Tivicay (dolutegravir sodium 25 mg) Dailymed
Generic: dolutegravir sodium is used for the treatment of HIV Infections

SHAPE: round
All Imprints
dolutegravir sodium 50 mg - sv 572 50 round yellow
dolutegravir sodium 25 mg - sv 572 25 round yellow
dolutegravir 50 mg oral tablet [tivicay] - sv 572 50 round yellow
dolutegravir 50 mg - 50 sv 572 round yellow
dolutegravir sodium 10 mg - sv 572 10 round white

Go PRO for all pill images
Recent Major Changes Section
Dosage and Administration, Pregnancy Testing Before Initiation (2.1 ).
Removed 4/2024
Warnings and Precautions, Embryo-Fetal Toxicity (5.3 )
Removed 4/2024
1 Indications And Usage
TIVICAY and TIVICAY PD are indicated in combination with other antiretroviral agents for the treatment of HIV‚ÄĎ1 infection in adults (treatment-na√Įve or -experienced) and in pediatric patients (treatment-na√Įve or -experienced but integrase strand transfer inhibitor [INSTI]-na√Įve) aged at least 4 weeks and weighing at least 3¬†kg [see Microbiology (12.4)].
TIVICAY is indicated in combination with rilpivirine as a complete regimen for the treatment of HIV-1 infection in adults to replace the current antiretroviral regimen in those who are virologically suppressed (HIV-1 RNA less than 50 copies/ mL) on a stable antiretroviral regimen for at least 6 months with no history of treatment failure or known substitutions associated with resistance to either antiretroviral agent.
TIVICAY and TIVICAY PD are an HIV-1 integrase strand transfer inhibitor (INSTI) indicated in combination with other antiretroviral agents for the treatment of HIV-1 infection in adults (treatment-na√Įve or -experienced) and in pediatric patients (treatment-na√Įve or -experienced but INSTI- na√Įve) aged at least 4 weeks and weighing at least 3¬†kg. (1 )
TIVICAY is indicated in combination with rilpivirine as a complete regimen for the treatment of HIV-1 infection in adults to replace the current antiretroviral regimen in those who are virologically suppressed (HIV-1 RNA less than 50 copies/mL) on a stable antiretroviral regimen for at least 6 months with no history of treatment failure or known substitutions associated with resistance to either antiretroviral agent. (1 )
2 Dosage And Administration
‚ÄĘ May be taken without regard to food. (2.2 , 2.6)
UGT = uridine diphosphate glucuronosyltransferase; CYP = cytochrome P450. a  Rilpivirine dose is 25 mg once daily for those switching to dolutegravir plus rilpivirine. b  Alternative combinations that do not include metabolic inducers should be considered where possible.
Adult Population
Recommended Dose
Treatment-na√Įve or treatment-experienced INSTI-na√Įve or virologically suppressed (HIV-1 RNA <50 copies per mL) adults switching to dolutegravir plus rilpivirinea (2.1 )
50 mg once daily
Treatment-na√Įve or treatment-experienced INSTI-na√Įve when coadministered with certain UGT1A or CYP3A inducers (2.1 ,7.2 ,7.3 )
50 mg twice daily
INSTI-experienced with certain INSTI-associated resistance substitutions or clinically suspected INSTI resistanceb (2.1 ,12.4 )
50 mg twice daily
Pediatric Patients: Treatment-na√Įve or treatment-experienced INSTI-na√Įve patients aged at least 4 weeks and weighing at least 3¬†kg. See Tables2 ,3 , and4 for complete pediatric dosing recommendations. (2.3 ,2.4 ,2.5 ). TIVICAY and TIVICAY PD are not bioequivalent and are not substitutable on a milligram-per-milligram basis.
a  If certain UGT1A or CYP3A inducers are coadministered, then adjust the weight-based dose of TIVICAY to twice daily. ( 2.3 ,2.4 ,7.2 ,7.3 )
Pediatric Population
Body Weight
Recommended Dosea
TIVICAY PD Tablets for Oral Suspension
3 kg to less than 6 kg
5 mg once daily
6 kg to less than 10 kg
15 mg once daily
10 kg to less than 14 kg
20 mg once daily
14 kg to less than 20 kg
25 mg once daily
20 kg and greater
30 mg once daily
Alternative dosing recommendations for TIVICAY tablets for patients weighing at least 14 kg (Table 4 ):
‚ÄĘ 14¬†kg to less than 20¬†kg: 40¬†mg once daily.‚ÄĘ 20¬†kg and greater: 50¬†mg once daily.2.1 Recommended Dosage in Adults
TIVICAY tablets may be taken with or without food.
Table 1. Dosing Recommendations for TIVICAY Tablets in Adult Patients INSTI = integrase strand transfer inhibitor. a  Rilpivirine dose is 25 mg once daily for those switching to dolutegravir plus rilpivirine. b  Alternative combinations that do not include metabolic inducers should be considered where possible [see Drug Interactions (7.3)].
Population
Recommended Dosage
Treatment-na√Įve or treatment-experienced INSTI-na√Įve or virologically suppressed (HIV‚ÄĎ1 RNA <50¬†copies/mL) adults switching to dolutegravir plus rilpivirinea
50 mg once daily
Treatment-na√Įve or treatment-experienced INSTI-na√Įve when coadministered with certain uridine diphosphate (UDP)-glucuronosyl transferase 1A1 (UGT1A) or cytochrome P450 (CYP)3A inducers [see Drug Interactions (7.2, 7.3)]
50 mg twice daily
INSTI-experienced with certain INSTI-associated resistance substitutions or clinically suspected INSTI resistanceb [see Microbiology (12.4)]
50 mg twice daily
2.2 General Dosing and Administration Instructions for Pediatric Patients
Do not substitute TIVICAY tablets and TIVICAY PD tablets for oral suspension on a milligram-per-milligram basis due to differing pharmacokinetic profiles [see Warnings and Precautions (5.6), Clinical Pharmacology (12.3)]. If switching from the tablets to the tablets for oral suspension, follow the recommended dosage in Table 3. If switching from the tablets for oral suspension to the tablets, follow the recommended dosage in Table 4. See administration instructions in Dosage and Administration (2.5).
2.3 Recommended Dosage in Pediatric Patients Weighing 3 to 14 kg
The recommended weight-based dosage of TIVICAY PD tablets for oral suspension in pediatric patients weighing 3 to 14¬†kg (4¬†weeks and older, treatment-na√Įve, or treatment-experienced but na√Įve to INSTI treatment) is described in Table 2.
Do not use TIVICAY tablets in patients weighing 3 to 14 kg. See administration instructions in Dosage and Administration (2.5).
Table 2. Recommended Dosage of TIVICAY PD in Pediatric Patients 4 Weeks and Older Weighing 3 to 14 kg a  If certain uridine diphosphate glucuronosyltransferase (UGT)1A or cytochrome P450 (CYP)3A inducers are coadministered, then administer TIVICAY PD twice daily [see Drug Interactions (7.2, 7.3)].
Body Weight
TIVICAY PD Tablets for Oral Suspension
Daily Dosea
Number of
5-mg Tablets
3 kg to less than 6 kg
5 mg once daily
1
6 kg to less than 10 kg
15 mg once daily
3
10 kg to less than 14 kg
20 mg once daily
4
2.4 Recommended Dosage in Pediatric Patients Weighing 14 kg or Greater
For pediatric patients weighing 14¬†kg or greater (4¬†weeks and older, treatment-na√Įve, or treatment-experienced but na√Įve to INSTI treatment) administer either:
‚ÄĘ TIVICAY PD tablets for oral suspension (preferred in pediatric patients weighing less than 20¬†kg) (Table 3), or‚ÄĘ TIVICAY tablets for oral use (Table 4)
Table 3. Recommended Dosage of TIVICAY PD Tablets for Oral Suspension in Pediatric Patients Weighing 14 kg or Greater a  If certain UGT1A or CYP3A inducers are coadministered, then administer TIVICAY PD twice daily [see Drug Interactions (7.2, 7.3)].
Body Weight
TIVICAY PD Tablets for Oral Suspension
Daily Dosea
Number of
5-mg Tablets
14 kg to less than 20 kg
25 mg once daily
5
20 kg and greater
30 mg once daily
6
Table 4. Recommended Dosage of TIVICAY Tablets in Pediatric Patients Weighing 14 kg or Greater a  If certain UGT1A or CYP3A inducers are coadministered, then administer TIVICAY twice daily [see Drug Interactions (7.2, 7.3)].
Body Weight
TIVICAY Tablets
Daily Dosea
Number of Tablets
14 kg to less than 20 kg
40 mg once daily
4 x 10-mg
20 kg and greater
50 mg once daily
1 x 50-mg
2.5 Additional Administration Instructions
Administer TIVICAY tablets and TIVICAY PD tablets for oral suspension with or without food.
Administration Instructions for TIVICAY PD
Do not chew, cut, or crush TIVICAY PD [see Instructions for Use]. Instruct patients (or instruct caregivers) to either:
‚ÄĘ Swallow the tablets for oral suspension whole (if more than one tablet is required, swallow one tablet at a time to reduce the risk of choking), or‚ÄĘ Fully disperse the tablets for oral suspension in 5 mL of drinking water (if using 1 or 3 tablets for oral suspension) or 10 mL (if using 4, 5, or 6 tablets for oral suspension) in the supplied cup; swirl the suspension so that no lumps remain. After full dispersion, administer the oral suspension within 30 minutes of mixing [see Instructions for Use].
3 Dosage Forms And Strengths
TIVICAY Tablets:
10¬†mg: Each tablet contains 10¬†mg of dolutegravir (as dolutegravir sodium). Tablets are white, round, film-coated, biconvex tablets debossed with ‚ÄúSV¬†572‚ÄĚ on one side and ‚Äú10‚ÄĚ on the other side.
25¬†mg: Each tablet contains 25¬†mg of dolutegravir (as dolutegravir sodium). Tablets are pale yellow, round, film-coated, biconvex tablets debossed with ‚ÄúSV¬†572‚ÄĚ on one side and ‚Äú25‚ÄĚ on the other side.
50¬†mg: Each tablet contains 50¬†mg of dolutegravir (as dolutegravir sodium). Tablets are yellow, round, film-coated, biconvex tablets debossed with ‚ÄúSV¬†572‚ÄĚ on one side and ‚Äú50‚ÄĚ on the other side.
TIVICAY PD Tablets for Oral Suspension:
Each tablet contains 5¬†mg of dolutegravir (as dolutegravir sodium). Tablets are white, round, strawberry cream flavored, film-coated, biconvex tablets debossed with ‚ÄúSV H7S‚ÄĚ on one side and ‚Äú5‚ÄĚ on the other side.
‚ÄĘ TIVICAY tablets: 10¬†mg, 25¬†mg, and 50¬†mg (3 )‚ÄĘ TIVICAY PD tablets for oral suspension: 5¬†mg (3 )
4 Contraindications
TIVICAY and TIVICAY PD are contraindicated in patients:
‚ÄĘ Previous hypersensitivity reaction to dolutegravir. (4 )‚ÄĘ Coadministration with dofetilide. (4 )
5 Warnings And Precautions
‚ÄĘ Hypersensitivity reactions characterized by rash, constitutional findings, and sometimes organ dysfunction, including liver injury, have been reported. Discontinue TIVICAY or TIVICAY PD and other suspect agents immediately if signs or symptoms of hypersensitivity reactions develop, as a delay in stopping treatment may result in a life-threatening reaction. (5.1 )‚ÄĘ Hepatotoxicity has been reported in patients receiving dolutegravir-containing regimens. Patients with underlying hepatitis B or C may be at increased risk for worsening or development of transaminase elevations. Monitoring for hepatotoxicity is recommended. (5.2 )‚ÄĘ Immune reconstitution syndrome has been reported in patients treated with combination antiretroviral therapy. (5.4 )‚ÄĘ TIVICAY tablets and TIVICAY PD tablets for oral suspension are not substitutable. (2.2 ,5.5 )5.1 Hypersensitivity Reactions
Hypersensitivity reactions have been reported and were characterized by rash, constitutional findings, and sometimes organ dysfunction, including liver injury. The events were reported in less than 1% of subjects receiving TIVICAY in Phase 3 clinical trials. Discontinue TIVICAY or TIVICAY PD and other suspect agents immediately if signs or symptoms of hypersensitivity reactions develop (including, but not limited to, severe rash or rash accompanied by fever, general malaise, fatigue, muscle or joint aches, bulers or peeling of the skin, oral bulers or lesions, conjunctivitis, facial edema, hepatitis, eosinophilia, angioedema, difficulty breathing). Clinical status, including liver aminotransferases, should be monitored and appropriate therapy initiated. Delay in stopping treatment with TIVICAY or TIVICAY PD or other suspect agents after the onset of hypersensitivity may result in a life-threatening reaction. TIVICAY and TIVICAY PD are contraindicated in patients who have experienced a previous hypersensitivity reaction to dolutegravir.
5.2 Hepatotoxicity
Hepatic adverse events have been reported in patients receiving a dolutegravir-containing regimen. Patients with underlying hepatitis B or C may be at increased risk for worsening or development of transaminase elevations with use of TIVICAY or TIVICAY PD [see Adverse Reactions (6.1)]. In some cases, the elevations in transaminases were consistent with immune reconstitution syndrome or hepatitis B reactivation particularly in the setting where anti-hepatitis therapy was withdrawn. Cases of hepatic toxicity, including elevated serum liver biochemistries, hepatitis, and acute liver failure have been reported in patients receiving a dolutegravir-containing regimen without pre-existing hepatic disease or other identifiable risk factors. Drug-induced liver injury leading to liver transplant has been reported with TRIUMEQ (abacavir, dolutegravir, and lamivudine). Monitoring for hepatotoxicity is recommended.
5.3 Risk of Adverse Reactions or Loss of Virologic Response Due to Drug Interactions
The concomitant use of TIVICAY or TIVICAY PD and other drugs may result in known or potentially significant drug interactions, some of which may lead to [see Contraindications (4), Drug Interactions (7.3)]:
‚ÄĘ Loss of therapeutic effect of TIVICAY or TIVICAY PD and possible development of resistance.‚ÄĘ Possible clinically significant adverse reactions from greater exposures of concomitant drugs.
For concomitant drugs for which the interaction can be mitigated, please see Table 8 for steps to prevent or manage these possible and known significant drug interactions, including dosing recommendations. Consider the potential for drug interactions prior to and during therapy with TIVICAY or TIVICAY PD; review concomitant medications during therapy with TIVICAY or TIVICAY PD; and monitor for the adverse reactions associated with the concomitant drugs.
5.4 Immune Reconstitution Syndrome
Immune reconstitution syndrome has been reported in patients treated with combination antiretroviral therapy, including TIVICAY or TIVICAY PD. During the initial phase of combination antiretroviral treatment, patients whose immune systems respond may develop an inflammatory response to indolent or residual opportunistic infections (such as Mycobacterium avium infection, cytomegalovirus, Pneumocystis jirovecii pneumonia [PCP], or tuberculosis), which may necessitate further evaluation and treatment.
Autoimmune disorders (such as Graves’ disease, polymyositis, and Guillain-Barré syndrome) have also been reported to occur in the setting of immune reconstitution; however, the time to onset is more variable and can occur many months after initiation of treatment.
5.5 Different Formulations Are Not Substitutable
TIVICAY and TIVICAY PD are not bioequivalent and are not substitutable on a milligram-per-milligram basis [see Clinical Pharmacology (12.3)]. If a pediatric patient switches from one formulation to the other, the dose must be adjusted for the new dosage formulation [see Dosage and Administration (2.2)]. Incorrect dosing of a given formulation may result in underdosing and loss of therapeutic effect and possible development of resistance or possible clinically significant adverse reactions from greater exposure of dolutegravir.
