MYDAYIS (dextroamphetamine sulfate 9.375 mg dextroamphetamine saccharate 9.375 mg amphetamine aspartate monohydrate 9.375 mg amphetamine sulfate 9.375 mg) Dailymed
Generic: dextroamphetamine sulfate, dextroamphetamine saccharate, amphetamine aspartate monohydrate, and amphetamine sulfate is used for the treatment of Arteriosclerosis Attention Deficit Disorder with Hyperactivity Glaucoma Hypertension Hyperthyroidism Narcolepsy Substance Abuse, Intravenous Substance-Related Disorders Cardiovascular Diseases

All Imprints
mydayis (dextroamphetamine sulfate, dextroamphetamine saccharate, amphetamine aspartate monohydrate, and amphetamine sulfate) capsule, extended release - shire 465 50 mg capsule blue
mydayis (dextroamphetamine sulfate, dextroamphetamine saccharate, amphetamine aspartate monohydrate, and amphetamine sulfate) capsule, extended release - shire 465 37 5 mg capsule brown
Boxed Warning
Warning: Abuse, Misuse, And Addiction
- Before prescribing MYDAYIS, assess each patient’s risk for abuse, misuse, and addiction.
- Educate patients and their families about these risks, proper storage of the drug, and proper disposal of any unused drug.
- Throughout treatment, reassess each patient’s risk and frequently monitor for signs and symptoms of abuse, misuse, and addiction.

Go PRO for all pill images
Recent Major Changes Section
Boxed Warning 10/2023 Dosage and Administration ( 2.1 ,2.2 )10/2023 Warnings and Precautions ( 5.1 ,5.2 ,5.3 ,5.4 ,5.6 ,5.10 )10/2023
Warning: Abuse, Misuse, And Addiction
MYDAYIS has a high potential for abuse and misuse, which can lead to the development of a substance use disorder, including addiction. Misuse and abuse of CNS stimulants, including MYDAYIS, can result in overdose and death [see Overdosage (10)], and this risk is increased with higher doses or unapproved methods of administration, such as snorting or injection.
Before prescribing MYDAYIS, assess each patient’s risk for abuse, misuse, and addiction. Educate patients and their families about these risks, proper storage of the drug, and proper disposal of any unused drug. Throughout MYDAYIS treatment, reassess each patient’s risk of abuse, misuse, and addiction and frequently monitor for signs and symptoms of abuse, misuse, and addiction [see Warnings and Precautions (5.1), Drug Abuse and Dependence (9.2)].
WARNING: ABUSE, MISUSE, AND ADDICTION
See full prescribing information for complete boxed warning.
MYDAYIS has a high potential for abuse and misuse, which can lead to the development of a substance use disorder, including addiction. Misuse and abuse of CNS stimulants, including MYDAYIS, can result in overdose and death (5.1 ,9.2 ,10 ):
- Before prescribing MYDAYIS, assess each patient’s risk for abuse, misuse, and addiction.
- Educate patients and their families about these risks, proper storage of the drug, and proper disposal of any unused drug.
- Throughout treatment, reassess each patient’s risk and frequently monitor for signs and symptoms of abuse, misuse, and addiction.
1 Indications And Usage
MYDAYIS is indicated for the treatment of Attention Deficit Hyperactivity Disorder (ADHD) in patients 13 years and older [see Clinical Studies (14)].
MYDAYIS is a central nervous system (CNS) stimulant indicated for the treatment of Attention Deficit Hyperactivity Disorder (ADHD) in patients 13 years and older. (1 )
Limitations of Use:
Pediatric patients 12 years and younger experienced higher plasma exposure than patients 13 years and older at the same dose and experienced higher rates of adverse reactions, mainly insomnia and decreased appetite. (8.4 )
Limitations of Use:
Pediatric patients 12 years and younger experienced higher plasma exposure than patients 13 years and older at the same dose, and experienced higher rates of adverse reactions, mainly insomnia and decreased appetite [see Use in Specific Populations (8.4)].
2 Dosage And Administration
- MYDAYIS should be administered once daily upon awakening.
Recommended Starting Dose Titration Schedule Maximum Daily Dose Adults 12.5 mg 12.5 mg weekly 50 mg Pediatrics (13 to 17) 12.5 mg 12.5 mg weekly 25 mg
- In adult patients with severe renal impairment the maximum dose should not exceed 25 mg daily. Use in adult patients with ESRD is not recommended. (
2.6 ,8.6 )- The maximum dose in pediatric patients with severe renal impairment is 12.5 mg daily. Use in pediatric patients with ESRD is not recommended. (
2.6 ,8.6 )- Patients are advised to take consistently either with or without food. (
2.2 )- Administer upon awakening because the effects may last up to 16 hours and there is the potential for insomnia. (
2.2 )- Prior to treatment, assess for presence of cardiac disease. (
2.1 )- To avoid substitution errors and overdosage, do not substitute for other amphetamine products on a milligram-per-milligram basis because of different amphetamine base compositions and differing pharmacokinetic profiles. (
2.7 )2.1 Pretreatment Screening
Prior to treating patients with MYDAYIS, assess:
for the presence of cardiac disease (i.e., perform a careful history, family history of sudden death or ventricular arrhythmia, and physical exam) [see Warnings and Precautions (5.2)] the family history and clinically evaluate patients for motor or verbal tics or Tourette’s syndrome before initiating MYDAYIS [see Warnings and Precautions (5.10)] 2.2 General Administration Information
Because the effects of MYDAYIS may last up to 16 hours and there is potential for insomnia, administer once daily in the morning upon awakening. In the event of a missed dose, do not administer later in the day. Do not administer additional medication to make up for the missed dose [see Adverse Reactions (6.1), Clinical Studies (14)].
