose of SPRYCEL in clinical studies is limited to isolated cases. The highest overdosage of 280 mg per day for 1 week was reported in two patients and both developed severe myelosuppression and bleeding. Since SPRYCEL is associated with severe myelosuppression [see Warnings and Precautions (5.1) and Adverse Reactions (6.1)], monitor patients who ingest more than the recommended dosage closely for myelosuppression and give appropriate supportive treatment.
Acute overdose in animals was associated with cardiotoxicity. Evidence of cardiotoxicity included ventricular necrosis and valvular/ventricular/atrial hemorrhage at single doses ‚Č•100 mg/kg (600 mg/m2) in rodents. There was a tendency for increased systolic and diastolic blood pressure in monkeys at single doses ‚Č•10 mg/kg (120 mg/m2).
11 Description
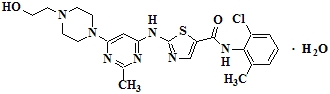
SPRYCEL (dasatinib) is a kinase inhibitor. The chemical name for dasatinib is N-(2-chloro-6-methylphenyl)-2-[[6-[4-(2-hydroxyethyl)-1-piperazinyl]-2-methyl-4-pyrimidinyl]amino]-5-thiazolecarboxamide, monohydrate. The molecular formula is C22H26ClN7O2S ‚ÄĘ H2O, which corresponds to a formula weight of 506.02 (monohydrate). The anhydrous free base has a molecular weight of 488.01. Dasatinib has the following chemical structure:
Dasatinib is a white to off-white powder. The drug substance is insoluble in water and slightly soluble in ethanol and methanol.
SPRYCEL tablets are white to off-white, biconvex, film-coated tablets containing dasatinib, with the following inactive ingredients: lactose monohydrate, microcrystalline cellulose, croscarmellose sodium, hydroxypropyl cellulose, and magnesium stearate. The tablet coating consists of hypromellose, titanium dioxide, and polyethylene glycol.
12 Clinical Pharmacology
12.1 Mechanism of Action
Dasatinib, at nanomolar concentrations, inhibits the following kinases: BCR-ABL, SRC family (SRC, LCK, YES, FYN), c-KIT, EPHA2, and PDGFRő≤. Based on modeling studies, dasatinib is predicted to bind to multiple conformations of the ABL kinase.
In vitro, dasatinib was active in leukemic cell lines representing variants of imatinib mesylate-sensitive and resistant disease. Dasatinib inhibited the growth of chronic myeloid leukemia (CML) and acute lymphoblastic leukemia (ALL) cell lines overexpressing BCR-ABL. Under the conditions of the assays, dasatinib could overcome imatinib resistance resulting from BCR-ABL kinase domain mutations, activation of alternate signaling pathways involving the SRC family kinases (LYN, HCK), and multi-drug resistance gene overexpression.
12.2 Pharmacodynamics
Cardiac Electrophysiology
Of 2440 patients treated with SPRYCEL at all doses tested in clinical trials, 16 patients (<1%) had QTc prolongation reported as an adverse reaction. Twenty-two patients (1%) experienced a QTcF > 500 ms. In 865 patients with leukemia treated with SPRYCEL 70 mg BID in five Phase 2 studies, the maximum mean changes in QTcF (90% upper bound CI) from baseline ranged from 7 ms to 13.4 ms.
An analysis of the data from five Phase 2 studies in patients (70 mg BID) and a Phase 1 study in healthy subjects (100 mg single dose) suggests that there is a maximum increase of 3 to 6 milliseconds in Fridericia corrected QTc interval from baseline for subjects receiving therapeutic doses of dasatinib, with associated upper 95% confidence intervals <10 msec.
12.3 Pharmacokinetics
The pharmacokinetics of dasatinib exhibits dose proportional increases in AUC and linear elimination characteristics over the dose range of 15 mg/day (0.15 times the lowest approved recommended dose) to 240 mg/day (1.7 times the highest approved recommended dose).
At 100 mg QD, the maximum concentration at steady state (Cmax) is 82.2 ng/mL (CV% 69%), area under the plasma drug concentration time curve (AUC) is 397 ng/mL*hr (CV% 55%). The clearance of dasatinib is found to be time-invariant. When administered to adult healthy subjects as dispersed tablets in juice, the adjusted geometric mean ratio was 0.97 (90% CI: 0.85, 1.10) for Cmax and 0.84 (90% CI: 0.78, 0.91) for AUC as compared to intact tablets.
The maximum plasma concentrations (Cmax) of dasatinib are observed between 0.5 hours and 6 hours (Tmax) following oral administration.
Food Effect
A high-fat meal increased the mean AUC of dasatinib following a single dose of 100 mg by 14%. The total calorie content of the high-fat meal was 985 kcal. The calories derived from fat, carbohydrates, and protein were 52%, 34%, and 14% for the high-fat meal.
The apparent volume of distribution is 2505 L (CV% 93%).
Binding of dasatinib to human plasma proteins in vitro was approximately 96% and of its active metabolite was 93%, with no concentration dependence over the range of 100 ng/mL to 500 ng/mL.
Dasatinib is a P-gp substrate in vitro.
Elimination
The mean terminal half-life of dasatinib is 3 hours to 5 hours. The mean apparent oral clearance is 363.8 L/hr (CV% 81.3%).
Dasatinib is metabolized in humans, primarily by CYP3A4. CYP3A4 is the primary enzyme responsible for the formation of the active metabolite. Flavin-containing monooxygenase 3 (FMO-3) and uridine diphosphate-glucuronosyltransferase (UGT) enzymes are also involved in the formation of dasatinib metabolites.
The exposure of the active metabolite, which is equipotent to dasatinib, represents approximately 5% of the AUC of dasatinib. The active metabolite of dasatinib is unlikely to play a major role in the observed pharmacology of the drug. Dasatinib also has several other inactive oxidative metabolites.
Elimination is primarily via the feces. Following a single radiolabeled dose of oral dasatinib, 4% of the administered radioactivity was recovered in the urine and 85% in the feces within 10 days. Unchanged dasatinib accounted for 0.1% of the administered dose in the urine and 19% of the administered dose in the feces with the remainder of the dose being metabolites.