6 Adverse Reactions
The following serious adverse drug reactions are discussed in other sections of the labeling:
The most common adverse reactions of moderate to severe intensity and incidence at least 2% (in those receiving TIVICAY in any one adult trial) are insomnia, fatigue, and headache. (6.1 )
To report SUSPECTED ADVERSE REACTIONS, contact ViiV Healthcare at 1-877-844-8872 or FDA at 1-800-FDA-1088 orwww.fda.gov/medwatch .
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared with rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Clinical Trials Experience in Adult Subjects
Treatment-Na√Įve Subjects: The safety assessment of TIVICAY in HIV‚ÄĎ1‚Äďinfected treatment-na√Įve subjects is based on the analyses of data from 2 international, multicenter, double-blind trials, SPRING-2 (ING113086) and SINGLE (ING114467) and data from the international, multicenter, open-label FLAMINGO (ING114915) trial.
In SPRING-2, 822 subjects were randomized and received at least 1 dose of either TIVICAY 50 mg once daily or raltegravir 400 mg twice daily, both in combination with fixed-dose dual nucleoside reverse transcriptase inhibitor (NRTI) treatment (either abacavir sulfate and lamivudine [EPZICOM] or emtricitabine/tenofovir [TRUVADA]). There were 808 subjects included in the efficacy and safety analyses. Through 96 weeks, the rate of adverse events leading to discontinuation was 2% in both treatment arms.
In SINGLE, 833 subjects were randomized and received at least 1 dose of either TIVICAY 50 mg with fixed-dose abacavir sulfate and lamivudine (EPZICOM) once daily or fixed-dose efavirenz/emtricitabine/tenofovir (ATRIPLA) once daily (study treatment was blinded through Week 96 and open-label from Week 96 through Week 144). Through 144 weeks, the rates of adverse events leading to discontinuation were 4% in subjects receiving TIVICAY 50 mg once daily + EPZICOM and 14% in subjects receiving ATRIPLA once daily.
Treatment‚ÄĎemergent adverse reactions of moderate to severe intensity observed in at least 2% of subjects in either treatment arm in SPRING-2 and SINGLE trials are provided in Table 5. Side-by-side tabulation is to simplify presentation; direct comparisons across trials should not be made due to differing trial designs.
Table 5. Treatment-Emergent Adverse Reactions of at Least Moderate Intensity (Grades 2 to 4) and at Least 2% Frequency in Treatment-Na√Įve Subjects in SPRING-2 (Week 96 Analysis) and SINGLE Trials (Week 144 Analysis) NRTI¬†=¬†Nucleoside Reverse Transcriptase Inhibitor. a¬†¬†Includes pooled terms: rash, rash generalized, rash macular, rash maculo-papular, rash pruritic, and drug eruption.
System Organ Class/
Preferred Term
SPRING-2
SINGLE
TIVICAY
50 mg Once Daily + 2 NRTIs
(n = 403)
Raltegravir
400 mg Twice Daily + 2 NRTIs
(n = 405)
TIVICAY
50 mg + EPZICOM Once Daily
(n = 414)
ATRIPLA
Once Daily
(n = 419)
Psychiatric
   Insomnia
<1%
<1%
3%
3%
   Depression
<1%
<1%
1%
2%
   Abnormal dreams
<1%
<1%
<1%
2%
Nervous System
   Dizziness
<1%
<1%
<1%
5%
   Headache
<1%
<1%
2%
2%
Gastrointestinal
   Nausea
1%
1%
<1%
3%
   Diarrhea
<1%
<1%
<1%
2%
Skin and Subcutaneous Tissue
   Rasha
0
<1%
<1%
6%
General Disorders
   Fatigue
<1%
<1%
2%
2%
Ear and Labyrinth
   Vertigo
0
<1%
0
2%
In addition, Grade 1 insomnia was reported by 1% and less than 1% of subjects receiving TIVICAY and raltegravir, respectively, in SPRING-2; whereas in SINGLE the rates were 7% and 4% for TIVICAY and ATRIPLA, respectively. These events were not treatment limiting.
In a multicenter, open-label trial (FLAMINGO), 243 subjects received TIVICAY 50 mg once daily versus 242 subjects who received darunavir 800 mg/ritonavir 100 mg once daily, both in combination with investigator-selected NRTI background regimen (either EPZICOM or TRUVADA). There were 484 subjects included in the efficacy and safety analyses. Through 96 weeks, the rates of adverse events leading to discontinuation were 3% in subjects receiving TIVICAY and 6% in subjects receiving darunavir/ritonavir. The adverse reactions observed in FLAMINGO were generally consistent with those seen in SPRING-2 and SINGLE.
Treatment-Experienced, Integrase Strand Transfer Inhibitor-Na√Įve Subjects: In an international, multicenter, double-blind trial (ING111762, SAILING), 719 HIV‚ÄĎ1‚Äďinfected, antiretroviral treatment-experienced adults were randomized and received either TIVICAY 50¬†mg once daily or raltegravir 400¬†mg twice daily with investigator-selected background regimen consisting of up to 2¬†agents, including at least one fully active agent. At 48 weeks, the rates of adverse events leading to discontinuation were 3% in subjects receiving TIVICAY 50¬†mg once daily + background regimen and 4% in subjects receiving raltegravir 400¬†mg twice daily + background regimen.
The only treatment‚ÄĎemergent adverse reaction of moderate to severe intensity with at least 2% frequency in either treatment group was diarrhea, 2% (6 of 354) in subjects receiving TIVICAY 50¬†mg once daily + background regimen and 1% (5 of 361) in subjects receiving raltegravir 400¬†mg twice daily + background regimen.
Treatment-Experienced, Integrase Strand Transfer Inhibitor-Experienced Subjects: In a multicenter, open-label, single‚ÄĎarm trial (ING112574, VIKING-3), 183¬†HIV‚ÄĎ1‚Äďinfected, antiretroviral treatment-experienced adults with virological failure and current or historical evidence of raltegravir and/or elvitegravir resistance received TIVICAY 50¬†mg twice daily with the current failing background regimen for 7¬†days and with optimized background therapy from Day 8. The rate of adverse events leading to discontinuation was 4% of subjects at Week 48.
Treatment‚ÄĎemergent adverse reactions in VIKING-3 were generally similar compared with observations with the 50-mg once-daily dose in adult Phase 3 trials.
Virologically Suppressed Subjects: The adverse reactions observed for TIVICAY plus rilpivirine in the Week 48 analysis of pooled data from 2 identical, international, multicenter, open-label trials (SWORD-1 and SWORD-2) of 513¬†HIV-1‚Äďinfected, virologically suppressed subjects switching from their current antiretroviral regimen to TIVICAY plus rilpivirine, were consistent with the adverse reaction profiles and severities for the individual components when administered with other antiretroviral agents. There were no adverse reactions (Grades 2 to 4) with an incidence of at least 2% in either treatment arm at Week 48. The safety profile during the additional follow-up period through Week 148 were consistent with Week 48. The rate of adverse events leading to discontinuation through Week 48 was 4% in subjects receiving TIVICAY plus rilpivirine once daily and less than 1% in subjects who remained on their current antiretroviral regimen. In the pooled analyses, the proportion of subjects receiving TIVICAY plus rilpivirine who discontinued treatment due to an adverse event through Week 148 was 8%.
Less Common Adverse Reactions Observed in Treatment-Na√Įve and Treatment-Experienced Trials: The following adverse reactions occurred in less than 2% of treatment-na√Įve or treatment-experienced subjects receiving TIVICAY in a combination regimen in any one trial. These events have been included because of their seriousness and assessment of potential causal relationship.
           Gastrointestinal Disorders: Abdominal pain, abdominal discomfort, flatulence, upper abdominal pain, vomiting.
           Hepatobiliary Disorders: Hepatitis.
           Musculoskeletal Disorders: Myositis.
           Psychiatric Disorders: Suicidal ideation, attempt, behavior, or completion. These events were observed primarily in subjects with a pre-existing history of depression or other psychiatric illness.
           Renal and Urinary Disorders: Renal impairment.
           Skin and Subcutaneous Tissue Disorders: Pruritus.
Laboratory Abnormalities:
¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†Treatment-Na√Įve Subjects: Selected laboratory abnormalities (Grades 2 to 4) with a worsening grade from baseline and representing the worst-grade toxicity in at least 2% of subjects are presented in Table 6. The mean change from baseline observed for selected lipid values is presented in Table 7. Side-by-side tabulation is to simplify presentation; direct comparisons across trials should not be made due to differing trial designs.
Table 6. Selected Laboratory Abnormalities (Grades 2 to 4) in Treatment-Na√Įve Subjects in SPRING-2 (Week 96 Analysis) and SINGLE Trials (Week 144 Analysis) ALT¬†=¬†Alanine aminotransferase; AST¬†=¬†Aspartate aminotransferase; NRTI¬†=¬†Nucleoside Reverse Transcriptase Inhibitor; ULN¬†=¬†Upper limit of normal.
Laboratory Parameter
Preferred Term
SPRING-2
SINGLE
TIVICAY 50 mg Once Daily + 2 NRTIs
(n = 403)
Raltegravir
400 mg Twice Daily + 2 NRTIs
(n = 405)
TIVICAY 50 mg + EPZICOM Once Daily
(n = 414)
ATRIPLA
Once Daily
(n = 419)
ALT
   Grade 2 (>2.5-5.0 x ULN)
4%
4%
3%
5%
   Grade 3 to 4 (>5.0 x ULN)
2%
2%
1%
<1%
AST
   Grade 2 (>2.5-5.0 x ULN)
5%
3%
3%
4%
   Grade 3 to 4 (>5.0 x ULN)
3%
2%
1%
3%
Total Bilirubin
   Grade 2 (1.6-2.5 x ULN)
3%
2%
<1%
<1%
   Grade 3 to 4 (>2.5 x ULN)
<1%
<1%
<1%
<1%
Creatine kinase
   Grade 2 (6.0-9.9 x ULN)
2%
5%
5%
3%
¬†¬†¬†Grade 3 to 4 (‚Č•10.0 x ULN)
7%
4%
7%
8%
Hyperglycemia
   Grade 2 (126-250 mg/dL)
6%
6%
9%
6%
   Grade 3 (>250 mg/dL)
<1%
2%
2%
<1%
Lipase
   Grade 2 (>1.5-3.0 x ULN)
7%
7%
11%
11%
   Grade 3 to 4 (>3.0 x ULN)
2%
5%
5%
4%
Total neutrophils
   Grade 2 (0.75-0.99 x 109)
4%
3%
4%
5%
   Grade 3 to 4 (<0.75 x 109)
2%
2%
3%
3%
Table 7. Mean Change from Baseline in Fasted Lipid Values in Treatment-Na√Įve Subjects in SPRING-2 (Week 96 Analysisa) and SINGLE Trials (Week 144 Analysisa) HDL = High density lipoprotein; LDL = Low density lipoprotein; NRTI¬†=¬†Nucleoside Reverse Transcriptase Inhibitor. a¬†¬†Subjects on lipid-lowering agents at baseline were excluded from these analyses (19 subjects in each arm in SPRING-2, and in SINGLE: TIVICAY¬†+¬†EPZICOM n¬†=¬†30 and ATRIPLA n¬†=¬†27). Ninety-four subjects initiated a lipid-lowering agent post-baseline; their last fasted on-treatment values (prior to starting the agent) were used regardless of whether they discontinued the agent (SPRING-2: TIVICAY n¬†=¬†9, raltegravir n¬†=¬†13; SINGLE: TIVICAY¬†+¬†EPZICOM n¬†=¬†36, ATRIPLA n¬†=¬†36).
Laboratory Parameter
Preferred Term
SPRING-2
SINGLE
TIVICAY
50 mg Once Daily + 2 NRTIs
(n = 403)
Raltegravir
400 mg Twice Daily + 2 NRTIs
(n = 405)
TIVICAY
50 mg + EPZICOM Once Daily
(n = 414)
ATRIPLA
Once Daily
(n = 419)
Cholesterol (mg/dL)
8.1
10.1
24.0
26.7
HDL cholesterol (mg/dL)
2.0
2.3
5.4
7.2
LDL cholesterol (mg/dL)
5.1
6.1
16.0
14.6
Triglycerides (mg/dL)
6.7
6.6
13.6
31.9
Laboratory abnormalities observed in the FLAMINGO trial were generally consistent with observations in SPRING-2 and SINGLE.
¬†¬†¬†¬†¬†¬†¬†¬†¬†Treatment-Experienced, Integrase Strand Transfer Inhibitor-Na√Įve Subjects: Laboratory abnormalities observed in SAILING were generally similar compared with observations seen in the treatment-na√Įve (SPRING-2 and SINGLE) trials.
         Treatment-Experienced, Integrase Strand Transfer Inhibitor-Experienced Subjects: The most common treatment-emergent laboratory abnormalities (greater than 5% for Grades 2 to 4 combined) observed in VIKING-3 at Week 48 were elevated ALT (9%), AST (8%), cholesterol (10%), creatine kinase (6%), hyperglycemia (14%), and lipase (10%). Two percent (4 of 183) of subjects had a Grade 3 to 4 treatment-emergent hematology laboratory abnormality, with neutropenia (2% [3 of 183]) being the most frequently reported.
         Virologically Suppressed Adults: Laboratory abnormalities observed in SWORD-1 and SWORD-2 were generally similar compared with observations seen in the other Phase 3 trials.
         Hepatitis B and/or Hepatitis C Virus Co-infection: In Phase 3 trials, subjects with hepatitis B and/or C virus co-infection were permitted to enroll provided that baseline liver chemistry tests did not exceed 5 times the upper limit of normal. Overall, the safety profile in subjects with hepatitis B and/or C virus co-infection was similar to that observed in subjects without hepatitis B or C co-infection, although the rates of AST and ALT abnormalities were higher in the subgroup with hepatitis B and/or C virus co-infection for all treatment groups. Grades 2 to 4 ALT abnormalities in hepatitis B and/or C co-infected compared with HIV mono-infected subjects receiving TIVICAY were observed in 18% vs. 3% with the 50-mg once-daily dose and 13% vs. 8% with the 50-mg twice-daily dose. Liver chemistry elevations consistent with immune reconstitution syndrome were observed in some subjects with hepatitis B and/or C at the start of therapy with TIVICAY, particularly in the setting where anti-hepatitis therapy was withdrawn [see Warnings and Precautions (5.2)].
Changes in Serum Creatinine: Dolutegravir has been shown to increase serum creatinine due to inhibition of tubular secretion of creatinine without affecting renal glomerular function [see Clinical Pharmacology (12.2)]. Increases in serum creatinine occurred within the first 4 weeks of treatment and remained stable through 96 weeks. In treatment-na√Įve subjects, a mean change from baseline of 0.15¬†mg/dL (range: -0.32¬†mg/dL to 0.65¬†mg/dL) was observed after 96¬†weeks of treatment. Creatinine increases were comparable by background NRTIs and were similar in treatment-experienced subjects.
Clinical Trials Experience in Pediatric Subjects
The safety and pharmacokinetics of TIVICAY and TIVICAY PD in HIV-1‚Äďinfected pediatric subjects aged at least 4 weeks and weighing at least 3 kg was evaluated in the IMPAACT P1093 trial and 2 weight-band-based pharmacokinetic substudies of the ODYSSEY trial [see Use in Specific Populations (8.4), Clinical Pharmacology (12.3)]. Overall, the safety data in these pediatric studies were similar to those seen in adults, and there was no clinically significant difference in dolutegravir exposure [see Clinical Pharmacology (12.3)].
IMPAACT P1093 is an ongoing, multicenter, open-label, non-comparative trial of HIV‚ÄĎ1‚Äďinfected pediatric subjects aged 4 weeks to less than 18 years [see Use in Specific Populations (8.4), Clinical Pharmacology (12.3), Clinical Studies (14.3)].
The safety analysis based on subjects (n = 75) who received the recommended dose (determined by weight and age) through Week 24 showed that 11% of subjects experienced drug-related clinical adverse reactions. The only Grade 1 to 2 drug-related clinical adverse reactions reported by more than one subject was immune reconstitution inflammatory syndrome (IRIS) (n = 2). There were no Grade 3 or 4 drug-related adverse reactions reported. No adverse reactions led to discontinuation.