2.3Administration Instructions
Administer MYDAYIS orally with or without food. Advise patients to take MYDAYIS consistently either with food or without food [see Clinical Pharmacology (12.3)].
MYDAYIS may be administered in one of the following ways:
- Swallow MYDAYIS capsules whole, or
- Open capsule and sprinkle the entire contents over a spoonful of applesauce. The sprinkled applesauce should be consumed immediately; it should not be stored. Patients should take the sprinkled applesauce in its entirety without chewing.
- The dose of a single capsule should not be divided.
2.4 Recommended Dosage
Adults (18 to 55 years)
The recommended starting dose of MYDAYIS is 12.5 mg once daily in the morning upon awakening. Initial doses of 25 mg once daily may be considered for some patients. Dosage may be adjusted in increments of 12.5 mg no sooner than weekly, up to a maximum dose of 50 mg once daily, based on the therapeutic needs and response of the patient. Doses above 50 mg daily have shown no additional clinically meaningful benefit.
Pediatric Patients (13 to 17 years)
The recommended starting dose is 12.5 mg once daily in the morning upon awakening. Dosage may be adjusted in increments of 12.5 mg no sooner than weekly, up to a recommended maximum dose of 25 mg once daily. The dose should be individualized according to the needs and response of the patient. Doses higher than 25 mg have not been evaluated in clinical trials in pediatric patients.
2.5 Dosage Modifications Due to Drug Interactions
Agents that alter gastrointestinal and urinary pH can impact urinary excretion and alter blood levels of amphetamine. Acidifying agents (e.g., ascorbic acid) decrease blood levels, while alkalinizing agents (e.g., sodium bicarbonate) increase blood levels. Adjust MYDAYIS dosage accordingly [see Drug Interactions (7.1)].
2.6 Dosage in Patients with Renal Impairment
In adult patients with severe renal impairment (GFR between 15 to <30 mL/min/1.73 m2), the recommended starting dose of MYDAYIS is 12.5 mg daily with a maximum recommended dose of 25 mg daily. MYDAYIS is not recommended for use in patients with end stage renal disease (ESRD <15 mL/min/1.73 m2). In pediatric patients (13 to 17 years) with severe renal impairment, the maximum dose is 12.5 mg, if tolerated [see Use in Specific Populations (8.6), Clinical Pharmacology (12.3)].
2.7 Switching From Other Amphetamine Products
For patients switching from another medication or any other amphetamine products, discontinue that treatment, and titrate with MYDAYIS using the titration schedule [see Dosage and Administration (2.4)].
Do not substitute for other amphetamine products on a milligram-per-milligram basis because of different amphetamine base compositions and differing pharmacokinetic profiles [see Warnings and Precautions (5.9), Description (11), Clinical Pharmacology (12.3)].
3 Dosage Forms And Strengths
- Extended-release capsules 12.5 mg: green body/green cap (imprinted with SHIRE 465 and 12.5 mg)
- Extended-release capsules 25 mg: ivory body/green cap (imprinted with SHIRE 465 and 25 mg)
- Extended-release capsules 37.5 mg: ivory body/light caramel cap (imprinted with SHIRE 465 and 37.5 mg)
- Extended-release capsules 50 mg: ivory body/purple cap (imprinted with SHIRE 465 and 50 mg)
Extended-release capsules: 12.5 mg, 25 mg, 37.5 mg, 50 mg (3 )
4 Contraindications
MYDAYIS is contraindicated in patients with:
- Known hypersensitivity to amphetamine, or other components of MYDAYIS. Hypersensitivity reactions such as angioedema and anaphylactic reactions have been reported in patients treated with other amphetamine products [see Adverse Reactions (6.2)].
- Concomitant treatment with monoamine oxidase inhibitors (MAOIs), and also within 14 days following discontinuation of treatment with a monoamine oxidase inhibitor, because of an increased risk of hypertensive crisis [see Drug Interactions (7.1)].
- Known hypersensitivity to amphetamine products or other ingredients in MYDAYIS. (
4 )- Use with monoamine oxidase (MAO) inhibitors, or within 14 days of the last MAO inhibitor dose. (
4 ,7.1 )
5 Warnings And Precautions
- Risks to Patients with Serious Cardiac Disease: Avoid use in patients with known structural cardiac abnormalities, cardiomyopathy, serious cardiac arrhythmia, coronary artery disease, or other serious cardiac disease. (
5.2 )- Increased Blood Pressure and Heart Rate: Monitor blood pressure and pulse. (
5.3 )- Psychiatric Adverse Reactions: Prior to initiating MYDAYIS, screen patients for risk factors for developing a manic episode. If new psychotic or manic symptoms occur, consider discontinuing MYDAYIS. (
5.4 )- Long-Term Suppression of Growth in Pediatric Patients: Closely monitor growth (height and weight) in pediatric patients. Pediatric patients not growing or gaining height or weight as expected may need to have their treatment interrupted. (
5.5 )- Peripheral Vasculopathy, Including Raynaud’s Phenomenon: Careful observation for digital changes is necessary during MYDAYIS treatment. Further clinical evaluation (e.g., rheumatology referral) may be appropriate for patients who develop signs or symptoms of peripheral vasculopathy. (
5.6 )- Seizures: May lower the convulsive threshold. If a seizure occurs, discontinue MYDAYIS. (
5.7 )- Serotonin Syndrome: Increased risk when coadministered with serotonergic agents (e.g., SSRIs, SNRIs, triptans), but also during overdosage situations. If it occurs, discontinue MYDAYIS and initiate supportive treatment. (
5.8 )- Motor and Verbal Tics, and Worsening of Tourette’s Syndrome: Before initiating MYDAYIS, assess the family history and clinically evaluate patients for tics or Tourette’s syndrome. Regularly monitor patients for the emergence or worsening of tics or Tourette’s syndrome. Discontinue treatment if clinically appropriate. (
5.10 )5.1 Abuse, Misuse, and Addiction
MYDAYIS has a high potential for abuse and misuse. The use of MYDAYIS exposes individuals to the risks of abuse and misuse, which can lead to the development of a substance use disorder, including addiction. MYDAYIS can be diverted for non-medical use into illicit channels or distribution [see Drug Abuse and Dependence (9.2)]. Misuse and abuse of CNS stimulants, including MYDAYIS, can result in overdose and death [see Overdosage (10)], and this risk is increased with higher doses or unapproved methods of administration, such as snorting or injection.