Age (15 to 86 years old), sex, and renal impairment (creatinine clearance 21.6 mL/min to 342.3 mL/min as estimated by Cockcroft Gault) have no clinically relevant effect on the pharmacokinetics of dasatinib.
Pediatric Patients
The pharmacokinetics of dasatinib were evaluated in 43 pediatric patients with leukemia or solid tumors at oral doses ranging from 60 mg/m2 to 120 mg/m2 once daily, taken with or without food. The pharmacokinetics showed dose proportionality with a dose-related increase in exposure. The mean Tmax was observed between 0.5 hours and 6 hours and the mean half-life was 2 hours to 5 hours. The geometric mean (CV%) of body weight normalized clearance in these 43 pediatric patients is 5.98 (41.5%) L/h/kg. In pediatric patients with a dosing regimen of 60 mg/m2, the model simulated geometric mean (CV%) steady-state plasma average concentrations of dasatinib were 14.7 (64.6%) ng/mL (for 2 to <6 years old), 16.3 (97.5%) ng/mL (for 6 to <12 years old), and 18.2 (67.7%) ng/mL (for 12 years and older) [see Dosage and Administration (2.2)]. Dasatinib clearance and volume of distribution change with body weight in pediatric patients. Dasatinib has not been studied in patients < 1 year old.
The bioavailability of dispersed tablets in pediatric patients was estimated to be 36% lower than that of intact tablets.
Patients with Hepatic Impairment
Compared to subjects with normal liver function, patients with moderate hepatic impairment (Child Pugh B) had decreases in mean Cmax by 47% and mean AUC by 8%. Patients with severe hepatic impairment (Child Pugh C) had decreases in mean Cmax by 43% and in mean AUC by 28% compared to the subjects with normal liver function.
Drug Interaction Studies
Cytochrome P450 Enzymes
The coadministration of ketoconazole (strong CYP3A4 inhibitor) twice daily increased the mean Cmax of dasatinib by 4-fold and the mean AUC of dasatinib by 5-fold following a single oral dose of 20 mg.
The coadministration of rifampin (strong CYP3A4 inducer) once daily decreased the mean Cmax of dasatinib by 81% and the mean AUC of dasatinib by 82%.
Dasatinib is a time-dependent inhibitor of CYP3A4. Dasatinib does not inhibit CYP1A2, 2A6, 2B6, 2C8, 2C9, 2C19, 2D6, or 2E1. Dasatinib does not induce CYP enzymes.
Gastric Acid Reducing Agents
The administration of 30 mL of aluminum hydroxide/magnesium hydroxide 2 hours prior to a single dose of SPRYCEL was associated with no relevant change in the mean AUC of dasatinib; however, the mean Cmax of dasatinib was increased by 26%.
The simultaneous administration of 30 mL of aluminum hydroxide/magnesium hydroxide with a single dose of SPRYCEL was associated with a 55% reduction in the mean AUC of dasatinib and a 58% reduction in the mean Cmax of dasatinib.
The administration of a single dose of SPRYCEL 10 hours following famotidine (H2 antagonist) reduced the mean AUC of dasatinib by 61% and the mean Cmax of dasatinib by 63%.
The administration of a single 100 mg dose of SPRYCEL 22 hours following a 40 mg dose of omeprazole (proton pump inhibitor) at steady state reduced the mean AUC of dasatinib by 43% and the mean Cmax of dasatinib by 42%.
Transporters
Dasatinib is not an inhibitor of P-gp in vitro.
13 Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
In a 2-year carcinogenicity study, rats were administered oral doses of dasatinib at 0.3, 1, and 3 mg/kg/day. The highest dose resulted in a plasma drug exposure (AUC) level approximately 60% of the human exposure at 100 mg once daily. Dasatinib induced a statistically significant increase in the combined incidence of squamous cell carcinomas and papillomas in the uterus and cervix of high-dose females and prostate adenoma in low-dose males.
Dasatinib was clastogenic when tested in vitro in Chinese hamster ovary cells, with and without metabolic activation. Dasatinib was not mutagenic when tested in an in vitro bacterial cell assay (Ames test) and was not genotoxic in an in vivo rat micronucleus study.
Dasatinib did not affect mating or fertility in male and female rats at plasma drug exposure (AUC) similar to the human exposure at 100 mg daily. In repeat dose studies, administration of dasatinib resulted in reduced size and secretion of seminal vesicles, and immature prostate, seminal vesicle, and testis. The administration of dasatinib resulted in uterine inflammation and mineralization in monkeys, and cystic ovaries and ovarian hypertrophy in rodents.
14 Clinical Studies
14.1 Newly Diagnosed Chronic Phase CML in Adults
DASISION (Dasatinib vs Imatinib Study in Treatment-Naive Chronic Myeloid Leukemia Patients) (NCT00481247) was an open-label, multicenter, international, randomized trial conducted in adult patients with newly diagnosed chronic phase CML. A total of 519 patients were randomized to receive either SPRYCEL 100 mg once daily or imatinib 400 mg once daily. Patients with a history of cardiac disease were included in this trial except those who had a myocardial infarction within 6 months, congestive heart failure within 3 months, significant arrhythmias, or QTc prolongation. The primary endpoint was the rate of confirmed complete cytogenetic response (CCyR) within 12 months. Confirmed CCyR was defined as a CCyR noted on two consecutive occasions (at least 28 days apart).
Median age was 46 years in the SPRYCEL group and 49 years in the imatinib groups, with 10% and 11% of patients ‚Č•65 years of age, respectively. There were slightly more male than female patients in both groups (59% vs 41%). Fifty-three percent of all patients were Caucasian and 39% were Asian. At baseline, the distribution of Hasford scores was similar in the SPRYCEL and imatinib treatment groups (low risk: 33% and 34%; intermediate risk: 48% and 47%; high risk: 19% and 19%, respectively). With a minimum of 12 months follow-up, 85% of patients randomized to SPRYCEL and 81% of patients randomized to imatinib were still on study.
With a minimum of 24 months follow-up, 77% of patients randomized to SPRYCEL and 75% of patients randomized to imatinib were still on study and with a minimum of 60 months follow-up, 61% and 62% of patients, respectively, were still on treatment at the time of study closure.
Efficacy results are summarized in Table 16.