The Grade 3 or 4 laboratory abnormalities reported in more than one subject were decreased neutrophil count (n = 11), decreased blood bicarbonate (n = 4), decreased hemoglobin (n = 3), increased lipase (n = 2), and increased blood potassium (n = 2). These laboratory events were not considered to be drug-related. Median laboratory values were similar at baseline and Week 24. Changes in median serum creatinine were similar to those observed in adults.
6.2 Postmarketing Experience
In addition to adverse reactions reported from clinical trials, the following adverse reactions have been identified during postmarketing use. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Hepatobiliary Disorders
Acute liver failure, hepatotoxicity.
Investigations
Weight increased.
Musculoskeletal
Arthralgia, myalgia.
Psychiatric
Anxiety.
7 Drug Interactions
‚ÄĘ Refer to the full prescribing information for important drug interactions with TIVICAY or TIVICAY PD. (4 ,7 )‚ÄĘ Drugs that are metabolic inducers may decrease the plasma concentrations of dolutegravir. (7.2 ,7.3 )‚ÄĘ TIVICAY or TIVICAY PD should be taken 2 hours before or 6 hours after taking cation-containing antacids or laxatives, sucralfate, oral supplements containing iron or calcium, or buffered medications. When taken with food, TIVICAY and supplements containing calcium or iron can be taken at the same time. (7.3 )7.1 Effect of Dolutegravir on the Pharmacokinetics of Other Agents
In vitro, dolutegravir inhibited the renal organic cation transporters, OCT2 (IC50 = 1.93 microM) and multidrug and toxin extrusion transporter (MATE) 1 (IC50 = 6.34 microM). In vivo, dolutegravir inhibits tubular secretion of creatinine by inhibiting OCT2 and potentially MATE1. Dolutegravir may increase plasma concentrations of drugs eliminated via OCT2 or MATE1 (dofetilide, dalfampridine, and metformin, Table 8) [see Contraindications (4), Drug Interactions (7.3)].
In vitro, dolutegravir inhibited the basolateral renal transporters, organic anion transporter (OAT) 1 (IC50 = 2.12 microM) and OAT3 (IC50 = 1.97 microM). However, in vivo, dolutegravir did not alter the plasma concentrations of tenofovir or para-amino hippurate, substrates of OAT1 and OAT3.
In vitro, dolutegravir did not inhibit (IC50 greater than 50 microM) the following: cytochrome P450 (CYP)1A2, CYP2A6, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, CYP3A, uridine diphosphate glucuronosyltransferase (UGT)1A1, UGT2B7, P-glycoprotein (P-gp), breast cancer resistance protein (BCRP), bile salt export pump (BSEP), organic anion transporter polypeptide (OATP)1B1, OATP1B3, OCT1, multidrug resistance protein (MRP)2, or MRP4. In vitro, dolutegravir did not induce CYP1A2, CYP2B6, or CYP3A4. Based on these data and the results of drug interaction trials, dolutegravir is not expected to affect the pharmacokinetics of drugs that are substrates of these enzymes or transporters.
7.2 Effect of Other Agents on the Pharmacokinetics of Dolutegravir
Dolutegravir is metabolized by UGT1A1 with some contribution from CYP3A. Dolutegravir is also a substrate of UGT1A3, UGT1A9, BCRP, and P-gp in vitro. Drugs that induce those enzymes and transporters may decrease dolutegravir plasma concentration and reduce the therapeutic effect of dolutegravir.
Coadministration of dolutegravir and other drugs that inhibit these enzymes may increase dolutegravir plasma concentration.
Etravirine significantly reduced plasma concentrations of dolutegravir, but the effect of etravirine was mitigated by coadministration of lopinavir/ritonavir or darunavir/ritonavir and is expected to be mitigated by atazanavir/ritonavir (Table 8) [see Drug Interactions (7.3), Clinical Pharmacology (12.3)].
In vitro, dolutegravir was not a substrate of OATP1B1 or OATP1B3.
7.3 Established and Other Potentially Significant Drug Interactions
Table 8 provides clinical recommendations as a result of drug interactions with TIVICAY or TIVICAY PD. These recommendations are based on either drug interaction trials or predicted interactions due to the expected magnitude of interaction and potential for serious adverse events or loss of efficacy [see Dosage and Administration (2), Clinical Pharmacology (12.3)].
Table 8. Established and Other Potentially Significant Drug Interactions: Alterations in Dose or Regimen May Be Recommended Based on Drug Interaction Trials or Predicted Interactions [see Dosage and Administration (2)] INSTI = integrase strand transfer inhibitor. a  See Clinical Pharmacology (12.3) Table 11 or Table 12 for magnitude of interaction. b  The lower dolutegravir exposures observed in INSTI-experienced patients (with certain INSTI-associated resistance substitutions or clinically suspected INSTI resistance [see Microbiology (12.4)]) upon coadministration with certain inducers may result in loss of therapeutic effect and development of resistance to TIVICAY or other coadministered antiretroviral agents.
Concomitant Drug Class: Drug Name
Effect on Concentration of Dolutegravir and/or Concomitant Drug
Clinical Comment
HIV-1 Antiviral Agents
Non-nucleoside reverse transcriptase inhibitor:
  Etravirinea
‚ÜďDolutegravir
Use of TIVICAY or TIVICAY PD with etravirine without coadministration of atazanavir/ritonavir, darunavir/ritonavir, or lopinavir/ritonavir is not recommended.
Non-nucleoside reverse transcriptase inhibitor:
  Efavirenza
‚ÜďDolutegravir
Adjust dose of TIVICAY to twice daily for treatment-na√Įve and treatment-experienced, INSTI-na√Įve adult patients.
In pediatric patients, increase the weight-based dose of TIVICAY or TIVICAY PD to twice daily (Tables 2, 3, and 4).
Use alternative combinations that do not include metabolic inducers where possible for INSTI-experienced patients with certain INSTI-associated resistance substitutions or clinically suspected INSTI resistance.b
Non-nucleoside reverse transcriptase inhibitor:
  Nevirapine
‚ÜďDolutegravir
Avoid coadministration with nevirapine because there are insufficient data to make dosing recommendations.
Protease inhibitors:
  Fosamprenavir/ritonavira
  Tipranavir/ritonavira
‚ÜďDolutegravir
Adjust dose of TIVICAY to twice daily for treatment-na√Įve and treatment-experienced, INSTI-na√Įve adult patients.
In pediatric patients, increase the weight-based dose of TIVICAY or TIVICAY PD to twice daily (Tables 2, 3, and 4).
Use alternative combinations that do not include metabolic inducers where possible for INSTI-experienced patients with certain INSTI-associated resistance substitutions or clinically suspected INSTI resistance.b
Other Agents
  Dofetilide
‚ÜĎDofetilide
Coadministration is contraindicated with TIVICAY or TIVICAY PD [see Contraindications (4)].
  Carbamazepinea
‚ÜďDolutegravir
Adjust dose of TIVICAY to twice daily in treatment-na√Įve or treatment-experienced, INSTI-na√Įve adult patients.
In pediatric patients, increase the weight-based dose of TIVICAY or TIVICAY PD to twice daily (Tables 2, 3, and 4).
Use alternative treatment that does not include carbamazepine where possible for INSTI-experienced patients with certain INSTI-associated resistance substitutions or clinically suspected INSTI resistance.b
  Oxcarbazepine  Phenytoin  Phenobarbital  St. John’s wort (Hypericum perforatum)
‚ÜďDolutegravir
Avoid coadministration with TIVICAY or TIVICAY PD because there are insufficient data to make dosing recommendations.
Medications containing polyvalent cations (e.g., Mg or Al):
  Cation-containing antacidsa or laxatives  Sucralfate  Buffered medications
‚ÜďDolutegravir
Administer TIVICAY or TIVICAY PD 2 hours before or 6 hours after taking medications containing polyvalent cations.
Oral calcium or iron supplements, including multivitamins containing calcium or iron a
‚ÜďDolutegravir
When taken with food, TIVICAY and supplements or multivitamins containing calcium or iron can be taken at the same time. Under fasting conditions, TIVICAY or TIVICAY PD should be taken 2 hours before or 6 hours after taking supplements containing calcium or iron.
Potassium channel blocker:
Dalfampridine
‚ÜĎDalfampridine
Elevated levels of dalfampridine increase the risk of seizures. The potential benefits of taking dalfampridine concurrently with TIVICAY or TIVICAY PD should be considered against the risk of seizures in these patients.
Metformin
‚ÜĎMetformin
Refer to the prescribing information for metformin for assessing the benefit and risk of concomitant use of TIVICAY or TIVICAY PD and metformin.
Rifampina
‚ÜďDolutegravir
Adjust dose of TIVICAY to twice daily for treatment-na√Įve and treatment-experienced, INSTI-na√Įve adult patients.
In pediatric patients, increase the weight-based dose of TIVICAY or TIVICAY PD to twice daily (Tables 2, 3, and 4).
Use alternatives to rifampin where possible for INSTI-experienced patients with certain INSTI-associated resistance substitutions or clinically suspected INSTI resistance.b
7.4 Drugs without Clinically Significant Interactions with Dolutegravir
Based on drug interaction trial results, the following drugs can be coadministered with dolutegravir without a dose adjustment: atazanavir/ritonavir, darunavir/ritonavir, elbasvir/grazoprevir, methadone, midazolam, omeprazole, oral contraceptives containing norgestimate and ethinyl estradiol, prednisone, rifabutin, rilpivirine, sofosbuvir/velpatasvir, and tenofovir [see Clinical Pharmacology (12.3)].
8 Use In Specific Populations
8.1 Pregnancy
Pregnancy Exposure Registry
There is a pregnancy exposure registry that monitors pregnancy outcomes in individuals exposed to TIVICAY or TIVICAY PD during pregnancy. Healthcare providers are encouraged to register patients by calling the Antiretroviral Pregnancy Registry (APR) at 1-800-258-4263.
Risk Summary
Data from two, ongoing birth outcome surveillance studies in Botswana and Eswatini which together include over 14,000 individuals evaluated during pregnancy show similar prevalence of neural tube defects among infants born to individuals taking dolutegravir at the time of conception compared to those born to individuals taking non-dolutegravir-containing regimens at conception or infants born to HIV-negative individuals. (see Data).
There are insufficient human data on the use of dolutegravir during pregnancy to definitively assess a drug-associated risk for birth defects and miscarriage. However, available human data from the APR do not indicate an increased risk of birth defects (see Data). The background risk for major birth defects for the indicated population is unknown. In the U.S. general population, the estimated background rate for major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
In animal reproduction studies, no evidence of adverse developmental outcomes (including neural tube defects) was observed with dolutegravir at systemic exposures (AUC) less than (rabbits) and approximately 27 times (rats) the exposure in humans at the maximum recommended human dose (MRHD) of TIVICAY (see Data).
Data
Human Data:
            Observational studies: The first interim analysis from an ongoing birth outcome surveillance study in Botswana identified an association between dolutegravir and an increased risk of neural tube defects when dolutegravir was administered at the time of conception and in early pregnancy. A subsequent analysis was conducted based on a larger cohort from the birth outcome surveillance study in Botswana and included over 9,460 individuals exposed to dolutegravir at conception, 23,664 individuals exposed to non-dolutegravir-containing regimens, and 170,723 HIV-negative pregnant individuals. The prevalence of neural tube defects in infants delivered to individuals taking dolutegravir at conception was 0.11% (95% CI: 0.05-0.19%). The observed prevalence rate did not differ significantly from that of infants delivered to individuals taking non-dolutegravir-containing regimens (0.11%, 95% CI: 0.07-0.16%), or to HIV-negative individuals (0.06%, 95% CI: 0.05-0.08%).
The Eswatini birth outcome surveillance study includes 9,743 individuals exposed to dolutegravir at conception, 1,838 individuals exposed to non-dolutegravir-containing regimens, and 32,259 HIV-negative pregnant individuals. The prevalence of neural tube defects in infants delivered to individuals taking dolutegravir at conception was 0.08% (95% CI: 0.04-0.16%). The observed prevalence rate did not differ significantly from that of infants delivered to individuals taking non-dolutegravir-containing regimens (0.22%, 95% CI: 0.06-0.56%) or to HIV-negative individuals (0.08%, 95% CI: 0.06-0.12%). The observed prevalence of neural tube defects in infants delivered to individuals taking non-dolutegravir-containing regimens had a wide confidence interval due to low sample size.
Limitations of these birth outcome surveillance studies include insufficient data to determine if baseline characteristics were balanced between the study groups or to assess other factors such as the use of folic acid during the preconception or first trimester periods.
            Antiretroviral Pregnancy Registry: Based on prospective reports to the APR, of 1,377 exposures to dolutegravir during pregnancy resulting in live births (including 874 exposed in the first trimester), the prevalence of defects in live births was 3.3% (95% CI: 2.2% to 4.7%) following first-trimester exposure to dolutegravir-containing regimens and 5.0% (95% CI: 3.2% to 7.3%) following second-/third-trimester exposure to dolutegravir-containing regimens. In the U.S. reference population of the Metropolitan Atlanta Congenital Defects Program (MACDP), the background birth defect rate was 2.7%.
Dolutegravir has been shown to cross the placenta. In a clinical trial in Uganda and South Africa in women during the last trimester of pregnancy receiving dolutegravir 50 mg once daily, the ratio of median dolutegravir concentration in fetal umbilical cord to that in maternal peripheral plasma was 1.21 (range 0.51-2.11) (n = 15).
Animal Data:
Dolutegravir was administered orally at up to 1,000 mg/kg daily to pregnant rats and rabbits on Gestation Days 6 to 17 and 6 to 18, respectively, and to rats on Gestation Day 6 to Lactation/Postpartum Day 20. No adverse effects on embryo-fetal (rats and rabbits) or pre/postnatal (rats) development were observed at up to the highest dose tested. During organogenesis, systemic exposures (AUC) to dolutegravir in rabbits were less than the exposure in humans at the MRHD and in rats were approximately 27 times the exposure in humans at the MRHD. In the rat pre/postnatal development study, decreased body weight of the developing offspring was observed during lactation at a maternally toxic dose (approximately 27 times human exposure at the MRHD).
8.2 Lactation
Risk Summary
Dolutegravir is present in human milk. It is not known whether dolutegravir affects human milk production or has effects on the breastfed infant.
Potential risks of breastfeeding include: (1) HIV‚ÄĎ1 transmission (in HIV-1‚Äďnegative infants), (2) developing viral resistance (in HIV-1‚Äďpositive infants), and (3) adverse reactions in a breastfed infant similar to those seen in adults.
8.4 Pediatric Use
The safety, pharmacokinetics, and effectiveness of TIVICAY and TIVICAY PD were evaluated in 75 HIV-1‚Äďinfected, treatment-na√Įve or treatment-experienced, INSTI-na√Įve pediatric and adolescent subjects aged 4 weeks to less than 18¬†years weighing at least 3¬†kg in an ongoing, open-label, multicenter, dose-finding clinical trial, IMPAACT P1093 [see Adverse Reactions (6.1), Clinical Pharmacology (12.3), Clinical Studies (14.3)]. Additional pharmacokinetics data were evaluated in 2 pharmacokinetic substudies in ODYSSEY, an ongoing open-label, randomized, non-inferiority trial to evaluate the safety, efficacy, and pharmacokinetic parameters of TIVICAY or TIVICAY PD plus two NRTIs compared with standard of care in HIV-1‚Äďinfected pediatric subjects younger than 18¬†years [see Clinical Pharmacology (12.3)].
Overall, the safety data in pediatric subjects from the IMPAACT P1093 trial were comparable to those observed in adults [see Adverse Reactions (6.1)]. The pharmacokinetic parameters of TIVICAY or TIVICAY PD in pediatric subjects from IMPAACT P1093 and ODYSSEY were comparable to those of adults receiving 50 mg once daily or twice daily [see Clinical Pharmacology (12.3)]. The effectiveness observed in IMPAACT P1093 is comparable to that of treatment-experienced adult subjects.