Before prescribing MYDAYIS, assess each patient’s risk for abuse, misuse, and addiction. Educate patients and their families about these risks and proper disposal of any unused drug. Advise patients to store MYDAYIS in a safe place, preferably locked, and instruct patients to not give MYDAYIS to anyone else. Throughout MYDAYIS treatment, reassess each patient’s risk of abuse, misuse, and addiction and frequently monitor for signs and symptoms of abuse, misuse, and addiction.
5.2 Risks to Patients with Serious Cardiac Disease
Sudden death has been reported in patients with structural cardiac abnormalities or other serious cardiac disease who were treated with CNS stimulants at the recommended ADHD dosage.
Avoid MYDAYIS use in patients with known structural cardiac abnormalities, cardiomyopathy, serious cardiac arrhythmia, coronary artery disease, or other serious cardiac disease.
5.3 Increased Blood Pressure and Heart Rate
CNS stimulants cause an increase in blood pressure (mean increase about 2 to 4 mmHg) and heart rate (mean increase about 3 to 6 bpm). Some patients may have larger increases.
Monitor all MYDAYIS-treated patients for potential tachycardia and hypertension [see Adverse Reactions (6.1)].
5.4 Psychiatric Adverse Reactions
Exacerbation of Pre-Existing Psychosis
CNS stimulants may exacerbate symptoms of behavior disturbance and thought disorder in patients with a pre-existing psychotic disorder.
Induction of a Manic Episode in Patients with Bipolar Disorder
CNS stimulants may induce a manic or mixed episode in patients with bipolar disorder. Prior to initiating MYDAYIS treatment, screen patients for risk factors for developing a manic episode (e.g., comorbid or history of depressive symptoms or a family history of suicide, bipolar disorder, and depression).
New Psychotic or Manic Symptoms
CNS stimulants, at the recommended dosage, may cause psychotic or manic symptoms, e.g., hallucinations, delusional thinking, or mania in patients without a prior history of psychotic illness or mania. In a pooled analysis of multiple short-term, placebo-controlled studies of CNS stimulants, psychotic or manic symptoms occurred in approximately 0.1% of CNS stimulant-treated patients compared to 0% of placebo-treated patients. If such symptoms occur, consider discontinuing MYDAYIS.
5.5 Long-Term Suppression of Growth in Pediatric Patients
CNS stimulants have been associated with weight loss and slowing of growth rate in pediatric patients.
In a 4 week, placebo-controlled trial of MYDAYIS in patients ages 6 to 17 years old with ADHD, there was a decrease in weight in the MYDAYIS groups compared to weight gain in the placebo group [see Adverse Reactions (6.1)].
Closely monitor growth (weight and height) in MYDAYIS-treated pediatric patients. Pediatric patients who are not growing or gaining weight as expected may need to have their treatment interrupted. MYDAYIS is not approved for use in pediatric patients 12 years and younger [see Use in Specific Populations (8.4)].
5.6 Peripheral Vasculopathy, Including Raynauds Phenomenon
CNS stimulants, including MYDAYIS, used to treat ADHD are associated with peripheral vasculopathy, including Raynaud’s phenomenon. Signs and symptoms are usually intermittent and mild; however, sequelae have included digital ulceration and/or soft tissue breakdown. Effects of peripheral vasculopathy, including Raynaud’s phenomenon, were observed in post-marketing reports and at the therapeutic dosage of CNS stimulants in all age groups throughout the course of treatment. Signs and symptoms generally improved after dosage reduction or discontinuation of the CNS stimulant.
Careful observation for digital changes is necessary during MYDAYIS treatment. Further clinical evaluation (e.g., rheumatology referral) may be appropriate for MYDAYIS-treated patients who develop signs or symptoms of peripheral vasculopathy.
5.7Seizures
MYDAYIS may lower the convulsive threshold in patients with prior history of seizure, in patients with prior EEG abnormalities in the absence of seizures, and in patients without a history of seizures and no prior EEG evidence of seizures. In the presence of seizures, MYDAYIS should be discontinued.
5.8Serotonin Syndrome
Serotonin syndrome, a potentially life-threatening reaction, may occur when amphetamines are used in combination with other drugs that affect the serotonergic neurotransmitter systems such as monoamine oxidase inhibitors (MAOIs), selective serotonin reuptake inhibitors (SSRIs), serotonin norepinephrine reuptake inhibitors (SNRIs), triptans, tricyclic antidepressants, fentanyl, lithium, tramadol, tryptophan, buspirone, and St. John's Wort [see Drug Interactions (7.1)]. The coadministration with cytochrome P450 2D6 (CYP2D6) inhibitors may also increase the risk with increased exposure to MYDAYIS. In these situations, consider an alternative nonserotonergic drug or an alternative drug that does not inhibit CYP2D6 [see Drug Interactions (7.1)].