Table 16: Efficacy Results in a Randomized Newly Diagnosed Chronic Phase CML Trial SPRYCEL (n=259) Imatinib (n=260) a Confirmed CCyR is defined as a CCyR noted on two consecutive occasions at least 28 days apart. b Major molecular response (at any time) was defined as BCR-ABL ratios ‚ȧ0.1% by RQ-PCR in peripheral blood samples standardized on the International scale. These are cumulative rates representing minimum follow up for the time frame specified.* Adjusted for Hasford score and indicated statistical significance at a pre-defined nominal level of significance.CI = confidence interval.
Confirmed CCyRa
   Within 12 months (95% CI)
76.8% (71.2‚Äď81.8)
66.2% (60.1‚Äď71.9)
   P-value
0.007*
Major Molecular Responseb
   12 months (95% CI)
52.1% (45.9‚Äď58.3)
33.8% (28.1‚Äď39.9)
   P-value
<0.0001
   60 months (95% CI)
76.4% (70.8‚Äď81.5)
64.2% (58.1‚Äď70.1)
The confirmed CCyR within 24, 36, and 60 months for SPRYCEL versus imatinib arms were 80% versus 74%, 83% versus 77%, and 83% versus 79%, respectively. The MMR at 24 and 36 months for SPRYCEL versus imatinib arms were 65% versus 50% and 69% versus 56%, respectively.
After 60 months follow-up, median time to confirmed CCyR was 3.1 months in 215 SPRYCEL responders and 5.8 months in 204 imatinib responders. Median time to MMR after 60 months follow-up was 9.3 months in 198 SPRYCEL responders and 15.0 months in 167 imatinib responders.
At 60 months, 8 patients (3%) on the dasatinib arm progressed to either accelerated phase or blast crisis while 15 patients (6%) on the imatinib arm progressed to either accelerated phase or blast crisis.
The estimated 60-month survival rates for SPRYCEL- and imatinib-treated patients were 90.9% (CI: 86.6%‚Äď93.8%) and 89.6% (CI: 85.2%‚Äď92.8%), respectively. Based on data 5 years after the last patient was enrolled in the trial, 83% and 77% of patients were known to be alive in the dasatinib and imatinib treatment groups, respectively, 10% were known to have died in both treatment groups, and 7% and 13% had unknown survival status in the dasatinib and imatinib treatment groups, respectively.
At 60 months follow-up in the SPRYCEL arm, the rate of MMR at any time in each risk group determined by Hasford score was 90% (low risk), 71% (intermediate risk) and 67% (high risk). In the imatinib arm, the rate of MMR at any time in each risk group determined by Hasford score was 69% (low risk), 65% (intermediate risk), and 54% (high risk).
BCR-ABL sequencing was performed on blood samples from patients in the newly diagnosed trial who discontinued dasatinib or imatinib therapy. Among dasatinib-treated patients the mutations detected were T315I, F317I/L, and V299L.
Dasatinib does not appear to be active against the T315I mutation, based on in vitro data.
14.2 Imatinib-Resistant or -Intolerant CML or Ph+ ALL in Adults
The efficacy and safety of SPRYCEL were investigated in adult patients with CML or Ph+¬†ALL whose disease was resistant to or who were intolerant to imatinib: 1158 patients had chronic phase CML, 858 patients had accelerated phase, myeloid blast phase, or lymphoid blast phase CML, and 130 patients had Ph+¬†ALL. In a clinical trial in chronic phase CML, resistance to imatinib was defined as failure to achieve a complete hematologic response (CHR; after 3 months), major cytogenetic response (MCyR; after 6 months), or complete cytogenetic response (CCyR; after 12 months); or loss of a previous molecular response (with concurrent ‚Č•10% increase in Ph+ metaphases), cytogenetic response, or hematologic response. Imatinib intolerance was defined as inability to tolerate 400 mg or more of imatinib per day or discontinuation of imatinib because of toxicity.
Results described below are based on a minimum of 2 years follow-up after the start of SPRYCEL therapy in patients with a median time from initial diagnosis of approximately 5 years. Across all studies, 48% of patients were women, 81% were white, 15% were black or Asian, 25% were 65 years of age or older, and 5% were 75 years of age or older. Most patients had long disease histories with extensive prior treatment, including imatinib, cytotoxic chemotherapy, interferon, and stem cell transplant. Overall, 80% of patients had imatinib-resistant disease and 20% of patients were intolerant to imatinib. The maximum imatinib dose had been 400‚Äď600 mg/day in about 60% of the patients and >600 mg/day in 40% of the patients.
The primary efficacy endpoint in chronic phase CML was MCyR, defined as elimination (CCyR) or substantial diminution (by at least 65%, partial cytogenetic response) of Ph+ hematopoietic cells. The primary efficacy endpoint in accelerated phase, myeloid blast phase, lymphoid blast phase CML, and Ph+ ALL was major hematologic response (MaHR), defined as either a CHR or no evidence of leukemia (NEL).
Dose-Optimization Trial: A randomized, open-label trial (NCT00123474) was conducted in adult patients with chronic phase CML to evaluate the efficacy and safety of SPRYCEL administered once daily compared with SPRYCEL administered twice daily. Patients with significant cardiac diseases, including myocardial infarction within 6 months, congestive heart failure within 3 months, significant arrhythmias, or QTc prolongation were excluded from the trial. The primary efficacy endpoint was MCyR in patients with imatinib-resistant CML. A total of 670 patients, of whom 497 had imatinib-resistant disease, were randomized to the SPRYCEL 100 mg once-daily, 140 mg once-daily, 50 mg twice-daily, or 70 mg twice-daily group. Median duration of treatment was 22 months.
Efficacy was achieved across all SPRYCEL treatment groups with the once-daily schedule demonstrating comparable efficacy (non-inferiority) to the twice-daily schedule on the primary efficacy endpoint (difference in MCyR 1.9%; 95% CI [‚ąí6.8%‚Äď10.6%]); however, the 100-mg once-daily regimen demonstrated improved safety and tolerability.
Efficacy results are presented in Tables 17 and 18 for adult patients with chronic phase CML who received the recommended starting dose of 100 mg once daily.