Safety and effectiveness of TIVICAY or TIVICAY PD have not been established in pediatric patients aged less than 4 weeks or weighing less than 3 kg or in any pediatric patients who are INSTI-experienced with documented or clinically suspected resistance to other INSTIs (e.g., raltegravir, elvitegravir).
8.5 Geriatric Use
Clinical trials of TIVICAY did not include sufficient numbers of subjects aged 65 and older to determine whether they respond differently from younger subjects. In general, caution should be exercised in the administration of TIVICAY in elderly patients reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy [see Clinical Pharmacology (12.3)].
8.6 Hepatic Impairment
No clinically important pharmacokinetic differences between subjects with moderate hepatic impairment and matching healthy subjects were observed. No dosage adjustment is necessary for patients with mild to moderate hepatic impairment (Child-Pugh Score A or B). The effect of severe hepatic impairment (Child-Pugh Score C) on the pharmacokinetics of dolutegravir has not been studied. Therefore, TIVICAY and TIVICAY PD are not recommended for use in patients with severe hepatic impairment [see Clinical Pharmacology (12.3)].
8.7 Renal Impairment
Dolutegravir plasma concentrations were decreased in subjects with severe renal impairment compared with those in matched healthy controls. However, no dosage adjustment is necessary for treatment-na√Įve or treatment-experienced and INSTI-na√Įve patients with mild, moderate, or severe renal impairment or for INSTI-experienced patients (with certain INSTI-associated resistance substitutions or clinically suspected INSTI resistance) with mild or moderate renal impairment. Caution is warranted for INSTI-experienced patients (with certain INSTI-associated resistance substitutions or clinically suspected INSTI resistance [see Microbiology (12.4)]) with severe renal impairment, as the decrease in dolutegravir concentrations may result in loss of therapeutic effect and development of resistance to TIVICAY, TIVICAY PD, or other coadministered antiretroviral agents [see Clinical Pharmacology (12.3)]. There is inadequate information to recommend appropriate dosing of dolutegravir in patients requiring dialysis.
10 Overdosage
There is no known specific treatment for overdose with TIVICAY or TIVICAY PD. If overdose occurs, the patient should be monitored, and standard supportive treatment applied as required. As dolutegravir is highly bound to plasma proteins, it is unlikely that it will be significantly removed by dialysis.
11 Description
TIVICAY contains dolutegravir, as dolutegravir sodium, an HIV INSTI. The chemical name of dolutegravir sodium is sodium (4R,12aS)-9-{[(2,4-difluorophenyl)methyl]carbamoyl}-4-methyl-6,8-dioxo-3,4,6,8,12,12a-hexahydro-2H-pyrido[1',2':4,5]pyrazino[2,1-b][1,3]oxazin-7-olate. The empirical formula is C20H18F2N3NaO5, and the molecular weight is 441.36 g/mol. It has the following structural formula:
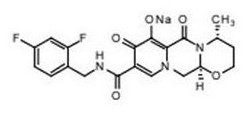
Dolutegravir sodium is a white to light yellow powder and is slightly soluble in water.
Each film-coated tablet of TIVICAY for oral administration contains 10.5, 26.3, or 52.6¬†mg of dolutegravir sodium, which is equivalent to 10, 25, or 50¬†mg dolutegravir free acid, respectively, and the following inactive ingredients: D-mannitol, microcrystalline cellulose, povidone K29/32, sodium starch glycolate, and sodium stearyl fumarate. The tablet film‚ÄĎcoating contains the inactive ingredients iron oxide yellow (25-mg and 50-mg tablets only), macrogol/PEG, polyvinyl alcohol-part hydrolyzed, talc, and titanium dioxide.
Each TIVICAY PD tablet for oral suspension contains 5.26 mg of dolutegravir sodium, which is equivalent to 5 mg dolutegravir free acid, and the following inactive ingredients: calcium sulfate dihydrate, crospovidone, mannitol, microcrystalline cellulose, povidone K29/32, silicified microcrystalline cellulose, sodium starch glycolate, strawberry cream flavor, sucralose, and sodium stearyl fumarate. The tablet film-coating contains hypromellose, polyethylene glycol, and titanium dioxide.
12 Clinical Pharmacology
12.1 Mechanism of Action
Dolutegravir is an HIV-1 antiretroviral agent [see Microbiology (12.4)].
12.2 Pharmacodynamics
Effects on Electrocardiogram
In a randomized, placebo-controlled, cross-over trial, 42¬†healthy subjects received single-dose oral administrations of placebo, dolutegravir 250-mg suspension (exposures approximately 3‚Äďfold of the 50-mg once-daily dose at steady state), and moxifloxacin 400¬†mg (active control) in random sequence. After baseline and placebo adjustment, the maximum mean QTc change based on Fridericia correction method (QTcF) for dolutegravir was 2.4¬†msec (1-sided 95% upper CI: 4.9¬†msec). TIVICAY did not prolong the QTc interval over 24¬†hours postdose.
Effects on Renal Function
The effect of dolutegravir on renal function was evaluated in an open-label, randomized, 3‚ÄĎarm, parallel, placebo-controlled trial in healthy subjects (n¬†=¬†37) who received dolutegravir 50¬†mg once daily (n¬†=¬†12), dolutegravir 50¬†mg twice daily (n¬†=¬†13), or placebo once daily (n¬†=¬†12) for 14¬†days. A decrease in creatinine clearance, as determined by 24-hour urine collection, was observed with both doses of dolutegravir after 14 days of treatment in subjects who received 50¬†mg once daily (9% decrease) and 50¬†mg twice daily (13% decrease). Neither dose of dolutegravir had a significant effect on the actual glomerular filtration rate (determined by the clearance of probe drug, iohexol) or effective renal plasma flow (determined by the clearance of probe drug, para-amino hippurate) compared with the placebo.
12.3 Pharmacokinetics
The pharmacokinetic properties of dolutegravir have been evaluated in healthy adult subjects and HIV‚ÄĎ1‚Äďinfected adult subjects. Exposure to dolutegravir was generally similar between healthy subjects and HIV‚ÄĎ1‚Äďinfected subjects. The non-linear exposure of dolutegravir following 50¬†mg twice daily compared with 50¬†mg once daily in HIV‚ÄĎ1‚Äďinfected subjects (Table 9) was attributed to the use of metabolic inducers in the background antiretroviral regimens of subjects receiving dolutegravir 50¬†mg twice daily in clinical trials.
Table 9. Dolutegravir Steady-State Pharmacokinetic Parameter Estimates in HIV-1‚ÄďInfected Adults a¬†¬†Based on population pharmacokinetic analyses using data from SPRING-1 and SPRING-2. b¬†¬†Based on population pharmacokinetic analyses using data from VIKING (ING112961) and VIKING-3.
Parameter
50 mg Once Daily
Geometric Meana (%CV)
50 mg Twice Daily
Geometric Meanb (%CV)
AUC(0-24) (mcg‚ąôh/mL)
53.6 (27)
75.1 (35)
Cmax (mcg/mL)
3.67 (20)
4.15 (29)
Cmin (mcg/mL)
1.11 (46)
2.12 (47)
TIVICAY tablets and TIVICAY PD tablets for oral suspension are not bioequivalent. The relative bioavailability of TIVICAY PD is approximately 1.6-fold higher than TIVICAY; therefore, the 2 dosage forms are not substitutable on a milligram-per-milligram basis [see Dosage and Administration (2.3)].
Absorption
Following oral administration of dolutegravir, peak plasma concentrations were observed 1 to 3 hours postdose. With once-daily dosing, pharmacokinetic steady state is achieved within approximately 5 days with average accumulation ratios for AUC, Cmax, and C24 h ranging from 1.2 to 1.5.
Dolutegravir plasma concentrations increased in a less than dose-proportional manner above 50¬†mg. Dolutegravir is a P‚ÄĎgp substrate in vitro. The absolute bioavailability of dolutegravir has not been established.
Effect of Food: TIVICAY or TIVICAY PD may be taken with or without food. Food increased the extent of absorption and slowed the rate of absorption of dolutegravir following a 50-mg dose of TIVICAY. Low-, moderate-, and high-fat meals increased dolutegravir AUC(0-‚ąě) by 33%, 41%, and 66%; increased Cmax by 46%, 52%, and 67%; and prolonged Tmax to 3, 4, and 5¬†hours from 2¬†hours under fasted conditions, respectively.
Distribution
Dolutegravir is highly bound (greater than or equal to 98.9%) to human plasma proteins based on in vivo data and binding is independent of plasma concentration of dolutegravir. The apparent volume of distribution (Vd/F) following 50-mg once-daily administration is estimated at 17.4 L based on a population pharmacokinetic analysis.
Cerebrospinal Fluid (CSF): In 12 treatment-na√Įve subjects on dolutegravir 50¬†mg daily plus abacavir/lamivudine, the median dolutegravir concentration in CSF was 13.2¬†ng/mL (range: 3.74 ng/mL to 18.3 ng/mL) 2 to 6¬†hours postdose after 16¬†weeks of treatment. The clinical relevance of this finding has not been established.
Elimination
Dolutegravir has a terminal half-life of approximately 14 hours and an apparent clearance (CL/F) of 1.0 L/h based on population pharmacokinetic analyses.
Metabolism: Dolutegravir is primarily metabolized via UGT1A1 with some contribution from CYP3A.
Polymorphisms in Drug‚ÄĎMetabolizing Enzymes: In a meta-analysis of healthy subject trials, subjects with UGT1A1 (n¬†=¬†7) genotypes conferring poor dolutegravir metabolism had a 32% lower clearance of dolutegravir and 46% higher AUC compared with subjects with genotypes associated with normal metabolism via UGT1A1 (n¬†=¬†41).
Excretion: After a single oral dose of [14C] dolutegravir, 53% of the total oral dose was excreted unchanged in feces. Thirty-one percent of the total oral dose was excreted in urine, represented by an ether glucuronide of dolutegravir (18.9% of total dose), a metabolite formed by oxidation at the benzylic carbon (3.0% of total dose), and its hydrolytic N-dealkylation product (3.6% of total dose). Renal elimination of unchanged drug was low (less than 1% of the dose).
Specific Populations
Pediatric Patients: The pharmacokinetics of dolutegravir were evaluated in the IMPAACT P1093 trial and in 2 weight-band-based pharmacokinetic substudies from the ODYSSEY trial. Steady-state plasma exposure at doses by weight band are summarized in Table 10  [see Clinical Studies (14.3)].
Mean dolutegravir AUC0-24h and C24h in HIV-1‚Äďinfected pediatric subjects were comparable to those in adults after 50¬†mg once daily or 50¬†mg twice daily. Mean Cmax is higher in pediatrics, but the increase is not considered clinically significant as the safety profiles were similar in pediatric and adult subjects [see Use in Specific Populations (8.4)].
Table 10. Summary of Pharmacokinetic Parameters in Pediatric HIV-1‚ÄďInfected Subjects (Pooled Analyses for IMPAACT P1093 and ODYSSEYa Trials) a¬†¬†Data from 2 weight-band-based pharmacokinetic substudies in the ODYSSEY trial. b¬†¬†The bioavailability of TIVICAY PD tablets for oral suspension is ~1.6-fold that of TIVICAY tablets.
Weight Band
Doseb of TIVICAY or TIVICAY PD
n
Pharmacokinetic Parameter Geometric Mean (%CV)
Cmax (mcg/mL)
AUC0-24h (mcg‚ąôh/mL)
C24h (ng/mL)
3 kg to <6 kg
TIVICAY PD5 mg once daily
8
3.80 (34)
49.37 (49)
962 (98)
6 kg to <10 kg
TIVICAY PD15 mg once daily
17
5.27 (50)
57.17 (76)
706 (177)
10 kg to <14 kg
TIVICAY PD20 mg once daily
13
5.99 (33)
68.75 (48)
977 (100)
14 kg to <20 kg
TIVICAY PD25 mg once daily
19
5.97 (42)
58.97 (44)
725 (75)
20 kg to <25 kg
TIVICAY PD30 mg once daily
9
7.16 (26)
71.53 (26)
759 (73)
‚Č•20¬†kg
TIVICAY50 mg once daily
49
4.92 (40)
54.98 (43)
778 (62)
Geriatric Patients: Population pharmacokinetic analysis indicated age had no clinically relevant effect on the pharmacokinetics of dolutegravir.
Patients with Hepatic Impairment: In a trial comparing 8 subjects with moderate hepatic impairment (Child-Pugh Score B) with 8 matched healthy controls, exposure of dolutegravir from a single 50-mg dose was similar between the 2 groups. The effect of severe hepatic impairment (Child-Pugh Score C) on the pharmacokinetics of dolutegravir has not been studied.
Patients with Renal Impairment: In a trial evaluating the pharmacokinetics of a single 50-mg tablet of dolutegravir comparing 8 subjects with severe renal impairment (CrCl less than 30 mL/min) with 8 matched healthy controls, AUC, Cmax, and C24 of dolutegravir were lower by 40%, 23%, and 43%, respectively, compared with those in matched healthy subjects. Population pharmacokinetic analysis using data from SAILING and VIKING-3 trials indicated that mild and moderate renal impairment had no clinically relevant effect on the exposure of dolutegravir. There is inadequate information to recommend appropriate dosing of dolutegravir in patients requiring dialysis.
HBV or HCV Co-infected Patients: Population analyses using pooled pharmacokinetic data from adult trials indicated no clinically relevant effect of HCV co-infection on the pharmacokinetics of dolutegravir. There were limited data on HBV co-infection.
Gender and Race: Population analyses using pooled pharmacokinetic data from adult trials indicated gender and race had no clinically relevant effect on the exposure of dolutegravir.
Drug Interaction Studies
Drug interaction trials were performed with TIVICAY and other drugs likely to be coadministered or commonly used as probes for pharmacokinetic interactions. The effects of dolutegravir on the exposure of coadministered drugs are summarized in Table 11 and the effects of coadministered drugs on the exposure of dolutegravir are summarized in Table 12.
Dosing or regimen recommendations as a result of established and other potentially significant drug-drug interactions with TIVICAY are provided in Table 8 [see Dosage and Administration (2.2), Drug Interactions (7.3)].
Table 11. Summary of Effect of Dolutegravir on the Pharmacokinetics of Coadministered Drugs a  The number of subjects represents the maximum number of subjects that were evaluated.
Coadministered Drug(s)
and Dose(s)
Dose of TIVICAY
n
Geometric Mean Ratio (90% CI) of Pharmacokinetic Parameters of Coadministered Drug with/without Dolutegravir No Effect = 1.00
Cmax
AUC
CŌĄ or C24
Elbasvir
  50 mg once daily
50 mg
single dose
12
0.97
(0.89, 1.05)
0.98
(0.93, 1.04)
0.98
(0.93, 1.03)
Ethinyl estradiol  0.035 mg
50 mgtwice daily
15
0.99(0.91 to 1.08)
1.03(0.96 to 1.11)
1.02(0.93 to 1.11)
Grazoprevir
  200 mg once daily
50 mg
single dose
12
0.64
(0.44, 0.93)
0.81
(0.67, 0.97)
0.86
(0.79, 0.93)
Metformin  500 mg twice daily
50 mgonce daily
15a
1.66(1.53 to 1.81)
1.79(1.65 to 1.93)
_
Metformin  500 mg twice daily
50 mgtwice daily
15a
2.11(1.91 to 2.33)
2.45(2.25 to 2.66)
_
Methadone  16 to 150 mg
50 mgtwice daily
11
1.00(0.94 to 1.06)
0.98(0.91 to 1.06)
0.99(0.91 to 1.07)
Midazolam   3 mg
25 mgonce daily
10
_
0.95(0.79 to 1.15)
_
Norelgestromin  0.25 mg
50 mgtwice daily
15
0.89(0.82 to 0.97)
0.98(0.91 to 1.04)
0.93(0.85 to 1.03)
Rilpivirine  25 mg once daily
50 mgonce daily
16
1.10(0.99 to 1.22)
1.06(0.98 to 1.16)
1.21(1.07 to 1.38)
Sofosbuvir
  400 mg once daily
  Metabolite (GS-331007)
50 mg
once daily
24
0.88
(0.80, 0.98)
0.92
(0.85, 0.99)
NA
1.01
(0.93, 1.10)
0.99
(0.97, 1.01)
0.99
(0.97, 1.01)
Tenofovir disoproxil fumarate  300 mg once daily
50 mgonce daily
15
1.09(0.97 to 1.23)
1.12(1.01 to 1.24)
1.19(1.04 to 1.35)
Velpatasvir
  100 mg once daily
50 mg
once daily
24
0.94
(0.86, 1.02)
0.91
(0.84, 0.98)
0.88
(0.82, 0.94)
Table 12. Summary of Effect of Coadministered Drugs on the Pharmacokinetics of Dolutegravir a  The number of subjects represents the maximum number of subjects that were evaluated. b  Comparison is rifampin taken with dolutegravir 50 mg twice daily compared with dolutegravir 50 mg twice daily. c  Comparison is rifampin taken with dolutegravir 50 mg twice daily compared with dolutegravir 50 mg once daily.