Serotonin syndrome symptoms may include mental status changes (e.g., agitation, hallucinations, delirium, and coma), autonomic instability (e.g., tachycardia, labile blood pressure, dizziness, diaphoresis, flushing, hyperthermia), neuromuscular symptoms (e.g., tremor, rigidity, myoclonus, hyperreflexia, incoordination), seizures, and/or gastrointestinal symptoms (e.g., nausea, vomiting, diarrhea).
Concomitant use of MYDAYIS with MAOI drugs is contraindicated [see Contraindications (4)].
Discontinue treatment with MYDAYIS and any concomitant serotonergic agents immediately if the above symptoms occur, and initiate supportive symptomatic treatment. If concomitant use of MYDAYIS with other serotonergic drugs or CYP2D6 inhibitors is clinically warranted, initiate MYDAYIS with lower doses, monitor patients for the emergence of serotonin syndrome during drug initiation or titration, and inform patients of the increased risk for serotonin syndrome.
5.9Potential for Overdose Due to Medication Errors
Medication errors, including substitution and dispensing errors, between MYDAYIS and other amphetamine products could occur, leading to possible overdosage. To avoid substitution errors and overdosage, do not substitute for other amphetamine products on a milligram-per-milligram basis because of different amphetamine base compositions and differing pharmacokinetic profiles [see Dosage and Administration (2.7), Overdosage (10)].
5.10 Motor and Verbal Tics, and Worsening of Tourettes Syndrome
CNS stimulants, including amphetamine, have been associated with the onset or exacerbation of motor and verbal tics. Worsening of Tourette’s syndrome has also been reported [see Adverse Reactions (6.2)] .
Before initiating MYDAYIS, assess the family history and clinically evaluate patients for tics or Tourette’s syndrome. Regularly monitor MYDAYIS-treated patients for the emergence or worsening of tics or Tourette’s syndrome, and discontinue treatment if clinically appropriate.
6 Adverse Reactions
The following adverse reactions are discussed in greater detail in other sections of the labeling:
- Abuse, Misuse, and Addiction [see Boxed Warning, Warnings and Precautions (5.1), Drug Abuse and Dependence (9.2, 9.3)]
- Hypersensitivity to amphetamine products or other ingredients of MYDAYIS [see Contraindications (4)]
- Hypertensive Crisis When Used Concomitantly with Monoamine Oxidase Inhibitors [see Contraindications (4), Drug Interactions (7.1)]
- Risks to Patients with Serious Cardiac Disease [see Warnings and Precautions (5.2)]
- Increased Blood Pressure and Heart Rate [see Warnings and Precautions (5.3)]
- Psychiatric Adverse Reactions [see Warnings and Precautions (5.4)]
- Long-Term Suppression of Growth in Pediatric Patients [see Warnings and Precautions (5.5)]
- Peripheral Vasculopathy, Including Raynaud's Phenomenon [see Warnings and Precautions (5.6)]
- Seizures [see Warnings and Precautions (5.7)]
- Serotonin Syndrome [see Warnings and Precautions (5.8)]
- Motor and Verbal Tics, and Worsening of Tourette's Syndrome [see Warnings and Precautions (5.10)]
Most common adverse reactions in patients with ADHD (incidence ≥5% and at a rate at least twice placebo) are:
- Pediatrics (13 years and older): insomnia, decreased appetite, decreased weight, irritability, and nausea. (
6.1 )- Adults: insomnia, decreased appetite, decreased weight, dry mouth, increased heart rate, and anxiety. (
6.1 )
To report SUSPECTED ADVERSE REACTIONS, contact Takeda Pharmaceuticals at 1-800-828-2088 or FDA at 1-800-FDA-1088 orwww.fda.gov/medwatch .
6.1Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
MYDAYIS was studied in adults (18 to 55 years) and pediatric patients (13 to 17 years) who met Diagnostic and Statistical Manual of Mental Disorders, 4th or 5th editions (DSM-IV-TR® or DSM-5) criteria for ADHD. The safety data for adults were pooled from three randomized, double-blind, placebo-controlled studies in doses of 12.5 mg to 75 mg per day (1.5 times the maximum recommended dosage). Doses higher than 50 mg per day did not demonstrate additional clinical benefit and are not recommended.
The safety data for pediatric patients (13 to 17 years) is from 1 randomized, double-blind, placebo-controlled study of doses of 12.5 mg to 25 mg. The total exposure in patients treated with MYDAYIS totalled 704; this included pediatric patients, 78 adolescent patients and 626 adult patients from multiple well-controlled trials. The duration of use ranged from 4 to 7 weeks [see Clinical Studies (14)].
Adverse Reactions Leading to Discontinuation of Treatment
In pooled controlled trials of adult patients, 9% (54/626) of MYDAYIS-treated patients discontinued due to adverse reactions compared to 2% (7/328) of placebo-treated patients. The most frequent adverse reactions leading to discontinuation (i.e., leading to discontinuation in at least 1% of MYDAYIS-treated patients and at a rate at least twice that of placebo) were insomnia (2%, n=15), blood pressure increased (2%, n=10), decreased appetite (1%, n=5), and headache (1%, n=4).
In a controlled trial including adolescent patients (13 to 17 years), 5% (4/78) of MYDAYIS-treated patients discontinued due to adverse reactions compared to 0% (0/79) of placebo-treated patients. The most frequent adverse reaction leading to discontinuation (i.e., leading to discontinuation in at least 1% of MYDAYIS-treated patients and at a rate at least twice that of placebo) were dizziness (1%, n=1), depression (1%, n=1), abdominal pain upper (1%, n=1), and viral infection (1%, n=1).