Table 17: Efficacy of SPRYCEL in Adult Patients with Imatinib-Resistant or -Intolerant Chronic Phase CML (minimum of 24 months follow-up) All Patients 100 mg Once Daily (n=167) a CHR (response confirmed after 4 weeks): WBC ‚ȧ institutional ULN, platelets <450,000/mm3, no blasts or promyelocytes in peripheral blood, <5% myelocytes plus metamyelocytes in peripheral blood, basophils in peripheral blood <20%, and no extramedullary involvement. b MCyR combines both complete (0% Ph+ metaphases) and partial (>0%‚Äď35%) responses.
Hematologic Response Rate % (95% CI)
   CHRa
92% (86‚Äď95)
Cytogenetic Response Rate % (95% CI)
   MCyRb
63% (56‚Äď71)
   CCyR
50% (42‚Äď58)
Table 18: Long-Term MMR of SPRYCEL in the Dose Optimization Trial: Adult Patients with Imatinib-Resistant or -Intolerant Chronic Phase CMLa Minimum Follow-up Period 2 Years 5 Years 7 Years a¬†¬†Results reported in recommended starting dose of 100 mg once daily. b¬†¬†Major molecular response criteria: Defined as BCR-ABL/control transcripts ‚ȧ0.1% by RQ-PCR in peripheral blood samples.
Major Molecular Responseb % (n/N)
   All Patients Randomized
34% (57/167)
43% (71/167)
44% (73/167)
   Imatinib-Resistant Patients
33% (41/124)
40% (50/124)
41% (51/124)
   Imatinib-Intolerant Patients
37% (16/43)
49% (21/43)
51% (22/43)
Based on data 7 years after the last patient was enrolled in the trial, 44% were known to be alive, 31% were known to have died, and 25% had an unknown survival status.
By 7 years, transformation to either accelerated or blast phase occurred in nine patients on treatment in the 100 mg once-daily treatment group.
Dose-Optimization Trial: One randomized open-label trial (NCT00123487) was conducted in patients with advanced phase CML (accelerated phase CML, myeloid blast phase CML, or lymphoid blast phase CML) to evaluate the efficacy and safety of SPRYCEL administered once daily compared with SPRYCEL administered twice daily. The primary efficacy endpoint was MaHR. A total of 611 patients were randomized to either the SPRYCEL 140 mg once-daily or 70 mg twice-daily group. Median duration of treatment was approximately 6 months for both treatment groups. The once-daily schedule demonstrated comparable efficacy (non-inferiority) to the twice-daily schedule on the primary efficacy endpoint; however, the 140-mg once-daily regimen demonstrated improved safety and tolerability.
Response rates for patients in the 140 mg once-daily group are presented in Table 19.
Table 19: Efficacy of SPRYCEL in Imatinib-Resistant or -Intolerant Advanced Phase CML and Ph+ ALL (2-Year Results) 140 mg Once Daily Accelerated (n=158) Myeloid Blast (n=75) Lymphoid Blast (n=33) Ph+ ALL (n=40) a¬†¬†Hematologic response criteria (all responses confirmed after 4 weeks): Major hematologic response: (MaHR) = complete hematologic response (CHR) + no evidence of leukemia (NEL). ¬†¬†¬†CHR: WBC ‚ȧ institutional ULN, ANC ‚Č•1000/mm3, platelets ‚Č•100,000/mm3, no blasts or promyelocytes in peripheral blood, bone marrow blasts ‚ȧ5%, <5% myelocytes plus metamyelocytes in peripheral blood, basophils in peripheral blood <20%, and no extramedullary involvement. ¬†¬†¬†NEL: same criteria as for CHR but ANC ‚Č•500/mm3 and <1000/mm3,¬†or platelets ‚Č•20,000/mm3 and ‚ȧ100,000/mm3. b¬†¬†MCyR combines both complete (0% Ph+ metaphases) and partial (>0%‚Äď35%) responses.CI = confidence interval¬†¬†¬†¬†¬†ULN = upper limit of normal range.
MaHR a
66%
28%
42%
38%
     (95% CI)
(59‚Äď74)
(18‚Äď40)
(26‚Äď61)
(23‚Äď54)
CHRa
47%
17%
21%
33%
     (95% CI)
(40‚Äď56)
(10‚Äď28)
(9‚Äď39)
(19‚Äď49)
NELa
19%
11%
21%
5%
     (95% CI)
(13‚Äď26)
(5‚Äď20)
(9‚Äď39)
(1‚Äď17)
MCyR b
39%
28%
52%
70%
     (95% CI)
(31‚Äď47)
(18‚Äď40)
(34‚Äď69)
(54‚Äď83)
CCyR
32%
17%
39%
50%
     (95% CI)
(25‚Äď40)
(10‚Äď28)
(23‚Äď58)
(34‚Äď66)
In the SPRYCEL 140 mg once-daily group, the median time to MaHR was 1.9 months (min-max: 0.7-14.5) for patients with accelerated phase CML, 1.9 months (min-max: 0.9-6.2) for patients with myeloid blast phase CML, and 1.8 months (min-max: 0.9-2.8) for patients with lymphoid blast phase CML.
In patients with myeloid blast phase CML, the median duration of MaHR was 8.1 months (min-max: 2.7-21.1) and 9.0 (min-max: 1.8-23.1) months for the 140 mg once-daily group and the 70 mg twice-daily group, respectively. In patients with lymphoid blast phase CML, the median duration of MaHR was 4.7 months (min-max: 3.0-9.0) and 7.9 months (min-max: 1.6-22.1) for the 140 mg once-daily group and the 70 mg twice-daily group, respectively. In patients with Ph+ ALL who were treated with SPRYCEL 140 mg once-daily, the median duration of MaHR was 4.6 months (min-max: 1.4-10.2). The medians of progression-free survival for patients with Ph+ ALL treated with SPRYCEL 140 mg once-daily and 70 mg twice-daily were 4.0 months (min-max: 0.4-11.1) and 3.1 months (min-max: 0.3-20.8), respectively.
14.3 CML in Pediatric Patients
The efficacy of SPRYCEL in pediatric patients was evaluated in two pediatric studies of 97 patients with chronic phase CML. Among 97 patients with chronic phase CML treated in two pediatric studies, an open-label, non-randomized dose-ranging trial (NCT00306202) and an open-label, non-randomized, single-arm trial (NCT00777036), 51 patients (exclusively from the single-arm trial) had newly diagnosed chronic phase CML and 46 patients (17 from the dose-ranging trial and 29 from the single-arm trial) were resistant or intolerant to previous treatment with imatinib. Ninety-one of the 97 pediatric patients were treated with SPRYCEL tablets 60 mg/m2 once daily (maximum dose of 100 mg once daily for patients with high BSA). Patients were treated until disease progression or unacceptable toxicity.