Coadministered Drug(s)
and Dose(s)
Dose of TIVICAY
n
Geometric Mean Ratio (90% CI) of Dolutegravir Pharmacokinetic Parameters with/without Coadministered Drugs No Effect = 1.00
Cmax
AUC
CŌĄ or C24
Atazanavir   400 mg once daily
30 mg once daily
12
1.50(1.40 to 1.59)
1.91(1.80 to 2.03)
2.80(2.52 to 3.11)
Atazanavir/ritonavir  300/100 mg once daily
30 mg once daily
12
1.34(1.25 to 1.42)
1.62(1.50 to 1.74)
2.21(1.97 to 2.47)
Darunavir/ritonavir  600/100 mg twice daily
30 mg once daily
15
0.89(0.83 to 0.97)
0.78(0.72 to 0.85)
0.62(0.56 to 0.69)
Efavirenz   600 mg once daily
50 mgonce daily
12
0.61(0.51 to 0.73)
0.43(0.35 to 0.54)
0.25(0.18 to 0.34)
Elbasvir/grazoprevir
  50/200 mg once daily
50 mg
single dose
12
1.22
(1.05, 1.40)
1.16
(1.00, 1.34)
1.14
(0.95, 1.36)
Etravirine  200 mg twice daily
50 mgonce daily
16
0.48(0.43 to 0.54)
0.29(0.26 to 0.34)
0.12(0.09 to 0.16)
Etravirine + darunavir/ritonavir   200 mg + 600/100 mg twice daily
50 mgonce daily
9
0.88(0.78 to 1.00)
0.75(0.69 to 0.81)
0.63(0.52 to 0.76)
Etravirine + lopinavir/ritonavir   200 mg + 400/100 mg twice daily
50 mg once daily
8
1.07(1.02 to 1.13)
1.11(1.02 to 1.20)
1.28(1.13 to 1.45)
Fosamprenavir/ritonavir  700 mg/100 mg twice daily
50 mg once daily
12
0.76(0.63 to 0.92)
0.65(0.54 to 0.78)
0.51(0.41 to 0.63)
Lopinavir/ritonavir  400/100 mg twice daily
30 mg once daily
15
1.00(0.94 to 1.07)
0.97(0.91 to 1.04)
0.94(0.85 to 1.05)
Rilpivirine  25 mg once daily
50 mg once daily
16
1.13 (1.06 to 1.21)
1.12 (1.05 to 1.19)
1.22 (1.15 to 1.30)
Tenofovir  300 mg once daily
50 mg once daily
15
0.97(0.87 to 1.08)
1.01(0.91 to 1.11)
0.92
(0.82 to 1.04)
Tipranavir/ritonavir  500/200 mg twice daily
50 mg once daily
14
0.54(0.50 to 0.57)
0.41(0.38 to 0.44)
0.24(0.21 to 0.27)
Antacid (MAALOX)  simultaneous administration
50 mg single dose
16
0.28(0.23 to 0.33)
0.26(0.22 to 0.32)
0.26(0.21 to 0.31)
Antacid (MAALOX)   2 h after dolutegravir
50 mg single dose
16
0.82(0.69 to 0.98)
0.74(0.62 to 0.90)
0.70(0.58 to 0.85)
Calcium carbonate 1,200 mg  simultaneous administration (fasted)
50 mgsingle dose
12
0.63(0.50 to 0.81)
0.61(0.47 to 0.80)
0.61(0.47 to 0.80)
Calcium carbonate 1,200 mg  simultaneous administration (fed)
50 mgsingle dose
11
1.07(0.83 to 1.38)
1.09(0.84 to 1.43)
1.08(0.81 to 1.42)
Calcium carbonate 1,200 mg  2 h after dolutegravir
50 mgsingle dose
11
1.00(0.78 to 1.29)
0.94(0.72 to 1.23)
0.90(0.68 to 1.19)
Carbamazepine  300 mg twice daily
50 mg once daily
16a
0.67(0.61 to 0.73)
0.51(0.48 to 0.55)
0.27(0.24 to 0.31)
Ferrous fumarate 324 mg  simultaneous administration (fasted)
50 mgsingle dose
11
0.43(0.35 to 0.52)
0.46(0.38 to 0.56)
0.44(0.36 to 0.54)
Ferrous fumarate 324 mg  simultaneous administration (fed)
50 mgsingle dose
11
1.03(0.84 to 1.26)
0.98(0.81 to 1.20)
1.00(0.81 to 1.23)
Ferrous fumarate 324 mg  2 h after dolutegravir
50 mgsingle dose
10
0.99(0.81 to 1.21)
0.95(0.77 to 1.15)
0.92(0.74 to 1.13)
Multivitamin (One-A-Day)  simultaneous administration
50 mgsingle dose
16
0.65(0.54 to 0.77)
0.67(0.55 to 0.81)
0.68(0.56 to 0.82)
Omeprazole  40 mg once daily
50 mg single dose
12
0.92(0.75 to 1.11)
0.97(0.78 to 1.20)
0.95(0.75 to 1.21)
Prednisone  60 mg once daily with taper
50 mg once daily
12
1.06(0.99 to 1.14)
1.11(1.03 to 1.20)
1.17(1.06 to 1.28)
Rifampinb   600 mg once daily
50 mg twice daily
11
0.57(0.49 to 0.65)
0.46(0.38 to 0.55)
0.28(0.23 to 0.34)
Rifampinc   600 mg once daily
50 mg twice daily
11
1.18(1.03 to 1.37)
1.33(1.15 to 1.53)
1.22(1.01 to 1.48)
Rifabutin  300 mg once daily
50 mg once daily
9
1.16(0.98 to 1.37)
0.95(0.82 to 1.10)
0.70(0.57 to 0.87)
12.4 Microbiology
Mechanism of Action
Dolutegravir inhibits HIV integrase by binding to the integrase active site and blocking the strand transfer step of retroviral DNA integration which is essential for the HIV replication cycle. Strand transfer biochemical assays using purified HIV-1 integrase and pre-processed substrate DNA resulted in IC50 values of 2.7 nM and 12.6 nM.
Antiviral Activity in Cell Culture
Dolutegravir exhibited antiviral activity against laboratory strains of wild-type HIV-1 with mean EC50 values of 0.5 nM (0.21 ng/mL) to 2.1 nM (0.85 ng/mL) in peripheral blood mononuclear cells (PBMCs) and MT-4 cells. Dolutegravir exhibited antiviral activity against 13 clinically diverse clade B isolates with a mean EC50 value of 0.52 nM in a viral integrase susceptibility assay using the integrase coding region from clinical isolates. Dolutegravir demonstrated antiviral activity in cell culture against a panel of HIV-1 clinical isolates (3 in each group of M clades A, B, C, D, E, F, and G, and 3 in group O) with EC50 values ranging from 0.02 nM to 2.14 nM for HIV-1. Dolutegravir EC50 values against 3 HIV-2 clinical isolates in PBMC assays ranged from 0.09 nM to 0.61 nM.
Antiviral Activity in Combination with Other Antiviral Agents
The antiviral activity of dolutegravir was not antagonistic when combined with the INSTI, raltegravir; non-nucleoside reverse transcriptase inhibitors (NNRTIs), efavirenz or nevirapine; the NRTIs, abacavir or stavudine; the protease inhibitors (PIs), amprenavir or lopinavir; the CCR5 co-receptor antagonist, maraviroc; or the fusion inhibitor, enfuvirtide. Dolutegravir antiviral activity was not antagonistic when combined with the HBV reverse transcriptase inhibitor, adefovir, or inhibited by the antiviral, ribavirin.
Resistance
Cell Culture: Dolutegravir-resistant viruses were selected in cell culture starting from different wild-type HIV-1 strains and clades. Amino acid substitutions E92Q, G118R, S153F or Y, G193E or R263K emerged in different passages and conferred decreased susceptibility to dolutegravir of up to 4-fold. Passage of mutant viruses containing the Q148R or Q148H substitutions selected for additional substitutions in integrase that conferred decreased susceptibility to dolutegravir (fold-change increase of 13 to 46). The additional integrase substitutions included T97A, E138K, G140S, and M154I. Passage of mutant viruses containing both G140S and Q148H selected for L74M, E92Q, and N155H.
Treatment-Na√Įve Subjects: No subject who received dolutegravir 50-mg once-daily in the treatment-na√Įve trials SPRING-2 (96 weeks) and SINGLE (144 weeks) had a detectable decrease in susceptibility to dolutegravir or background NRTIs in the resistance analysis subset (n¬†=¬†12 with HIV-1 RNA greater than 400 copies per mL at failure or last visit and having resistance data). Two virologic failure subjects in SINGLE had treatment-emergent G/D/E193D and G193G/E integrase substitutions at Week 84 and Week 108, respectively, and 1 subject with 275¬†copies/mL HIV-1 RNA had a treatment-emergent Q157Q/P integrase substitution detected at Week 24. None of these subjects had a corresponding decrease in dolutegravir susceptibility. No treatment-emergent genotypic resistance to the background regimen was observed in the dolutegravir arm in either the SPRING-2 or SINGLE trials. No treatment-emergent primary resistance substitutions were observed in either treatment group in the FLAMINGO trial through Week 96.
Treatment-Experienced, Integrase Strand Transfer Inhibitor-Na√Įve Subjects: In the dolutegravir arm of the SAILING trial for treatment-experienced and INSTI-na√Įve subjects (n¬†=¬†354), treatment-emergent integrase substitutions were observed in 6 of 28 (21%) subjects who had virologic failure and resistance data. In 5 of the 6 subjects‚Äô isolates emergent INSTI substitutions included L74L/M/I, Q95Q/L, V151V/I (n¬†=¬†1 each), and R263K (n¬†=¬†2). The change in dolutegravir phenotypic susceptibility for these 5 subject isolates was less than 2-fold. One subject isolate had pre-existing raltegravir resistance substitutions E138A, G140S, and Q148H at baseline and had additional emergent INSTI-resistance substitutions T97A and E138A/T with a corresponding 148-fold reduction in dolutegravir susceptibility at failure. In the comparator raltegravir arm, 21 of 49 (43%) subjects with post-baseline resistance data had evidence of emergent INSTI-resistance substitutions (L74M, E92Q, T97A, E138Q, G140S/A, Y143R/C, Q148H/R, V151I, N155H, E157Q, and G163K/R) and raltegravir phenotypic resistance.
Virologically Suppressed Subjects: SWORD-1 and SWORD-2 are identical trials in virologically suppressed subjects receiving 2 NRTIs plus either an INSTI, an NNRTI, or a PI, that switched to dolutegravir plus rilpivirine (n¬†=¬†513) or remained on their current antiviral regimen (n¬†=¬†511). In the pooled SWORD-1 and SWORD-2 trials, 12 subjects (7 in SWORD-1 and 5 in SWORD-2) had confirmed virologic failure (HIV-1 RNA greater than 200 copies/mL) while receiving dolutegravir plus rilpivirine at any time through Week 148. Ten of the confirmed virologic failures had post-baseline resistance data, with 6 isolates showing evidence of rilpivirine resistance, and 2 with evidence of dolutegravir resistance substitutions. Six isolates showed genotypic and/or phenotypic resistance to rilpivirine with emergent NNRTI-resistance substitutions E138E/A (rilpivirine 1.6-fold change), M230M/L (rilpivirine 2-fold change), L100L/I, K101Q, and E138A (rilpivirine 4.1-fold change), K101K/E (rilpivirine 1.2-fold change), K101K/E, M230M/L (rilpivirine 2-fold change), and L100L/V/M, M230M/L (rilpivirine 31-fold change). In addition, 1 virologic failure subject had NNRTI‚ÄĎresistance substitutions K103N and V179I at Week 88 with rilpivirine phenotypic fold change of 5.2 but had no baseline sample.
One virologic failure isolate had emergent INSTI‚ÄĎresistance substitution V151V/I present post-baseline with baseline INSTI‚ÄĎresistance substitutions N155N/H and G163G/R (by exploratory HIV proviral DNA archive sequencing); no integrase phenotypic data were available for this isolate at virologic failure. One other subject had the dolutegravir resistance substitution G193E at baseline and virologic failure, but no detectable phenotypic resistance (fold change¬†=¬†1.02) at Week¬†24.
No resistance-associated substitutions were observed for the 2 subjects meeting confirmed virologic failure in the comparative current antiretroviral regimen arms at Week 48.
Treatment-Experienced, Integrase Strand Transfer Inhibitor-Experienced Subjects: VIKING-3 examined the efficacy of dolutegravir 50 mg twice daily plus optimized background therapy in subjects with prior or current virologic failure on an INSTI- (elvitegravir or raltegravir) containing regimen. Use of TIVICAY in INSTI-experienced patients should be guided by the number and type of baseline INSTI substitutions. The efficacy of TIVICAY 50 mg twice daily is reduced in patients with an INSTI-resistance Q148 substitution plus 2 or more additional INSTI-resistance substitutions, including T66A, L74I/M, E138A/K/T, G140S/A/C, Y143R/C/H, E157Q, G163S/E/K/Q, or G193E/R.
Response by Baseline Genotype
Of the 183 subjects with baseline data, 30% harbored virus with a substitution at Q148, and 33% had no primary INSTI-resistance substitutions (T66A/I/K, E92Q/V, Y143R/C/H, Q148H/R/K, and N155H) at baseline, but had historical genotypic evidence of INSTI-resistance substitutions, phenotypic evidence of elvitegravir or raltegravir resistance, or genotypic evidence of INSTI-resistance substitutions at screening.
Response rates by baseline genotype were analyzed in an ‚Äúas-treated‚ÄĚ analysis at Week 48 (n¬†=¬†175) (Table 13). The response rate at Week 48 to dolutegravir-containing regimens was 47% (24 of 51) when Q148 substitutions were present at baseline; Q148 was always present with additional INSTI-resistance substitutions (Table 13). In addition, a diminished virologic response of 40% (6 of 15) was observed when the substitution E157Q or K was present at baseline with other INSTI-resistance substitutions but without a Q148H or R substitution.
Table 13. Response by Baseline Integrase Genotype in Subjects with Prior Experience to an Integrase Strand Transfer Inhibitor in VIKING-3 INSTI = integrase strand transfer inhibitor. a  Includes INSTI-resistance substitutions Y143R/C/H and N155H. b  INSTI-resistance substitutions included T66A, L74I/M, E138A/K/T, G140S/A/C, Y143R/C/H, E157Q, G163S/E/K/Q, or G193E/R. Two additional subjects had baseline genotypes of Q148Q/R plus L74L/I/M (virologic failure) and Q148R plus E138K (responder). c  The most common pathway with Q148H/R + greater than or equal to 2 INSTI-resistance substitutions had Q148+G140+E138 substitutions (n = 16).