Adverse Reactions Occurring at an Incidence of ≥2% and at Least Twice Placebo Among MYDAYIS-Treated Adults in Clinical Trials
The most common adverse reactions reported in adults were insomnia, decreased appetite, dry mouth, decreased weight, heart rate increased, and anxiety. Table 1 uls the adverse reactions that occurred ≥2% compared to placebo. The most common adverse reaction (insomnia) generally occurred early during treatment with MYDAYIS.
Table 1: Adverse Reactions Reported by 2% or More of Adults Taking MYDAYIS and at Least Twice the Incidence in Patients Taking Placebo in 3 Clinical Trials (4, 6, and 7 Weeks) Body System Adverse Reaction MYDAYIS Includes doses up to 75 mg (1.5 times the maximum recommended dosage). (N = 626)Placebo (N = 328) Nervous System Anxiety 7% 3% Feeling Jittery 2% 1% Agitation 2% 0% Bruxism 2% 0% Psychiatric Disorders Insomnia 31% 8% Depression 3% 0% Metabolism and Nutritional Disorders Decreased Appetite 30% 4% Weight Decreased 9% 0% Gastrointestinal System Dry Mouth 23% 4% Diarrhea 3% 1% Cardiovascular System Heart Rate Increased 9% 0% Palpitations 4% 2% Genitourinary System Dysmenorrhea Dysmenorrhea was observed in 11 females. 4% 2% Erectile Dysfunction Erectile dysfunction was observed in 6 males. 2% 1%
Adverse Reactions Occurring at an Incidence of 2% or More and at Least Twice Placebo Among MYDAYIS-Treated Adolescents (13 to 17 years) in a 4 Week Clinical Trial
The most common adverse reactions reported in adolescents were decreased appetite, nausea, insomnia, abdominal pain upper, irritability, and weight decreased. Table 2 uls the adverse reactions that occurred ≥2% compared to placebo.
Table 2: Adverse Reactions Reported by ≥2% or More of Adolescents Taking MYDAYIS and at Least Twice the Incidence in Patients Taking Placebo in a 4 Week Clinical Trial Body System Adverse Reaction MYDAYIS (N = 78) Placebo (N = 79) Nervous System Dizziness 4% 0% Metabolism and Nutrition Disorders Decreased appetite 22% 6% Weight decreased 5% 1% Psychiatric Disorders Irritability 6% 3% Insomnia Insomnia includes terms: initial insomnia, middle insomnia, terminal insomnia and insomnia. 8% 3% Gastrointestinal Disorders Nausea 8% 4% Abdominal pain upper 4% 1% 6.2Adverse Reactions Associated with the Use of Amphetamines
The following adverse reactions have been associated with the use of amphetamines. The following adverse reactions have been identified during postapproval use of amphetamines. Because these reactions are reported voluntarily from a population of uncertain size, it is not possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Allergic: Urticaria, rash, hypersensitivity reactions, including angioedema and anaphylaxis. Serious skin rashes, including Stevens-Johnson syndrome and toxic epidermal necrolysis have been reported.
Cardiovascular: Dyspnea, sudden death. There have been isolated reports of cardiomyopathy associated with chronic amphetamine use.
Central Nervous System: Psychotic episodes at recommended doses, overstimulation, restlessness, euphoria, dyskinesia, dysphoria, headache, tics, fatigue, aggression, anger, logorrhea, dermatillomania, and paresthesia (including formication), motor and verbal tics.
Endocrine: Impotence, changes in libido, frequent or prolonged erections.
Eye Disorders: Mydriasis.
Gastrointestinal: Unpleasant taste, constipation, intestinal ischemia.
Musculoskeletal and Connective Tissue Disorders: Rhabdomyolysis.
Skin: Alopecia.
Vascular Disorders: Raynaud’s phenomenon.
7 Drug Interactions
Acidifying and Alkalinizing Agents: Agents that alter GI and urinary pH can alter blood levels of amphetamine. Acidifying agents (GI and urinary) decrease amphetamine blood levels, while alkalinizing agents (GI and urinary) increase amphetamine blood levels. Adjust MYDAYIS dosage accordingly. (2.5 ,7.1 )
7.1Drugs Having Clinically Important Interactions with Amphetamines
Table 3: Drugs Having Clinically Important Interactions with Amphetamines Monoamine Oxidase Inhibitors (MAOIs) Clinical Impact MAOI antidepressants slow amphetamine metabolism, increasing amphetamines effect on the release of norepinephrine and other monoamines from adrenergic nerve endings causing headaches and other signs of hypertensive crisis. Toxic neurological effects and malignant hyperpyrexia can occur, sometimes with fatal results. Intervention Do not administer MYDAYIS during or within 14 days following the administration of MAOI [see Contraindications (4)]. Serotonergic Drugs Clinical Impact The concomitant use of amphetamines and serotonergic drugs increases the risk of serotonin syndrome. Intervention Initiate with lower doses and monitor patients for signs and symptoms of serotonin syndrome, particularly during MYDAYIS initiation or dosage increase. If serotonin syndrome occurs, discontinue MYDAYIS and concomitant serotonergic drug(s) [see Warnings and Precautions (5.7)]. Alkalinizing Agents Clinical Impact May increase exposure to amphetamine and exacerbate the action of amphetamine. Intervention Caution should be taken when coadministering MYDAYIS and gastrointestinal and urinary alkalinizing agents. Acidifying Agents Clinical Impact Lower blood levels and efficacy of amphetamines. Intervention Increase dose of MYDAYIS based on clinical response. Tricyclic Antidepressants Clinical Impact May enhance the activity of tricyclic or sympathomimetic agents causing sustained increases in the concentration of d-amphetamine in the brain; cardiovascular effects can be potentiated. Intervention Monitor frequently and adjust MYDAYIS dose or use alternative therapy based on clinical response. CYP2D6 Inhibitors Clinical Impact May increase the exposure of amphetamine. Intervention Start with lower doses and monitor frequently and adjust MYDAYIS dose or use alternative therapy based on clinical response. Gastric pH Modulators Clinical Impact Potential change in shape of PK profile and exposure may occur. Intervention Monitor patients for changes in clinical effect and use alternative therapy based on clinical response. 7.2Drug/Laboratory Test Interactions
Amphetamines can cause a significant elevation in plasma corticosteroid levels. This increase is greatest in the evening. Amphetamines may interfere with urinary steroid determinations.