Baseline demographic characteristics of the 46 imatinib resistant or intolerant patients were: median age 13.5 years (range 2 to 20 years), 78.3% White, 15.2% Asian, 4.4% Black, 2.2% other, and 52% female. Baseline characteristics of the 51 newly diagnosed patients were: median age 12.8 years (range 1.9 to 17.8 years), 60.8% White, 31.4% Asian, 5.9% Black, 2% Other, and 49% female.
Median duration of follow-up was 5.2 years (range 0.5 to 9.3 years) for the imatinib resistant or intolerant patients and 4.5 years (range 1.3 to 6.4 years) for the newly diagnosed patients, respectively. Efficacy results for the two pediatric studies are summarized in Table 20.
Table 20 shows increasing trend for response for CCyR, MCyR, and MMR across time (3 months to 24 months). The increasing trend in response for all three endpoints is seen in both the newly diagnosed and imatinib resistant or intolerant patients.
Table 20: Efficacy of SPRYCEL in Pediatric Patients with CP-CML Cumulative Response Over Time by Minimum Follow-Up Period
3 months 6 months 12 months 24 months a    Patients from pediatric study of newly diagnosed CP-CML receiving oral tablet formulation b    Patients from pediatric studies of imatinib-resistant or -intolerant CP-CML receiving oral tablet formulation
CCyR(95% CI)
Newly diagnosed(N = 51)a
43.1%(29.3, 57.8)
66.7%(52.1, 79.2)
96.1%(86.5, 99.5)
96.1%(86.5, 99.5)
Prior imatinib(N = 46)b
45.7%(30.9, 61.0)
71.7%(56.5, 84.0)
78.3%(63.6, 89.1)
82.6%(68.6, 92.2)
MCyR (95% CI)
Newly diagnosed(N = 51)a
60.8%(46.1, 74.2)
90.2%(78.6, 96.7)
98.0%(89.6, 100)
98.0%(89.6, 100)
Prior imatinib(N = 46)b
60.9%(45.4, 74.9)
82.6%(68.6, 92.2)
89.1%(76.4, 96.4)
89.1%(76.4, 96.4)
MMR(95% CI)
Newly diagnosed(N = 51)a
7.8%(2.2, 18.9)
31.4%(19.1, 45.9)
56.9%(42.2, 70.7)
74.5%(60.4, 85.7)
Prior imatinib(N = 46)b
15.2%(6.3, 28.9)
26.1%(14.3, 41.1)
39.1%(25.1, 54.6)
52.2%(36.9, 67.1)
With a median follow-up of 4.5 years in newly diagnosed patients, the median durations of CCyR, MCyR, MMR could not be estimated as more than half of the responding patients had not progressed at the time of data cut-off. Range of duration of response was (2.5+ to 66.5+ months for CCyR), (1.4 to 66.5+ months for MCyR), and (5.4+ to 72.5+ months for subjects who achieved MMR by month 24 and 0.03+ to 72.5+ months for subjects who achieved MMR at any time), where ‚Äė+‚Äô indicates a censored observation.
With a median follow-up of 5.2 years in imatinib-resistant or - intolerant patients, the median durations of CCyR, MCyR, and MMR could not be estimated as more than half the responding patients had not progressed at the time of data cut-off. Range of duration of response was (2.4 to 86.9+ months for CCyR), (2.4 to 86.9+ months for MCyR), and (2.6+ to 73.6+ months for MMR), where ‚Äė+‚Äô indicates a censored observation.
The median time to response for MCyR was 2.9 months (95% CI: 2.8 months, 3.5 months) in the pooled imatinib-resistant/intolerant CP-CML patients. The median time to response for CCyR was 3.3 months (95% CI: 2.8 months, 4.7 months) in the pooled imatinib-resistant/intolerant CP-CML patients. The median time to response for MMR was 8.3 months (95% CI: 5.0 months, 11.8 months) in the pooled imatinib- resistant/intolerant CP-CML patients.
The median time to response for MCyR was 3.0 months (95% CI: 2.8 months, 4.3 months) in the newly diagnosed treatment-na√Įve CP-CML patients. The median time to response for CCyR was 5.5 months (95% CI: 3.0 months, 5.7 months) in the newly diagnosed treatment-na√Įve CP-CML patients. The median time to response for MMR was 8.9 months (95% CI: 6.2 months, 11.7 months) in the newly diagnosed treatment-na√Įve CP-CML patients.
In the Phase II pediatric study, 1 newly diagnosed patient and 2 imatinib-resistant or -intolerant patients progressed to blast phase CML.
14.4 Ph+ ALL in Pediatric Patients
The efficacy of SPRYCEL in combination with chemotherapy was evaluated in a single cohort (cohort 1) of Study CA180372 (NCT01460160), a multicenter, multiple-cohort study of pediatric patients with newly diagnosed B-cell precursor Ph+ ALL. The 78 patients in cohort 1 received SPRYCEL at a daily dose of 60 mg/m2 for up to 24 months, in combination with chemotherapy. The backbone chemotherapy regimen was the AIEOP-BFM ALL 2000 multi-agent chemotherapy protocol.
Patients had a median age of 10.4 years (range 2.6 to 17.9 years) and included 20 patients (25%) 2 to 6 years of age, 37 patients (46%) 7 to 12 years of age, and 24 patients (30%) 13 to 17 years of age. Eighty-two percent of patients were white, and 55% were male. Thirty-two patients (41%) had a white blood cell count (WBC) of ‚Č•50,000 mcl at diagnosis, and 17 patients (22%) had extramedullary disease.
Efficacy was established on the basis of 3-year event-free survival (EFS), defined as the time from the start of SPRYCEL to lack of complete response at the end of the third high risk block, relapse, secondary malignancy, or death from any cause. The 3-year EFS binary rate for patients on Study CA180372 was 64.1% (95% CI: 52.4, 74.7). At the end of induction, 75 patients (96%) had a bone marrow with <5% lymphoblasts, and 76 patients (97%) achieved this by the end of consolidation.