Baseline Genotype
Week 48 (<50 copies/mL) n = 175
Overall Response
66% (116/175)
No Q148 substitutiona
74% (92/124)
Q148H/R + G140S/A/C without additional INSTI-resistance substitutionb
61% (17/28)
Q148H/R + ‚Č•2 INSTI-resistance substitutionsb,c
29% (6/21)
Response by Baseline Phenotype
Response rates by baseline phenotype were analyzed in an as-treated analysis using all subjects with available baseline phenotypes through Week 48 (n = 163) (Table 14). These baseline phenotypic groups are based on subjects enrolled in VIKING-3 and are not meant to represent definitive clinical susceptibility cut points for dolutegravir. The data are provided to guide clinicians on the likelihood of virologic success based on pretreatment susceptibility to dolutegravir in INSTI-resistant patients.
Table 14. Response by Baseline Dolutegravir Phenotype (Fold-Change from Reference) in Subjects with Prior Experience to an Integrase Strand Transfer Inhibitor in VIKING-3
Baseline Dolutegravir Phenotype (Fold-Change from Reference)
Response at Week 48 (<50 copies/mL) Subset n = 163
Overall Response
64% (104/163)
<3-fold change
72% (83/116)
3- <10-fold change
53% (18/34)
‚Č•10-fold change
23% (3/13)
Integrase Strand Transfer Inhibitor Treatment-Emergent Resistance
There were 50 subjects with virologic failure on the dolutegravir twice-daily regimen in VIKING-3 with HIV-1 RNA greater than 400 copies/mL at the failure timepoint, Week 48 or beyond, or the last timepoint on trial. Thirty-nine subjects with virologic failure had resistance data that were used in the Week 48 analysis. In the Week 48 resistance analysis 85% (33 of 39) of the subjects with virologic failure had treatment-emergent INSTI-resistance substitutions in their isolates. The most common treatment-emergent INSTI-resistance substitution was T97A. Other frequently emergent INSTI-resistance substitutions included L74M, I or V, E138K or A, G140S, Q148H, R or K, M154I, or N155H. Substitutions E92Q, Y143R or C/H, S147G, V151A, and E157E/Q each emerged in 1 to 3 subjects’ isolates. At failure, the median dolutegravir fold-change from reference was 61-fold (range: 0.75 to 209) for isolates with emergent INSTI-resistance substitutions (n = 33).
Resistance to one or more background drugs in the dolutegravir twice-daily regimen also emerged in 49% (19 of 39) of subjects in the Week 48 resistance analysis.
In VIKING-4 (ING116529), 30 subjects with current virological failure on an INSTI-containing regimen and genotypic evidence of INSTI-resistance substitutions at screening were randomized to receive either dolutegravir 50 mg twice daily or placebo with the current failing regimen for 7 days and then all subjects received open-label dolutegravir plus optimized background regimen from Day 8. Virologic responses at Week 48 by baseline genotypic and phenotypic INSTI-resistance categories and the INSTI resistance-associated substitutions that emerged on dolutegravir treatment in VIKING-4 were consistent with those seen in VIKING-3.
Cross-Resistance
Site-Directed Integrase Strand Transfer Inhibitor-Resistant Mutant HIV-1 and HIV-2 Strains: The susceptibility of dolutegravir was tested against 60 INSTI-resistant site-directed mutant HIV-1 viruses (28 with single substitutions and 32 with 2 or more substitutions) and 6 INSTI-resistant site-directed mutant HIV-2 viruses. The single INSTI-resistance substitutions T66K, I151L, and S153Y conferred a greater than 2-fold decrease in dolutegravir susceptibility (range: 2.3-fold to 3.6-fold from reference). Combinations of multiple substitutions T66K/L74M, E92Q/N155H, G140C/Q148R, G140S/Q148H, R or K, Q148R/N155H, T97A/G140S/Q148, and substitutions at E138/G140/Q148 showed a greater than 2-fold decrease in dolutegravir susceptibility (range: 2.5-fold to 21-fold from reference). In HIV-2 mutants, combinations of substitutions A153G/N155H/S163G and E92Q/T97A/N155H/S163D conferred 4-fold decreases in dolutegravir susceptibility, and E92Q/N155H and G140S/Q148R showed 8.5-fold and 17-fold decreases in dolutegravir susceptibility, respectively.
Reverse Transcriptase Inhibitor- and Protease Inhibitor-Resistant Strains: Dolutegravir demonstrated equivalent antiviral activity against 2 NNRTI-resistant, 3 NRTI-resistant, and 2 PI-resistant HIV-1 mutant clones compared with the wild-type strain.
13 Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
Two-year carcinogenicity studies in mice and rats were conducted with dolutegravir. Mice were administered doses of up to 500 mg/kg, and rats were administered doses of up to 50 mg/kg. In mice, no significant increases in the incidence of drug-related neoplasms were observed at the highest doses tested, resulting in dolutegravir AUC exposures approximately 14 times higher than those in humans at the maximum recommended dose. In rats, no increases in the incidence of drug-related neoplasms were observed at the highest dose tested, resulting in dolutegravir AUC exposures 10 times and 15 times higher in males and females, respectively, than those in humans at the maximum recommended dose.
Mutagenesis
Dolutegravir was not genotoxic in the bacterial reverse mutation assay, mouse lymphoma assay, or in the in vivo rodent micronucleus assay.
Impairment of Fertility
In a study conducted in rats, there were no effects on mating or fertility with dolutegravir up to 1,000 mg/kg/day. This dose is associated with an exposure that is approximately 24 times higher than the exposure in humans at the maximum recommended dose.
14 Clinical Studies
14.1 Description of Clinical Studies
The efficacy and safety of TIVICAY or TIVICAY PD were evaluated in the studies summarized in Table 15.
Table 15. Trials Conducted with TIVICAY or TIVICAY PD in HIV-1‚ÄďInfected Subjects NRTI = nucleoside reverse transcriptase inhibitor; BR¬†=¬†Background regimen; INSTI = integrase strand transfer inhibitor; OBT = Optimized background therapy; CAR¬†=¬†Current antiretroviral regimen.
Population
Trial
Trial Arms
Timepoint (Week)
Adults:
¬† Treatment-na√Įve
SPRING-2 (ING113086)(NCT01227824)
TIVICAY + 2 NRTIs (n = 403)
Raltegravir + 2 NRTIs (n = 405)
96
SINGLE (ING114467)(NCT01263015)
TIVICAY + EPZICOM (n = 414)
ATRIPLA (n = 419)
144
FLAMINGO (ING114915)(NCT01449929)
TIVICAY + NRTI BR (n = 243)
Darunavir/ritonavir + NRTI BR (n = 242)
96
¬† Treatment-experienced, INSTI-na√Įve
SAILING (ING111762)(NCT01231516)
TIVICAY + BR (n = 354)
Raltegravir + BR (n = 361)
48
  INSTI-experienced
VIKING-3 (ING112574)
(NCT01328041)
TIVICAY + OBT (n = 183)
48
  Virologically suppressed
SWORD-1 (NCT02429791)
SWORD-2 (NCT02422797)
Pooled presentation
TIVICAY + Rilpivirine (n = 513)
CAR (n = 511)
48
Pediatrics:
  4 weeks and older and weighing at least 3 kg without INSTI resistance
IMPAACT P1093 (NCT01302847)
TIVICAY or TIVICAY PD + BR (n = 75)
24
14.2 Adult Subjects
Treatment-Na√Įve Subjects
In SPRING-2, 822 subjects were randomized and received at least 1 dose of either TIVICAY 50 mg once daily or raltegravir 400 mg twice daily, both in combination with fixed-dose dual NRTI treatment (either abacavir sulfate and lamivudine [EPZICOM] or emtricitabine/tenofovir [TRUVADA]). There were 808 subjects included in the efficacy and safety analyses. At baseline, the median age of subjects was 36 years, 13% female, 15% non-white, 11% had hepatitis B and/or C virus co-infection, 2% were CDC Class C (AIDS), 28% had HIV-1 RNA greater than 100,000 copies per mL, 48% had CD4+ cell count less than 350 cells/mm3, and 39% received EPZICOM; these characteristics were similar between treatment groups.
In SINGLE, 833 subjects were randomized and received at least 1 dose of either TIVICAY 50 mg once daily with fixed-dose abacavir sulfate and lamivudine (EPZICOM) or fixed-dose efavirenz/emtricitabine/tenofovir (ATRIPLA). At baseline, the median age of subjects was 35 years, 16% female, 32% non-white, 7% had hepatitis C co-infection (hepatitis B virus co-infection was excluded), 4% were CDC Class C (AIDS), 32% had HIV-1 RNA greater than 100,000 copies/mL, and 53% had CD4+ cell count less than 350 cells/mm3; these characteristics were similar between treatment groups.
Outcomes for SPRING-2 (Week 96 analysis) and SINGLE (Week 144 open-label phase analysis which followed the Week 96 double-blind phase) are provided in Table 16. Side-by-side tabulation is to simplify presentation; direct comparisons across trials should not be made due to differing trial designs.
Table 16. Virologic Outcomes of Randomized Treatment in SPRING-2 at Week 96 and SINGLE at Week 144 (Snapshot Algorithm) NRTI = Nucleoside reverse transcriptase inhibitor. a  Adjusted for pre-specified stratification factors. b  The primary endpoint was assessed at Week 48 and the virologic success rate was 88% in the group receiving TIVICAY and 86% in the raltegravir group, with a treatment difference of 2.6% and 95% CI of (-1.9%, 7.2%). c  The primary endpoint was assessed at Week 48 and the virologic success rate was 88% in the group receiving TIVICAY and 81% in the ATRIPLA group, with a treatment difference of 7.4% and 95% CI of (2.5%, 12.3%). d  Includes subjects who discontinued due to an adverse event or death at any time point if this resulted in no virologic data on treatment during the analysis window. e  Other includes reasons such as withdrew consent, loss to follow-up, moved, and protocol deviation.
SPRING-2 Week 96
SINGLE Week 144
TIVICAY 50 mg Once Daily + 2 NRTIs (n = 403)
Raltegravir 400 mg Twice Daily + 2 NRTIs (n = 405)
TIVICAY 50 mg + EPZICOM Once Daily (n = 414)
ATRIPLA Once Daily (n = 419)
HIV-1 RNA <50 copies/mL
82%
78%
71%
63%
    Treatment differencea
4.9% (95% CI: -0.6%, 10.3%)b
8.3% (95% CI: 2.0%, 14.6%)c
Virologic nonresponse
5%
10%
10%
7%
    Data in window not <50 copies/mL
1%
3%
4%
<1%
    Discontinued for lack of efficacy
2%
3%
3%
3%
    Discontinued for other reasons while not suppressed
<1%
3%
3%
4%
    Change in ART regimen
<1%
<1%
0
0
No virologic data
12%
12%
18%
30%
Reasons
    Discontinued study/study drug due to adverse event or deathd
2%
2%
4%
14%
    Discontinued study/study drug for other reasonse
8%
9%
12%
13%
    Missing data during window but on study
2%
<1%
2%
3%
Proportion (%) of Subjects with HIV-1 RNA <50 copies/mL by Baseline Category
Plasma viral load (copies/mL)
¬†¬†¬†¬†‚ȧ100,000
84%
83%
73%
64%
    >100,000
79%
63%
69%
61%
Gender
    Male
84%
79%
72%
66%
    Female
70%
68%
69%
48%
Race
    White
83%
78%
72%
71%
    African-American/African Heritage/Other
77%
75%
71%
47%
SPRING-2: Virologic outcomes were also comparable across baseline characteristics including CD4+ cell count, age, and use of EPZICOM or TRUVADA as NRTI background regimen. The median change in CD4+ cell counts from baseline was 276 cells/mm3 in the group receiving TIVICAY and 264 cells/mm3 for the raltegravir group at 96 weeks.
There was no treatment-emergent resistance to dolutegravir or to the NRTI background.
SINGLE: Treatment differences were maintained across baseline characteristics including baseline viral load, CD4+ cell count, age, gender, and race.
The adjusted mean changes in CD4+ cell counts from baseline were 378 cells/mm3 in the group receiving TIVICAY + EPZICOM and 332 cells/mm3 for the ATRIPLA group at 144 weeks. The adjusted difference between treatment arms and 95% CI was 46.9 cells/mm3 (15.6 cells/mm3, 78.2 cells/mm3) (adjusted for pre-specified stratification factors: baseline HIV-1 RNA, and baseline CD4+ cell count).
There was no treatment-emergent resistance to dolutegravir, abacavir, or lamivudine.
FLAMINGO: In FLAMINGO, 485 subjects were randomized and received at least 1 dose of either TIVICAY 50¬†mg once daily (n¬†=¬†243) or darunavir¬†+¬†ritonavir 800¬†mg/100¬†mg once daily (n¬†=¬†242), both in combination with investigator-selected NRTI background regimen (either fixed-dose abacavir and lamivudine [EPZICOM] or fixed-dose emtricitabine/tenofovir disoproxil fumarate [TRUVADA]). There were 484 subjects included in the efficacy and safety analyses. At baseline, the median age of subjects was 34¬†years, 15% female, 28% non-white, 10% had hepatitis B and/or C virus co-infection, 3% were CDC Class C (AIDS), 25% had HIV‚ÄĎ1 RNA greater than 100,000¬†copies/mL, and 35% had CD4+ cell count less than 350¬†cells/mm3; these characteristics were similar between treatment groups. Overall response rates by Snapshot algorithm through Week 96 were 80% for TIVICAY and 68% for darunavir/ritonavir. The proportion of subjects who were non-responders (HIV-1 RNA greater than or equal to 50¬†copies per mL) at Week 96 was 8% and 12% in the arms receiving TIVICAY and darunavir¬†+¬†ritonavir, respectively; no virologic data were available for 12% and 21% for subjects treated with TIVICAY and darunavir¬†+¬†ritonavir, respectively. The adjusted overall response rate difference in proportion and 95% CI was 12.4% (4.7%, 20.2%). No treatment-emergent primary resistance substitutions were observed in either treatment group.
Treatment-Experienced, Integrase Strand Transfer Inhibitor-Na√Įve Subjects
In the international, multicenter, double-blind trial (SAILING), 719 HIV‚ÄĎ1‚Äďinfected, antiretroviral treatment-experienced adults were randomized and received either TIVICAY 50¬†mg once daily or raltegravir 400¬†mg twice daily with investigator-selected background regimen consisting of up to 2 agents, including at least 1 fully active agent. There were 715 subjects included in the efficacy and safety analyses. At baseline, the median age was 43¬†years, 32% were female, 50% non-white, 16% had hepatitis B and/or C virus co-infection, 46% were CDC Class C (AIDS), 20% had HIV-1 RNA greater than 100,000¬†copies/mL, and 72% had CD4+ cell count less than 350¬†cells/mm3; these characteristics were similar between treatment groups. All subjects had at least 2-class antiretroviral treatment resistance, and 49% of subjects had at least 3-class antiretroviral treatment resistance at baseline. Week 48 outcomes for SAILING are shown in Table 17.
Table 17. Virologic Outcomes of Randomized Treatment in SAILING at 48 Weeks (Snapshot Algorithm) a  BR = Background regimen. Background regimen was restricted to less than or equal to 2 antiretroviral treatments with at least 1 fully active agent. b  Adjusted for pre-specified stratification factors. c  Other includes reasons such as withdrew consent, loss to follow-up, moved, and protocol deviation.