8 Use In Specific Populations
- Pregnancy: Based on animal data, may cause fetal harm. (
8.1 )- Lactation: Breastfeeding not recommended. (
8.2 )- Pediatric: Safety and effectiveness have not been established in pediatric patients ages 12 years and younger. (
8.4 )- Renal Impairment: Dose adjustment is needed in patients with severe renal insufficiency. Use of MYDAYIS in patients with ESRD is not recommended. (
2.6 ,8.6 )8.1 Pregnancy
Pregnancy Exposure Registry
There is a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to MYDAYIS during pregnancy. Healthcare providers are encouraged to register patients by calling the National Pregnancy Registry for Psychiatric Medications at 1-866-961-2388 or visiting online at https://womensmentalhealth.org/research/pregnancyregistry/.
Risk Summary
The limited available data from published literature and postmarketing reports on use of amphetamine in pregnant women are not sufficient to inform a drug-associated risk for major birth defects and miscarriage. Adverse pregnancy outcomes, including premature delivery and low birth weight, have been seen in infants born to mothers dependent on amphetamines [see Clinical Considerations].
In an embryofetal development study, amphetamine (d- to l- enantiomer ratio of 3:1, the same as in MYDAYIS) had no effects on embryofetal morphological development or survival when administered to pregnant rats and rabbits throughout the period of organogenesis up to doses 10 times the maximum recommended human dose (MRHD) of 25 mg/day given to adolescents, on a mg/m2 body surface area basis. However, in a pre- and postnatal development study, amphetamine (d- to l- ratio of 3:1) administered orally to pregnant rats during gestation and lactation caused a decrease in pup survival and a decrease in pup body weight that correlated with a delay in developmental landmarks at clinically relevant doses of amphetamine. In addition, adverse effects on reproductive performance were observed in pups whose mothers were treated with amphetamine. Long-term neurochemical and behavioral effects have also been reported in animal developmental studies using clinically relevant doses of amphetamine [see Data].
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
Clinical Considerations
Fetal/Neonatal Adverse Reactions
Amphetamines, such as MYDAYIS, cause vasoconstriction and thereby may decrease placental perfusion. In addition, amphetamines can stimulate uterine contractions increasing the risk of premature delivery. Infants born to amphetamine-dependent mothers have an increased risk of premature delivery and low birth weight.
Monitor infants born to mothers taking amphetamines for symptoms of withdrawal such as feeding difficulties, irritability, agitation, and excessive drowsiness.
Data
Animal Data
Amphetamine (d- to l- enantiomer ratio of 3:1, the same as in MYDAYIS) had no apparent effects on embryofetal morphological development or survival when administered orally to pregnant rats and rabbits throughout the period of organogenesis at doses of up to 6 and 16 mg/kg/day, respectively. These doses are approximately 2 and 10 times, respectively, the maximum recommended human dose (MRHD) of 25 mg/day given to adolescents, on a mg/m2 body surface area basis. Fetal malformations and death have been reported in mice following parenteral administration of d-amphetamine doses of 50 mg/kg/day (approximately 8 times the MRHD given to adolescents on a mg/m2 basis) or greater to pregnant animals. Administration of these doses was also associated with severe maternal toxicity.
A pre- and postnatal development study was conducted with amphetamine (d- to l- enantiomer ratio of 3:1) in which pregnant rats received daily oral doses of 2, 6, and 10 mg/kg from gestation Day 6 to lactation Day 20. These doses are approximately 0.6, 2, and 3 times the MRHD of 25 mg/day amphetamine (d- to l- ratio of 3:1) given to adolescents, on a mg/m2 basis. All doses caused hyperactivity and decreased weight gain in the dams. A decrease in pup survival was seen at all doses. A decrease in pup body weight was seen at 6 and 10 mg/kg which correlated with delays in developmental landmarks, such as preputial separation and vaginal opening. Increased pup locomotor activity was seen at 10 mg/kg on Day 22 postpartum but not at 5 weeks postweaning. When pups were tested for reproductive performance at maturation, gestational weight gain, number of implantations, and number of delivered pups were decreased in the group whose mothers had been given 10 mg/kg.
A number of studies from the literature in rodents indicate that prenatal or early postnatal exposure to amphetamine (d- or d, l-) at doses similar to those used clinically can result in long-term neurochemical and behavioral alterations. Reported behavioral effects include learning and memory deficits, altered locomotor activity, and changes in sexual function.