15 References
1. http://www.osha.gov/SLTC/hazardousdrugs/index.html
16 How Supplied/storage And Handling
How Supplied
SPRYCEL¬ģ (dasatinib) tablets are available as described in Table 21.
Table 21: SPRYCEL Trade Presentations
NDC Number
Strength
Description
Tablets per Bottle
0003-0527-11
20 mg
white to off-white, biconvex, round, film-coated tablet with ‚ÄúBMS‚ÄĚ debossed on one side and ‚Äú527‚ÄĚ on the other side
60
0003-0528-11
50 mg
white to off-white, biconvex, oval, film-coated tablet with ‚ÄúBMS‚ÄĚ debossed on one side and ‚Äú528‚ÄĚ on the other side
60
0003-0524-11
70 mg
white to off-white, biconvex, round, film-coated tablet with ‚ÄúBMS‚ÄĚ debossed on one side and ‚Äú524‚ÄĚ on the other side
60
0003-0855-22
80 mg
white to off-white, biconvex, triangle, film-coated tablet with ‚ÄúBMS‚ÄĚ and ‚Äú80‚ÄĚ (BMS over 80) debossed on one side and ‚Äú855‚ÄĚ on the other side
30
0003-0852-22
100 mg
white to off-white, biconvex, oval, film-coated tablet with ‚ÄúBMS 100‚ÄĚ debossed on one side and ‚Äú852‚ÄĚ on the other side
30
0003-0857-22
140 mg
white to off-white, biconvex, round, film-coated tablet with ‚ÄúBMS‚ÄĚ and ‚Äú140‚ÄĚ (BMS over 140) debossed on one side and ‚Äú857‚ÄĚ on the other side
30
Storage
SPRYCEL tablets should be stored at 20¬įC to 25¬įC (68¬įF to 77¬įF); excursions permitted between 15¬įC and 30¬įC (59¬įF and 86¬įF) [see USP Controlled Room Temperature].
Handling and Disposal
SPRYCEL is an antineoplastic product. Follow special handling and disposal procedures.1
Personnel who are pregnant should avoid exposure to crushed or broken tablets.
SPRYCEL tablets consist of a core tablet, surrounded by a film coating to prevent exposure of healthcare professionals to the active substance. The use of latex or nitrile gloves for appropriate disposal when handling tablets that are inadvertently crushed or broken is recommended, to minimize the risk of dermal exposure.
17 Patient Counseling Information
Advise the patient to read the FDA-approved patient labeling (Patient Information).
Inform patients of the possibility of developing low blood cell counts. Advise patients to immediately report fever particularly in association with any suggestion of infection [see Warnings and Precautions (5.1)].
Inform patients of the possibility of serious bleeding and to report immediately any signs or symptoms suggestive of hemorrhage (unusual bleeding or easy bruising) [see Warnings and Precautions (5.2)].
Patients should be informed of the possibility of developing fluid retention (swelling, weight gain, dry cough, chest pain on respiration, or shortness of breath) and advised to seek medical attention promptly if those symptoms arise [see Warnings and Precautions (5.3)].
Cardiovascular Toxicity
Inform patients of the possibility of developing cardiovascular toxicity, including cardiac ischemic events, cardiac-related fluid retention, conduction abnormalities, and TIAs. Advise patients to seek immediate medical attention if symptoms suggestive of cardiovascular toxicity occur, such as chest pain, shortness of breath, palpitations, transient vision problems, or slurred speech [see Warnings and Precautions (5.4)].
Pulmonary Arterial Hypertension
Inform patients of the possibility of developing pulmonary arterial hypertension (dyspnea, fatigue, hypoxia, and fluid retention) and advise them to seek medical attention promptly if those symptoms arise [see Warnings and Precautions (5.5)].
Tumor Lysis Syndrome
Inform patients to immediately report and seek medical attention for any symptoms such as nausea, vomiting, weakness, edema, shortness of breath, muscle cramps, and seizures, which may indicate tumor lysis syndrome [see Warnings and Precautions (5.8)].
Growth and Development in Pediatric Patients
Inform pediatric patients and their caregivers of the possibility of developing bone growth abnormalities, bone pain, or gynecomastia and advise them to seek medical attention promptly if those symptoms arise [see Warnings and Precautions (5.10)].
‚ÄĘ Advise pregnant women of the potential risk to a fetus [see Warnings and Precautions (5.9) and Use in Specific Populations (8.1)].‚ÄĘ Advise females of reproductive potential and males with female partners of reproductive potential to use effective contraception during treatment with SPRYCEL and for 30 days after the last dose. Advise females to contact their healthcare provider if they become pregnant, or if pregnancy is suspected, while taking SPRYCEL [see Warnings and Precautions (5.9) and Use in Specific Populations (8.1, 8.3)].
‚ÄĘ Advise women that breastfeeding is not recommended during treatment with SPRYCEL and for 2 weeks after the final dose [see Use in Specific Populations (8.2)].
Inform patients that they may experience nausea, vomiting, or diarrhea with SPRYCEL. Advise patients to seek medical attention if these symptoms are bothersome or persistent.
Advise patients using antacids to avoid taking SPRYCEL and antacids less than 2 hours apart [see Drug Interactions (7.1)].
Inform patients that they may experience headache or musculoskeletal pain with SPRYCEL. Advise patients to seek medical attention if these symptoms are bothersome or persistent.
Inform patients that they may experience fatigue with SPRYCEL. Advise patients to seek medical attention if this symptom is bothersome or persistent.
Inform patients that they may experience skin rash with SPRYCEL. Advise patients to seek medical attention if this symptom is bothersome or persistent.
Inform patients that SPRYCEL contains 135 mg of lactose monohydrate in a 100-mg daily dose and 189 mg of lactose monohydrate in a 140-mg daily dose.
Advise patients that SPRYCEL can cause hepatotoxicity and that patients with previous history of liver diseases may be at risk. Advise patients to seek immediate medical attention if any symptoms suggestive of hepatotoxicity occur, such as abdominal pain, jaundice and scleral icterus, anorexia, bleeding, bruising, and dark-colored urine [see Warnings and Precautions (5.11)].