TIVICAY 50 mg Once Daily + BRa
(n = 354)
Raltegravir 400 mg Twice Daily + BRa
(n = 361)
HIV-1 RNA <50 copies/mL
71%
64%
    Adjustedb treatment difference
7.4% (95% CI: 0.7%, 14.2%)
Virologic nonresponse
20%
28%
No virologic data
9%
9%
Reasons
    Discontinued study/study drug due to adverse event or death
3%
4%
    Discontinued study/study drug for other reasonsc
5%
4%
    Missing data during window but on study
2%
1%
Proportion (%) with HIV-1 RNA <50 copies/mL by Baseline Category
Plasma viral load (copies/mL)
¬†¬†¬†¬†‚ȧ50,000 copies/mL
75%
71%
    >50,000 copies/mL
62%
47%
Background regimen
    No darunavir use
67%
60%
    Darunavir use with primary PI substitutions
85%
67%
    Darunavir use without primary PI substitutions
69%
70%
Gender
    Male
70%
66%
    Female
74%
60%
Race
    White
75%
71%
    African-American/African Heritage/Other
67%
57%
Treatment differences were maintained across the baseline characteristics including CD4+ cell count and age.
The mean changes in CD4+ cell counts from baseline were 162 cells/mm3 in the group receiving TIVICAY and 153 cells/mm3 in the raltegravir group.
Treatment-Experienced, Integrase Strand Transfer Inhibitor-Experienced Subjects
VIKING-3 examined the effect of TIVICAY 50 mg twice daily over 7 days of functional monotherapy, followed by OBT with continued treatment of TIVICAY 50 mg twice daily.
In the multicenter, open-label, single-arm VIKING-3 trial, 183 HIV‚ÄĎ1‚Äďinfected, antiretroviral treatment-experienced adults with virological failure and current or historical evidence of raltegravir and/or elvitegravir resistance received TIVICAY 50¬†mg twice daily with the current failing background regimen for 7¬†days, then received TIVICAY with OBT from Day 8. A total of 183¬†subjects enrolled: 133¬†subjects with INSTI resistance at screening and 50¬†subjects with only historical evidence of resistance (and not at screening). At baseline, median age of subjects was 48¬†years; 23% were female, 29% non-white, and 20% had hepatitis B and/or C virus co-infection. Median baseline CD4+ cell count was 140¬†cells/mm3, median duration of prior antiretroviral treatment was 13¬†years, and 56% were CDC Class C. Subjects showed multiple-class antiretroviral treatment resistance at baseline: 79% had greater than or equal to 2 NRTI, 75% greater than or equal to 1 NNRTI, and 71% greater than or equal to 2 PI major substitutions; 62% had non-R5 virus.
Mean reduction from baseline in HIV-1 RNA at Day 8 (primary endpoint) was 1.4 log10 (95% CI: 1.3 log10, 1.5 log10). Response at Week 48 was affected by baseline INSTI substitutions [see Microbiology (12.4)].
After the functional monotherapy phase, subjects had the opportunity to re-optimize their background regimen when possible. Week 48 virologic outcomes for VIKING-3 are shown in Table 18.
Table 18. Virologic Outcomes of Treatment of VIKING-3 at 48 Weeks (Snapshot Algorithm) OBT = Optimized Background Therapy.
TIVICAY 50 mg Twice Daily + OBT
(n = 183)
HIV-1 RNA <50 copies/mL
63%
Virologic nonresponse
32%
No virologic data
Reasons
    Discontinued study/study drug due to adverse event or death
3%
Proportion (%) with HIV-1 RNA <50 copies/mL by Baseline Category
Gender
    Male
63%
    Female
64%
Race
    White
63%
    African-American/African Heritage/Other
64%
Subjects harboring virus with Q148 and with additional Q148-associated secondary substitutions also had a reduced response at Week 48 in a stepwise fashion [see Microbiology (12.4)].
The median change in CD4+ cell count from baseline was 80 cells/mm3 at Week 48.
Virologically Suppressed Subjects
SWORD-1 and SWORD-2 are identical 148-week, Phase 3, randomized, multicenter, parallel-group, non-inferiority trials. A total of 1,024 adult HIV-1‚Äďinfected subjects who were on a stable suppressive antiretroviral regimen (containing 2 NRTIs plus either an INSTI, an NNRTI, or a PI) for at least 6¬†months (HIV-1 RNA less than 50¬†copies/mL), with no history of treatment failure and no known substitutions associated with resistance to dolutegravir or rilpivirine received treatment in the trials. Subjects were randomized 1:1 to continue their current antiretroviral regimen (n¬†=¬†511) or be switched to TIVICAY 50¬†mg plus rilpivirine 25¬†mg administered once daily (n¬†=¬†513). Subjects originally assigned to continue their current antiretroviral regimen and who remained virologically suppressed at Week 48 switched to TIVICAY plus rilpivirine at Week 52 (n¬†=¬†477).
The primary efficacy endpoint for the SWORD trial was the proportion of subjects with plasma HIV-1 RNA less than 50¬†copies/mL at Week¬†48. The proportion of subjects with HIV-1 RNA less than 50¬†copies/mL at Week¬†48 was 95% for both treatment groups; treatment difference and 95% CI was -0.2% (-3.0%, 2.5%). The proportion of subjects with HIV‚ÄĎ1 RNA greater than or equal to 50¬†copies/mL (virologic failure) at Week¬†48 was 0.6% and 1.2% for the dolutegravir plus rilpivirine treatment group and the current antiretroviral regimen treatment groups, respectively; treatment difference and 95% CI was -0.6% (-1.7%, 0.6%). At Week¬†148 in the pooled SWORD-1 and SWORD-2 trials, 84% of subjects who received TIVICAY plus rilpivirine from study start had plasma HIV-1 RNA less than 50¬†copies/mL (Snapshot algorithm). In subjects who initially remained on their current antiretroviral regimen and switched to TIVICAY plus rilpivirine at Week 52, 90% had plasma HIV-1 RNA less than 50¬†copies/mL at Week¬†148 (Snapshot algorithm), which was comparable to the response rate (89%) observed at Week¬†100 (similar exposure duration) in subjects receiving TIVICAY plus rilpivirine from study start.
Refer to the prescribing information for JULUCA (dolutegravir and rilpivirine) tablet for complete virologic outcome information.
14.3 Pediatric Subjects
IMPAACT P1093 is an ongoing Phase 1/2, multicenter, open-label trial to evaluate the pharmacokinetic parameters, safety, tolerability, and efficacy of TIVICAY or TIVICAY PD in combination treatment regimens in HIV-1‚Äďinfected infants, children, and adolescents aged at least 4¬†weeks to 18¬†years. Subjects were stratified by 5 age cohorts: Cohort¬†1, aged 12 to less than 18¬†years; Cohort¬†2A, aged 6 to less than 12¬†years; Cohort¬†3, aged 2 to less than 6¬†years; Cohort¬†4, aged 6 months to less than 2¬†years; and Cohort¬†5, aged 4¬†weeks to less than 6¬†months. Seventy-five subjects received the recommended dose (determined by weight and age) of TIVICAY or TIVICAY PD [see Dosage and Administration (2.2, 2.3, 2.4)].
These 75¬†subjects had a median age of 27¬†months (range: 1 to 214), were 59% female, and 68% were Black or African American. At baseline, mean plasma HIV-1 RNA was 4.4¬†log10¬†copies/mL, median CD4+ cell count was 1,225¬†cells/mm3 (range: 1 to 8,255), and median CD4+% was 23% (range: 0.3% to 49%). Overall, 33% had baseline plasma HIV-1 RNA greater than 50,000¬†copies/mL and 12% had a CDC HIV clinical classification of category C. The majority (80%) of subjects were treatment-experienced, but all were INSTI-na√Įve. Most subjects had previously used at least 1 NNRTI (44%) or 1 PI (76%).
Virologic outcomes from IMPAACT P1093 include subjects who received either TIVICAY tablets or TIVICAY PD tablets for oral suspension as per the dosing recommendations for their weight band and who had reached Week 24 (n = 58) or Week 48 (n = 42). At Week 24, 62% of subjects achieved HIV-1 RNA less than 50 copies/mL and 86% achieved HIV-1 RNA less than 400 copies/mL (Snapshot algorithm). The median CD4 count (percent) increase from baseline to Week 24 was 105 cells/mm3 (5%). At Week 48, 69% of subjects achieved HIV-1 RNA less than 50 copies/mL and 79% achieved HIV-1 RNA less than 400 copies/mL (Snapshot algorithm). The median CD4 count (percent) increase from baseline to Week 48 was 141 cells/mm3 (7%).
16 How Supplied/storage And Handling
TIVICAY tablets, 10¬†mg, are white, round, film-coated, biconvex tablets debossed with ‚ÄúSV¬†572‚ÄĚ on one side and ‚Äú10‚ÄĚ on the other side. Bottle of 30 tablets with child-resistant closure and containing a desiccant. NDC 49702-226-13.
Store and dispense the 10-mg tablets in the original package, protect from moisture, and keep the bottle tightly closed. Do not remove desiccant.
TIVICAY tablets, 25¬†mg, are pale yellow, round, film-coated, biconvex tablets debossed with ‚ÄúSV¬†572‚ÄĚ on one side and ‚Äú25‚ÄĚ on the other side. Bottle of 30 tablets with child-resistant closure. NDC 49702-227-13.
TIVICAY tablets, 50¬†mg, are yellow, round, film-coated, biconvex tablets debossed with ‚ÄúSV¬†572‚ÄĚ on one side and ‚Äú50‚ÄĚ on the other side. Bottle of 30 tablets with child-resistant closure. NDC 49702-228-13.
Store TIVICAY tablets at 25¬įC (77¬įF); excursions permitted 15¬į to 30¬įC (59¬į to 86¬įF) [See USP Controlled Room Temperature].
TIVICAY PD tablets for oral suspension, 5¬†mg, are white, round, strawberry cream flavored, film-coated, biconvex tablets debossed with ‚ÄúSV H7S‚ÄĚ on one side and ‚Äú5‚ÄĚ on the other side. Bottle of 60 tablets with child-resistant closure containing a desiccant. Each bottle is packaged with one 30-mL dosing cup and one 10-mL oral dosing syringe with 1-mL gradations. NDC¬†49702-255-37.
Store TIVICAY PD tablets for oral suspension below 30¬įC (86¬įF). Store and dispense the 5-mg tablets in the original bottle, protect from moisture, and keep the bottle tightly closed. Do not remove desiccant.
17 Patient Counseling Information
Advise the patient to read the FDA-approved patient labeling (Patient Information and Instructions for Use).
Drug Interactions
TIVICAY or TIVICAY PD may interact with other drugs; therefore, advise patients to report to their healthcare provider the use of any other prescription or nonprescription medication or herbal products, including St. John’s wort [see Contraindications (4), Warnings and Precautions (5.4), Drug Interactions (7)].
Hypersensitivity Reactions
Advise patients to immediately contact their healthcare provider if they develop rash. Instruct patients to immediately stop taking TIVICAY or TIVICAY PD and other suspect agents, and seek medical attention if they develop a rash associated with any of the following symptoms, as it may be a sign of a more serious reaction such as severe hypersensitivity: fever; generally ill feeling; extreme tiredness; muscle or joint aches; bulers or peeling of the skin; oral bulers or lesions; eye inflammation; facial swelling; swelling of the eyes, lips, tongue, or mouth; breathing difficulty; and/or signs and symptoms of liver problems (e.g., yellowing of the skin or whites of the eyes, dark or tea-colored urine, pale-colored stools or bowel movements, nausea, vomiting, loss of appetite, or pain, aching, or sensitivity on the right side below the ribs) [see Warnings and Precautions (5.1)].
Hepatotoxicity
Inform patients that hepatotoxicity has been reported with dolutegravir [see Warnings and Precautions (5.2)]. Advise patients that laboratory monitoring for hepatoxicity during therapy with TIVICAY or TIVICAY PD is recommended, especially for patients with liver disease, such as hepatitis B or C.
Immune Reconstitution Syndrome
Advise patients to inform their healthcare provider immediately of any signs or symptoms of infection as inflammation from previous infection may occur soon after combination antiretroviral therapy, including when TIVICAY or TIVICAY PD is started [see Warnings and Precautions (5.4)].
Different Formulations Are Not Bioequivalent
Advise patients that TIVICAY and TIVICAY PD are not bioequivalent and are not substitutable on a milligram-per-milligram basis. Advise patients or their care provider that patients switching from one formulation to the other must adjust the dose for the new dosage formulation [see Dosage and Administration (2.2) and Warnings and Precautions (5.5)].
Pregnancy Registry
Inform patients that there is an antiretroviral pregnancy registry to monitor fetal outcomes in those exposed to TIVICAY or TIVICAY PD during pregnancy [see Use in Specific Populations (8.1)].
Lactation
Inform individuals with HIV-1 infection that the potential risks of breastfeeding include: (1) HIV-1 transmission (in HIV-1‚Äďnegative infants), (2) developing viral resistance (in HIV-1‚Äďpositive infants), and (3) adverse reactions in a breastfed infant similar to those seen in adults [see¬†Use in Specific Populations (8.2)].
Administration Instructions
To avoid a dosing error from using the wrong formulation of dolutegravir, strongly advise patients and caregivers to visually inspect the tablets to verify the correct formulation each time the prescription is filled [see Dosage and Administration (2), Warnings and Precautions (5.5), How Supplied/Storage and Handling (16)].
Inform patients and caregivers that TIVICAY PD tablets for oral suspension may be swallowed whole or dispersed in drinking water and should not be chewed, cut or crushed. The amount of water needed to disperse the tablet will depend on the dose (number of tablets prescribed).
Instruct patients and caregivers that if a dose of TIVICAY or TIVICAY PD is missed, to take it as soon as they remember. Advise patients and caregivers not to double the next dose or take more than the prescribed dose [see Dosage and Administration (2)].
Storage
Instruct patients and caregivers to store the TIVICAY 10-mg tablets and TIVICAY PD 5-mg tablets for oral suspension in the original package, keep the bottle tightly closed, and protect from moisture. Do not remove desiccant [see How Supplied/Storage and Handling (16)].
TIVICAY, TIVICAY PD, EPZICOM, JULUCA, and TRIUMEQ are trademarks owned by or licensed to the ViiV Healthcare group of companies.
The other brands uled are trademarks owned by or licensed to their respective owners and are not owned by or licensed to the ViiV Healthcare group of companies. The makers of these brands are not affiliated with and do not endorse the ViiV Healthcare group of companies or its products.
Manufactured for:
ViiV Healthcare
Durham, NC 27701
©2024 ViiV Healthcare group of companies or its licensor.
TVC:19PI
PHARMACIST‚ÄĎDETACH HERE AND GIVE INSTRUCTIONS TO PATIENT_ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _ _
Spl Patient Package Insert Section
PATIENT INFORMATION
TIVICAY (TIV-eh-kay)
(dolutegravir)
tablets
TIVICAY PD (TIV-eh-kay Pe De)
(dolutegravir)
tablets for oral suspension
What is TIVICAY and TIVICAY PD?
TIVICAY and TIVICAY PD are prescription medicines used to treat Human Immunodeficiency Virus-1 (HIV-1) infection together with:
‚ÄĘ other HIV-1 medicines in adults who have not received HIV-1 medicines in the past or to replace their current HIV-1 medicines.‚ÄĘ other HIV-1 medicines in children, aged at least 4 weeks and weighing at least 6.6 pounds (3 kg), who have not received HIV-1 medicines in the past or to replace their current HIV-1 medicines when their healthcare provider determines that they meet certain requirements.
TIVICAY is used together with rilpivirine as a complete regimen to treat Human Immunodeficiency Virus-1 (HIV-1) infection in adults to replace their current HIV-1 medicines when their healthcare provider determines that they meet certain requirements.
HIV-1 is the virus that causes Acquired Immune Deficiency Syndrome (AIDS).
It is not known if TIVICAY or TIVICAY PD is safe and effective in children who are less than 4 weeks of age and weigh less than 6.6 pounds (3 kg) or in children who have received certain types of medicine for HIV-1 infection.
Do not take TIVICAY or TIVICAY PD if you:
‚ÄĘ have ever had an allergic reaction to a medicine that contains dolutegravir.‚ÄĘ take dofetilide.