8.2 Lactation
Risk Summary
Based on limited case reports in published literature, amphetamine (d- or d, l-) is present in human milk, at relative infant doses of 2 to 13.8% of the maternal weight-adjusted dosage and a milk/plasma ratio ranging between 1.9 and 7.5. There are no reports of adverse effects on the breastfed infant. Long-term neurodevelopmental effects on infants from amphetamine exposure are unknown. It is possible that large dosages of dextroamphetamine might interfere with milk production, especially in women whose lactation is not well established. Because of the potential for serious adverse reactions in nursing infants, including serious cardiovascular reactions, blood pressure and heart rate increase, suppression of growth, and peripheral vasculopathy, advise patients that breastfeeding is not recommended during treatment with MYDAYIS.
8.4 Pediatric Use
The safety and effectiveness of MYDAYIS in pediatric patients with ADHD ages 13 to 17 years have been established in two placebo-controlled clinical studies [see Adverse Reactions (6.1), Clinical Pharmacology (12.3), Clinical Studies (14)].
The safety and effectiveness of MYDAYIS have not been established in pediatric patients ages 12 years and younger.
MYDAYIS has been studied for the treatment of ADHD in pediatric patients 6 to 12 years in two placebo controlled safety and efficacy trials. In the first trial, pediatric patients 6 to 12 years experienced higher rates of adverse reactions in some cases compared to patients 13 years and older, including higher rates of insomnia (30% vs 8%) and appetite decreased (43% vs 22%). In addition, amphetamine systemic exposures (both d- and l-) in pediatric patients 6 to 12 years following a single dose were higher than those observed in adults at the same dose (72 to 79% higher Cmax and approximately 83% higher AUC). A second trial evaluated a lower dose than those approved for pediatric patients 13 to 17 years; efficacy was not demonstrated for the lower dose. Therefore, a safe and effective dose cannot be established in pediatric patients 12 years and younger.
Growth Suppression
Growth should be monitored during treatment with stimulants, including MYDAYIS, in pediatric patients 13 to 17 years who are not growing or gaining weight as expected may need to have their treatment interrupted [see Warnings and Precautions (5.5), Adverse Reactions (6.1)].
Juvenile Animal Toxicity Data
Juvenile rats treated with mixed amphetamine salts (same as in MYDAYIS) early in the postnatal period through sexual maturation demonstrated transient changes in motor activity. Learning and memory was impaired at approximately 8 times the maximum recommended human dose (MRHD) given to children on a mg/m2 basis. No recovery was seen following a drug-free period. A delay in sexual maturation was observed at a dose approximately 8 times the MRHD given to children on a mg/m2 basis, although there was no effect on fertility.
In a juvenile developmental study, rats received daily oral doses of amphetamine (d to l enantiomer ratio of 3:1, the same as in MYDAYIS) of 2, 6, or 20 mg/kg on days 7 to 13 of age; from Day 14 to approximately Day 60 of age these doses were given b.i.d. for total daily doses of 4, 12, or 40 mg/kg. The latter doses are approximately 0.8, 2, and 8 times the MRHD of 25 mg/day given to children on a mg/m2 basis. Postdosing hyperactivity was seen at all doses; motor activity measured prior to the daily dose was decreased during the dosing period but the decreased motor activity was largely absent after an 18 day drug-free recovery period. Performance in the Morris water maze test for learning and memory was impaired at the 40 mg/kg dose, and sporadically at the lower doses, when measured prior to the daily dose during the treatment period; no recovery was seen after a 19 day drug-free period. A delay in the developmental milestones of vaginal opening and preputial separation was seen at 40 mg/kg but there was no effect on fertility.
8.5 Geriatric Use
Clinical studies of MYDAYIS did not include sufficient numbers of subjects aged 65 and over to determine whether they respond differently from younger subjects. Other reported clinical experience has not identified differences in responses between the elderly and younger patients. In general, dose selection for an elderly patient should start at the low end of the dosing range, reflecting the greater frequency of decreased hepatic, renal, or cardiac function, and of concomitant disease or other drug therapy.
8.6Renal Impairment
Due to reduced clearance of amphetamine in patients with severe renal insufficiency (GFR 15 to <30 mL/min/1.73 m2), the maximum dose in adults should be reduced. Pediatric patients ages 13 to 17 years with severe renal insufficiency can be given the recommended starting dose if tolerated, but the dose should not be escalated. MYDAYIS is not recommended in patients with ESRD (GFR <15 mL/min/1.73 m2) [see Dosage and Administration (2.6), Clinical Pharmacology (12.3)].
d-Amphetamine is not dialyzable.
9 Drug Abuse And Dependence
9.1 Controlled Substance
MYDAYIS contains mixed amphetamine salts, a Schedule II controlled substance.
9.2 Abuse
MYDAYIS has a high potential for abuse and misuse which can lead to the development of a substance use disorder, including addiction [see Warnings and Precautions (5.1)]. MYDAYIS can be diverted for non-medical use into illicit channels or distribution.
Abuse is the intentional non-therapeutic use of a drug, even once, to achieve a desired psychological or physiological effect. Misuse is the intentional use, for therapeutic purposes, of a drug by an individual in a way other than prescribed by a healthcare provider or for whom it was not prescribed. Drug addiction is a cluster of behavioral, cognitive, and physiological phenomena that may include a strong desire to take the drug, difficulties in controlling drug use (e.g., continuing drug use despite harmful consequences, giving a higher priority to drug use than other activities and obligations), and possible tolerance or physical dependence.
Misuse and abuse of amphetamine may cause increased heart rate, respiratory rate, or blood pressure; sweating; dilated pupils; hyperactivity; restlessness; insomnia; decreased appetite; loss of coordination; tremors; flushed skin; vomiting; and/or abdominal pain. Anxiety, psychosis, hostility, aggression, and suicidal or homicidal ideation have also been observed with CNS stimulants abuse and/or misuse. Misuse and abuse of CNS stimulants, including MYDAYIS, can result in overdose and death [see Overdosage (10)], and this risk is increased with higher doses or unapproved methods of administration, such as snorting or injection.