‚ÄĘ Missed Dose¬† Advise patients that if they miss a dose of SPRYCEL, they should take the next scheduled dose at its regular time. The patient should not take two doses at the same time.‚ÄĘ Grapefruit Juice¬† Advise patients not to drink grapefruit juice as it may increase the amount of SPRYCEL in their blood and therefore increase their risk of adverse reactions. Distributed by:Bristol-Myers Squibb CompanyPrinceton, NJ 08543 USA
Patient Informationsprycel (spry-sell)(dasatinib)tablets
What is SPRYCEL?
SPRYCEL is a prescription medicine used to treat:
‚ÄĘ adults with newly diagnosed Philadelphia chromosome-positive (Ph+) chronic myeloid leukemia (CML) in chronic phase.‚ÄĘ adults with Ph+ CML who no longer benefit from, or did not tolerate, other treatment, including imatinib.‚ÄĘ adults with Ph+ acute lymphoblastic leukemia (Ph+¬†ALL) who no longer benefit from, or did not tolerate, other treatment.‚ÄĘ children 1 year of age and older with Ph+ CML in chronic phase.‚ÄĘ children 1 year of age and older with newly diagnosed Ph+ ALL in combination with chemotherapy.
It is not known if SPRYCEL is safe and effective in children under 1 year of age.
Before taking SPRYCEL, tell your healthcare provider about all of your medical conditions, including if you:
‚ÄĘ have problems with your immune system‚ÄĘ have heart problems, including a condition called congenital long QT syndrome‚ÄĘ have low potassium or low magnesium levels in your blood‚ÄĘ have liver problems‚ÄĘ are lactose (milk sugar) intolerant‚ÄĘ are pregnant or plan to become pregnant. SPRYCEL can harm your unborn baby.¬† Females who can become pregnant:¬†
o You should not become pregnant during treatment with SPRYCEL.o You should use effective birth control (contraception) during treatment and for 30 days after your last dose of SPRYCEL.o Talk to your healthcare provider right away if you become pregnant or think you may be pregnant during treatment with SPRYCEL.  Males with female partners who can become pregnant: 
o You should use effective birth control (contraception) during treatment and for 30 days after your last dose of SPRYCEL.o Your female partner should call her healthcare provider if she becomes pregnant or thinks she is pregnant during your treatment with SPRYCEL.‚ÄĘ are breastfeeding or plan to breastfeed. It is not known if SPRYCEL passes into your breast milk. You should not breastfeed during treatment and for 2 weeks after your last dose of SPRYCEL.
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, antacids, and herbal supplements. If you take an antacid medicine, take it 2 hours before or 2 hours after your dose of SPRYCEL.
How should I take SPRYCEL?
‚ÄĘ Take SPRYCEL exactly as your healthcare provider tells you to take it.‚ÄĘ Your healthcare provider may change your dose of SPRYCEL or temporarily stop treatment with SPRYCEL. Do not change your dose or stop taking SPRYCEL without first talking to your healthcare provider.‚ÄĘ Take SPRYCEL 1 time a day.‚ÄĘ Take SPRYCEL with or without food, either in the morning or in the evening.‚ÄĘ Swallow SPRYCEL tablets whole. Do not crush, cut or chew the tablets.
o If your child cannot swallow tablets whole, talk with your healthcare provider.‚ÄĘ You should not drink grapefruit juice during treatment with SPRYCEL.‚ÄĘ If you miss a dose of SPRYCEL, take your next scheduled dose at your regular time. Do not take two doses at the same time.‚ÄĘ If you take too much SPRYCEL, call your healthcare provider or go to the nearest hospital emergency room right away.
What are the possible side effects of SPRYCEL?
SPRYCEL may cause serious side effects, including:
‚ÄĘ Low blood cell counts. Low blood cell counts are common with SPRYCEL and can be severe, including low red blood cell counts (anemia), low white blood cell counts (neutropenia), and low platelet counts (thrombocytopenia). Your healthcare provider will do blood tests to check your blood cell counts regularly during your treatment with SPRYCEL. Call your healthcare provider right away if you have a fever or any signs of an infection during treatment with SPRYCEL.‚ÄĘ Bleeding problems. Bleeding problems are common with SPRYCEL. Sometimes these bleeding problems can be serious and lead to death. Call your healthcare provider right away if you have:
o unusual bleeding or bruising of your skino bright red or dark tar-like stoolso decreased alertness, headache, or change in speech‚ÄĘ Your body may hold too much fluid (fluid retention). Fluid retention is common with SPRYCEL and can sometimes be severe. In severe cases, fluid may build up in the lining of your lungs, the sac around your heart, or your stomach cavity. Call your healthcare provider right away if you get any of these symptoms during treatment with SPRYCEL:
o swelling all over your bodyo weight gaino shortness of breath, especially if this happens with low levels of physical activity or at resto dry cougho chest pain when taking a deep breath‚ÄĘ Heart and blood vessel (cardiovascular) problems. SPRYCEL may cause heart problems, including an abnormal heart rate, a heart attack, or small strokes that last only a few minutes or a few hours, called transient ischemic attacks (TIAs). TIAs are often a warning sign that you are at risk for a more serious stroke. Your healthcare provider will monitor the potassium and magnesium levels in your blood and your heart function.Get medical help right away if you develop any of the following symptoms during treatment with SPRYCEL:
o chest paino shortness of breatho feeling like your heart is beating too fast or you feel abnormal heart beatso vision changes that may last for a short timeo slurred speech‚ÄĘ Pulmonary Arterial Hypertension (PAH). SPRYCEL may cause high blood pressure in the vessels of your lungs. PAH may happen at any time during your treatment with SPRYCEL. Your healthcare provider should check your heart and lungs before and during treatment with SPRYCEL. Call your healthcare provider right away if you have shortness of breath, tiredness, or swelling all over your body (fluid retention).‚ÄĘ Severe skin reactions. SPRYCEL may cause skin reactions that can sometimes be severe. Get medical help right away if you get a skin reaction with fever, sore mouth or throat, or bulering or peeling of your skin or in the mouth.‚ÄĘ Tumor Lysis Syndrome (TLS). TLS is caused by a fast breakdown of cancer cells. TLS can cause you to have kidney failure and the need for dialysis treatment, and an abnormal heartbeat. Your healthcare provider may do blood tests to check you for TLS. Call your healthcare provider or get emergency medical help right away if you develop any of these symptoms during treatment with SPRYCEL:
o nauseao vomitingo weaknesso swelling
o shortness of breatho muscle crampso seizures
‚ÄĘ Slowing of growth and development in children. Effects on bone growth and development in children have happened with SPRYCEL and can sometimes be severe. Your healthcare provider will monitor your child‚Äôs bone growth and development during treatment with SPRYCEL. Get medical help right away if your child develops bone pain.‚ÄĘ Liver problems. SPRYCEL can cause liver problems. People who have had liver problems in the past may be at risk for getting liver problems with SPRYCEL. Your healthcare provider will monitor your liver function as needed during the treatment with SPRYCEL. Call your healthcare provider or get medical help right away if you develop any symptoms of liver problems, including:
o stomach-area (abdominal) paino yellowing of your skin or white part of your eyeso loss of appetite
o bleedingo bruisingo dark ‚Äútea-colored‚ÄĚ urine
The most common side effects of SPRYCEL in adults and children receiving SPRYCEL alone include:
‚ÄĘ diarrhea‚ÄĘ headache‚ÄĘ skin rash‚ÄĘ shortness of breath
‚ÄĘ tiredness‚ÄĘ nausea‚ÄĘ muscle pain
The most common side effects of SPRYCEL in children receiving SPRYCEL with chemotherapy include:
‚ÄĘ swelling, pain and redness of the lining of your mouth, throat, stomach and bowel (mucositis)‚ÄĘ low white blood cell counts with fever‚ÄĘ fever‚ÄĘ diarrhea‚ÄĘ nausea‚ÄĘ vomiting‚ÄĘ muscle pain‚ÄĘ stomach-area (abdominal) pain‚ÄĘ cough‚ÄĘ headache‚ÄĘ rash
‚ÄĘ tiredness‚ÄĘ constipation‚ÄĘ abnormal heart rate‚ÄĘ high blood pressure (hypertension)‚ÄĘ swelling‚ÄĘ infections‚ÄĘ low blood pressure‚ÄĘ decreased appetite‚ÄĘ allergic reactions‚ÄĘ shortness of breath‚ÄĘ nose bleed‚ÄĘ numbness or tingling of your hands and feet‚ÄĘ feeling confused or disoriented
SPRYCEL may cause fertility problems in males and females. Talk to your healthcare provider if this is a concern for you.
Tell your healthcare provider if you have any side effect that bothers you or that does not go away. These are not all of the possible side effects of SPRYCEL.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store SPRYCEL?
‚ÄĘ Store SPRYCEL at room temperature between 68¬įF to 77¬įF (20¬įC to 25¬įC).‚ÄĘ Ask your healthcare provider or pharmacist about the right way to throw away expired or unused SPRYCEL.‚ÄĘ Wear latex or nitrile gloves when handling tablets that have accidentally been crushed or broken.‚ÄĘ Females who are pregnant should not handle crushed or broken SPRYCEL tablets.
Keep SPRYCEL and all medicines out of the reach of children.
General information about the safe and effective use of SPRYCEL.
Medicines are sometimes prescribed for purposes other than those uled in a Patient Information leaflet. Do not use SPRYCEL for a condition for which it is not prescribed. Do not give SPRYCEL to other people even if they have the same symptoms you have. It may harm them. You can ask your healthcare provider or pharmacist for information about SPRYCEL that is written for health professionals.
What are the ingredients in SPRYCEL?
Active ingredient: dasatinib
Inactive ingredients: lactose monohydrate, microcrystalline cellulose, croscarmellose sodium, hydroxypropyl cellulose, and magnesium stearate. The tablet coating consists of hypromellose, titanium dioxide, and polyethylene glycol. Distributed by: Bristol-Myers Squibb Company, Princeton, NJ 08543 USAFor more information, go to www.sprycel.com or call 1-800-332-2056.
This Patient Information has been approved by the U.S. Food and Drug Administration.
Revised: February 2023
Sprycel 20 Mg Tablets Representative Packaging
See How Supplied section for a complete ul of available packages of SPRYCEL.
60 Tablets NDC 0003-0527-11SPRYCEL¬ģ (dasatinib)Tablets20 mgRx onlyDo not crush, cut or chew tablets.Swallow tablets whole.Bristol Myers Squibb

Sprycel 50 Mg Tablets Representative Packaging
60 Tablets NDC 0003-0528-11 SPRYCEL¬ģ (dasatinib)Tablets50 mgRx onlyDo not crush, cut or chew tablets.Swallow tablets whole.Bristol Myers Squibb
Sprycel 70 Mg Tablets Representative Packaging
60 Tablets NDC 0003-0524-11SPRYCEL¬ģ (dasatinib)Tablets70 mgRx onlyDo not crush, cut or chew tablets.Swallow tablets whole.Bristol Myers Squibb

Sprycel 80 Mg Tablets Representative Packaging
30 Tablets NDC 0003-0855-22SPRYCEL¬ģ (dasatinib)Tablets80 mgRx onlyDo not crush, cut or chew tablets.Swallow tablets whole.Bristol Myers Squibb
Sprycel 100 Mg Tablets Representative Packaging
30 Tablets NDC 0003-0852-22 SPRYCEL¬ģ (dasatinib)Tablets100 mgRx onlyDo not crush, cut or chew tablets.Swallow tablets whole.Bristol Myers Squibb

Sprycel 140 Mg Tablets Representative Packaging
30 Tablets NDC 0003-0857-22 SPRYCEL¬ģ (dasatinib) Tablets 140 mg Rx only Do not crush, cut or chew tablets.Swallow tablets whole.Bristol Myers Squibb
DISCLAIMER:
"This tool does not provide medical advice, and is for informational and educational purposes only, and is not a substitute for professional medical advice, treatment or diagnosis. Call your doctor to receive medical advice. If you think you may have a medical emergency, please dial 911."
"Do not rely on openFDA to make decisions regarding medical care. While we make every effort to ensure that data is accurate, you should assume all results are unvalidated. We may limit or otherwise restrict your access to the API in line with our Terms of Service."
"This product uses publicly available data from the U.S. National Library of Medicine (NLM), National Institutes of Health, Department of Health and Human Services; NLM is not responsible for the product and does not endorse or recommend this or any other product."
PillSync may earn a commission via links on our site