Before you take TIVICAY or TIVICAY PD, tell your healthcare provider about all of your medical conditions, including if you:
‚ÄĘ have or have had liver problems, including hepatitis B or C infection.‚ÄĘ are pregnant or plan to become pregnant. Talk to your healthcare provider about the benefits and risks of treatment with TIVICAY or TIVICAY PD during pregnancy.
Pregnancy Registry. There is a pregnancy registry for those who take TIVICAY or TIVICAY PD during pregnancy. The purpose of this registry is to collect information about the health of you and your baby. Talk with your healthcare provider about how you can take part in this registry.
‚ÄĘ are breastfeeding or plan to breastfeed. TIVICAY and TIVICAY PD pass to your baby in your breast milk. Talk with your healthcare provider about the following risks to your baby from breastfeeding during treatment with TIVICAY and TIVICAY PD:
o the HIV-1 virus may pass to your baby if your baby does not have HIV-1 infection.o the HIV-1 virus may become harder to treat if your baby has HIV-1 infection.o your baby may get side effects from TIVICAY.
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements.
Some medicines interact with TIVICAY or TIVICAY PD. Keep a ul of your medicines and show it to your healthcare provider and pharmacist when you get a new medicine.
‚ÄĘ You can ask your healthcare provider or pharmacist for a ul of medicines that interact with TIVICAY or TIVICAY PD.‚ÄĘ Do not start taking a new medicine without telling your healthcare provider. Your healthcare provider can tell you if it is safe to take TIVICAY or TIVICAY PD with other medicines.
How should I take TIVICAY or TIVICAY PD?
‚ÄĘ Take TIVICAY or TIVICAY PD exactly as your healthcare provider tells you to take it.‚ÄĘ Take TIVICAY or TIVICAY PD with or without food.‚ÄĘ For children who cannot swallow tablets, read the Instructions for Use at the end of this patient information for detailed instructions on how to prepare a dose of TIVICAY PD tablets for oral suspension.‚ÄĘ TIVICAY PD may be swallowed whole or dispersed in drinking water and should not be chewed, cut, or crushed.‚ÄĘ TIVICAY tablets are not the same as TIVICAY PD tablets for oral suspension and cannot be substituted for each other. Check to make sure you receive the correct form of TIVICAY each time you or your child‚Äôs prescription is filled to avoid using the wrong medicine.‚ÄĘ Do not change your dose, switch medicines or stop taking TIVICAY or TIVICAY PD without talking with your healthcare provider first.‚ÄĘ If you take antacids, laxatives, or other medicines that contain aluminum, magnesium, or buffered medicines, TIVICAY or TIVICAY PD should be taken at least 2 hours before or 6 hours after you take these medicines.‚ÄĘ If you need to take iron or calcium supplements by mouth during treatment with TIVICAY or TIVICAY PD:
o If you take TIVICAY with food, you may take these supplements at the same time that you take TIVICAY.o If you do not take TIVICAY or TIVICAY PD with food, take TIVICAY or TIVICAY PD at least 2 hours before or 6 hours after you take these supplements.‚ÄĘ Do not miss a dose of TIVICAY or TIVICAY PD.‚ÄĘ If you miss a dose of TIVICAY or TIVICAY PD, take it as soon as you remember. Do not take 2 doses at the same time or take more than your prescribed dose.‚ÄĘ Stay under the care of a healthcare provider during treatment with TIVICAY or TIVICAY PD.‚ÄĘ Do not run out of TIVICAY or TIVICAY PD. The virus in your blood may increase and the virus may become harder to treat. When your supply starts to run low, get more from your healthcare provider or pharmacy.‚ÄĘ If you take too much TIVICAY or TIVICAY PD, call your healthcare provider or go to the nearest hospital emergency room right away.
What are the possible side effects of TIVICAY or TIVICAY PD?
‚ÄĘ TIVICAY or TIVICAY PD can cause serious side effects including:‚ÄĘ Allergic reactions. Call your healthcare provider right away if you develop a rash with TIVICAY or TIVICAY PD. Stop taking TIVICAY or TIVICAY PD and get medical help right away if you develop a rash with any of the following signs or symptoms:
o fevero generally ill feelingo tirednesso muscle or joint acheso bulers or sores in mouth
o bulers or peeling of the skino redness or swelling of the eyeso swelling of the mouth, face, lips, or tongueo problems breathing
‚ÄĘ Liver problems. People with a history of hepatitis B or C virus may have an increased risk of developing new or worsening changes in certain liver tests during treatment with TIVICAY or TIVICAY PD. Liver problems, including liver failure, have also happened in people without a history of liver disease or other risk factors. Your healthcare provider may do blood tests to check your liver. Call your healthcare provider right away if you develop any of the following signs or symptoms of liver problems:
o your skin or the white part of your eyes turns yellow (jaundice)o dark or ‚Äútea-colored‚ÄĚ urineo light-colored stools (bowel movements)
o nausea or vomitingo loss of appetiteo pain, aching, or tenderness on the right side of your stomach area
‚ÄĘ Changes in your immune system (Immune Reconstitution Syndrome) can happen when you start taking HIV-1 medicines. Your immune system may get stronger and begin to fight infections that have been hidden in your body for a long time. Tell your healthcare provider right away if you start having new symptoms after you start taking TIVICAY or TIVICAY PD.‚ÄĘ The most common side effects of TIVICAY include:
o trouble sleeping
o tiredness
o headache
These are not all the possible side effects of TIVICAY or TIVICAY PD. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1‚ÄĎ800‚ÄĎFDA‚ÄĎ1088.
How should I store TIVICAY or TIVICAY PD?
‚ÄĘ Store TIVICAY 10-mg, 25-mg, and 50-mg tablets at room temperature between 68¬įF to 77¬įF (20¬įC to 25¬įC).‚ÄĘ Store TIVICAY 10-mg tablets in the original bottle. Keep the bottle tightly closed and protected from moisture. The bottle contains a desiccant packet to help keep your medicine dry (protect it from moisture). Do not remove the desiccant packet from the bottle.‚ÄĘ Store TIVICAY PD 5-mg tablets for oral suspension at room temperature below 86¬įF (30¬įC) in the original bottle. Keep the bottle tightly closed and protected from moisture. The bottle contains a desiccant packet to help keep your medicine dry (protect it from moisture). Do not remove the desiccant packet from the bottle.
Keep TIVICAY, TIVICAY PD, and all medicines out of the reach of children.
General information about the safe and effective use of TIVICAY or TIVICAY PD.
Medicines are sometimes prescribed for purposes other than those uled in a Patient Information leaflet. Do not use TIVICAY or TIVICAY PD for a condition for which it was not prescribed. Do not give TIVICAY or TIVICAY PD to other people, even if they have the same symptoms you have. It may harm them. You can ask your healthcare provider or pharmacist for information about TIVICAY that is written for health professionals.
What are the ingredients in TIVICAY and TIVICAY PD?
Active ingredient: dolutegravir.
Inactive ingredients:
TIVICAY tablets: D-mannitol, microcrystalline cellulose, povidone K29/32, sodium starch glycolate, and sodium stearyl fumarate. The tablet film‚ÄĎcoating contains the inactive ingredients iron oxide yellow (for the 25-mg and 50-mg tablets only), macrogol/PEG, polyvinyl alcohol-part hydrolyzed, talc, and titanium dioxide.
TIVICAY PD tablets for oral suspension: calcium sulfate dihydrate, crospovidone, mannitol, microcrystalline cellulose, povidone K29/32, silicified microcrystalline cellulose, sodium starch glycolate, strawberry cream flavor, sucralose, and sodium stearyl fumarate. The tablet film-coating contains hypromellose, polyethylene glycol, and titanium dioxide.
Manufactured for:
ViiV Healthcare
Durham, NC 27701
TIVICAY and TIVICAY PD are trademarks owned by or licensed to the ViiV Healthcare group of companies.
©2024 ViiV Healthcare group of companies or its licensor.
TVC:14PIL
For more information, go to www.TIVICAY.com or call 1-877-844-8872.
This Patient Information has been approved by the U.S. Food and Drug Administration.
Revised: 4/2024
INSTRUCTIONS FOR USE
TIVICAY PD (TIV-eh-kay Pe De)
(dolutegravir) tablets for oral suspension
5 mg
Read this Instructions for Use before giving a dose of medicine.
Follow the steps below, using clean drinking water to prepare and give a dose to an infant or a child who cannot swallow the tablets.
Important information
Always give this medicine exactly as your healthcare provider tells you. Talk to your healthcare provider if you are not sure.
Do not chew, cut, or crush the tablets.
If you forget to give a dose of medicine, give it as soon as you remember. Do not give 2 doses at the same time or give more than your healthcare provider has prescribed.
If your child does not or cannot take the full dose, call your healthcare provider.
If you give too much medicine, get emergency medical help right away.
If your child is able and prefers to swallow the tablets, then you may skip the following steps.
Your pack contains:
‚ÄĘ A bottle containing 60 TIVICAY PD tablets for oral suspension.‚ÄĘ Dosing kit:
‚ÄĘ Cup: Use this to prepare and give the medicine to children.‚ÄĘ Syringe: Use this to give the medicine to infants.
You will also need:
‚ÄĘ Clean drinking water.
Getting Ready
Step 1. Pour water

Figure A
‚ÄĘ Pour clean drinking water into the cup.The Water Volume Guide in Figure A shows the amount of water needed for the prescribed dose. See Figure A.
Use drinking water only.
Do not use any other drink or food to prepare the dose.
Step 2. Prepare the medicine
Figure B                                     Figure C
‚ÄĘ Add the prescribed number of tablet(s) to the water. See Figure B.‚ÄĘ Swirl the cup gently for 1 to 2 minutes to disperse the tablet(s). The medicine will become cloudy. Take care not to spill any of the medicine. See Figure C.‚ÄĘ Check that the medicine is ready. If there are any lumps of tablet, swirl the cup until they are gone.
If you spill any medicine, clean up the spill.
Throw away the rest of the prepared medicine and make a new dose.
You must give the dose of medicine within 30 minutes of preparing the dose. If it has been more than 30 minutes, wash away all the dose in the cup using water and prepare a new dose of medicine.
Giving the medicine
Step 3. Give the medicine
Figure D
‚ÄĘ Make sure that the child is upright. Give all the prepared medicine to the child. See Figure D.‚ÄĘ Add another 5 mL of drinking water to the cup, swirl, and give it all to the child.‚ÄĘ Repeat if any medicine remains in the cup to make sure the child gets the full dose.
Figure E                                    Figure F
‚ÄĘ Place the tip of the syringe into the prepared medicine and draw up all the medicine into the syringe by pulling up on the plunger. See Figure¬†E.‚ÄĘ Place the tip of the syringe against the inside of the infant‚Äôs cheek. Gently push down the plunger to give the dose slowly. See Figure F.‚ÄĘ Add another 5 mL of drinking water to the cup and swirl. Draw up the remaining medicine into the syringe and give it all to the infant.‚ÄĘ Repeat if any medicine remains in the syringe to make sure the infant gets the full dose.
Allow time for the medicine to be swallowed.
Cleaning
Step 4. Clean the dosing lis
Figure G                                   Figure H
‚ÄĘ Wash the cup with water. See Figure G.‚ÄĘ Pull the plunger out of the syringe and wash the syringe parts separately in water. Allow parts to dry completely before reassembling and storing. See Figure H.‚ÄĘ All parts will need to be clean before preparing the next dose.
Storage Information
Store TIVICAY PD tablets for oral suspension at room temperature below 86¬įF (30¬įC) in the original bottle. Keep the bottle tightly closed and protect from moisture. The bottle contains a desiccant packet to help keep your medicine dry (protect it from moisture). Do not remove the desiccant packet from the bottle.
Keep TIVICAY PD and all medicines out of the reach of children.
Disposal Information
When all the tablets in the bottle have been taken or are no longer needed, throw away the bottle, cup, and syringe. Dispose of them using your local household waste guidelines.
You will get a new cup and syringe in your next pack.
Manufactured for:
ViiV Healthcare
Durham, NC 27701
Trademarks are owned by or licensed to the ViiV Healthcare group of companies.
©2024 ViiV Healthcare group of companies or its licensor.
TVC: 3IFU
This Instructions for Use has been approved by the U.S. Food and Drug Administration.
Revised: 4/2024
Package Label.principal Display Panel
PRINCIPAL DISPLAY PANEL
NDC 49702-255-37
Tivicay PD
(dolutegravir) Tablets for Oral Suspension
5 mg
Rx Only
Tivicay and Tivicay PD are not interchangeable.
Each tablet contains 5 mg of dolutegravir equivalent to 5.26 mg of dolutegravir sodium.
Do not chew, cut, or crush tablet for oral suspension.
Do not accept if security seal is missing or broken.
See prescribing information for dosage information.
Contents:
‚ÄĘ 1 Bottle of 60 tablets‚ÄĘ 1 Oral dosing syringe‚ÄĘ 1 Dosing cup‚ÄĘ Prescribing Information‚ÄĘ Instruction for Use
60 tablets
©2024 ViiV Healthcare group of companies or its licensor.
Made in Japan
Rev. 1/2024
62000000091728

Package Label.principal Display Panel
PRINCIPAL DISPLAY PANEL
NDC 49702-226-13
Tivicay
(dolutegravir)
Tablets
10 mg
Rx Only
Each film-coated tablet contains dolutegravir sodium equivalent to 10 mg of dolutegravir.
30 Tablets
Keep out of reach of children.
Store in original package at controlled room temperature of 25¬įC (77¬įF); excursions permitted to 15¬į to 30¬įC (59¬į to 86¬įF) [See USP].
Keep bottle tightly closed; protect from moisture. Do not remove desiccant.
Do not accept if membrane seal under cap is missing or broken.
See prescribing information for dosage information.
Manufactured for:
ViiV Healthcare
Durham, NC 27701
by:
GlaxoSmithKline
Durham, NC 27701
Made in Japan
Rev. 2/23
62000000085469

Package Label.principal Display Panel
PRINCIPAL DISPLAY PANEL
NDC 49702-227-13
Tivicay
(dolutegravir)
Tablets
25 mg
Rx Only
Each film-coated tablet contains dolutegravir sodium equivalent to 25 mg of dolutegravir.
30 Tablets
Keep out of reach of children.
Store at controlled room temperature of 25¬įC (77¬įF); excursions permitted to 15¬į to 30¬įC (59¬į to 86¬įF) [See USP].
Do not accept if membrane seal under cap is missing or broken.
See prescribing information for dosage information.
Manufactured for:
ViiV Healthcare
Durham, NC 27701
by:
GlaxoSmithKline
Durham, NC 27701
Made in Japan
Rev. 2/23
62000000085470

Package Label.principal Display Panel
PRINCIPAL DISPLAY PANEL
NDC 49702-228-13
Tivicay
(dolutegravir)
Tablets
50 mg
Rx Only
Each film-coated tablet contains dolutegravir sodium equivalent to 50 mg of dolutegravir.
30 Tablets
Keep out of reach of children.
Store at controlled room temperature of 25¬įC (77¬įF); excursions permitted to 15¬į to 30¬įC (59¬į to 86¬įF) [see USP].
Do not accept if membrane seal under cap is missing or broken.
See prescribing information for dosage information.
Manufactured for:
ViiV Healthcare
Durham, NC 27701
by:
GlaxoSmithKline
Durham, NC 27701
Made in Japan
Rev. 2/23
62000000085468

DISCLAIMER:
"This tool does not provide medical advice, and is for informational and educational purposes only, and is not a substitute for professional medical advice, treatment or diagnosis. Call your doctor to receive medical advice. If you think you may have a medical emergency, please dial 911."
"Do not rely on openFDA to make decisions regarding medical care. While we make every effort to ensure that data is accurate, you should assume all results are unvalidated. We may limit or otherwise restrict your access to the API in line with our Terms of Service."
"This product uses publicly available data from the U.S. National Library of Medicine (NLM), National Institutes of Health, Department of Health and Human Services; NLM is not responsible for the product and does not endorse or recommend this or any other product."
PillSync may earn a commission via links on our site