9.3 Dependence
Physical Dependence
MYDAYIS may produce physical dependence. Physical dependence is a state that develops as a result of physiological adaptation in response to repeated drug use, manifested by withdrawal signs and symptoms after abrupt discontinuation or a significant dose reduction of a drug.
Withdrawal signs and symptoms after abrupt discontinuation or dose reduction following prolonged use of CNS stimulants including MYDAYIS include dysphoric mood; depression; fatigue; vivid, unpleasant dreams; insomnia or hypersomnia; increased appetite; and psychomotor retardation or agitation.
Tolerance
MYDAYIS may produce tolerance. Tolerance is a physiological state characterized by a reduced response to a drug after repeated administration (i.e., a higher dose of a drug is required to produce the same effect that was once obtained at a lower dose).
10 Overdosage
Clinical Effects of Overdose
Overdose of CNS stimulants is characterized by the following sympathomimetic effects:
- Cardiovascular effects including tachyarrhythmias, and hypertension or hypotension. Vasospasm, myocardial infarction, or aortic dissection may precipitate sudden cardiac death. Takotsubo cardiomyopathy may develop.
- CNS effects including psychomotor agitation, confusion, and hallucinations. Serotonin syndrome, seizures, cerebral vascular accidents, and coma may occur.
- Life-threatening hyperthermia (temperatures greater than 104°F) and rhabdomyolysis may develop.
Overdose Management
Consider the possibility of multiple drug ingestion. The pharmacokinetic profile of MYDAYIS should be considered when treating patients with overdose. D-amphetamine is not dialyzable. Consider contacting the Poison Help line (1-800-222-1222) or a medical toxicologist for additional overdose management recommendations.
11 Description
MYDAYIS extended-release capsules contain mixed salts of a single-entity amphetamine, a CNS stimulant. MYDAYIS contains equal amounts (by weight) of four salts: dextroamphetamine sulfate and amphetamine sulfate, dextroamphetamine saccharate and amphetamine aspartate monohydrate. This results in a 3:1 mixture of dextro- to levo- amphetamine base equivalent.
The 12.5 mg, 25 mg, 37.5 mg and 50 mg strength capsules are for oral administration. They contain three types of drug-releasing beads, an immediate release and two different types of delayed release (DR) beads. The first DR bead releases amphetamine at pH 5.5 and the other DR bead releases amphetamine at pH 7.0.
CAPSULE STRENGTHS EACH CAPSULE CONTAINS: 12.5 mg 25 mg 37.5 mg 50 mg Dextroamphetamine Saccharate 3.125 mg 6.250 mg 9.375 mg 12.500 mg Amphetamine Aspartate Monohydrate 3.125 mg 6.250 mg 9.375 mg 12.500 mg Dextroamphetamine Sulfate 3.125 mg 6.250 mg 9.375 mg 12.500 mg Amphetamine Sulfate 3.125 mg 6.250 mg 9.375 mg 12.500 mg Total mixed amphetamine salts 12.500 mg 25 mg 37.5 mg 50 mg Total amphetamine base equivalence 7.8 mg 15.6 mg 23.5 mg 31.3 mg
Inactive Ingredients and Colors
The inactive ingredients in MYDAYIS capsules include: hard gelatin capsules, ethylcellulose, hydroxypropyl methylcellulose, methacrylic acid copolymer, methyl acrylate, methyl methacrylate, methacrylic acid copolymer, opadry beige, sugar spheres, talc, and triethyl citrate. The gelatin capsules for all four strengths contain gelatin, titanium dioxide, yellow iron oxide, and edible inks. The 12.5 mg and 25 mg strength gelatin capsules also contain FD&C Blue #2. The 37.5 mg strength also contains red iron oxide. The 50 mg strength capsule also contains D&C Red #28, D&C Red #33, and FD&C Blue #1.
12 Clinical Pharmacology
12.1 Mechanism of Action
Amphetamines are non-catecholamine sympathomimetic amines with CNS stimulant activity. The exact mode of therapeutic action in ADHD is not known.
12.2 Pharmacodynamics
Amphetamines block the reuptake of norepinephrine and dopamine into the presynaptic neuron and increase the release of these monoamines into the extraneuronal space.
12.3 Pharmacokinetics
MYDAYIS contains d-amphetamine and l-amphetamine salts in the ratio of 3:1. Pharmacokinetic studies of d- and l-amphetamine after oral administration of MYDAYIS have been conducted in healthy adults (19 to 52 years) and pediatric patients (6 to 17 years) with ADHD. Following administration of MYDAYIS, the peak plasma concentrations occurred in about 7 to 10 hours in pediatric patients and about 8 hours in adults for both d-amphetamine and l-amphetamine. The mean plasma elimination half-life for d-amphetamine ranges from about 10 to 11 hours and l-amphetamine from 10 to 13 hours in both pediatric and adult patients.
Absorption
MYDAYIS exhibits linear dose proportionality over the range of 12.5 to 50 mg. Steady-state is achieved between Days 7 and 8 of dosing with mean accumulation ratio of 1.6. A single dose of MYDAYIS 37.5 mg capsules provided comparable plasma concentration profiles of both d- and l-amphetamine to mixed amphetamine salts extended release (MAS-ER) 25 mg followed by 12.5 mg immediate release amphetamine administered 8 hours later (Figure 1).
Figure 1: Mean Plasma Concentrations of d- and l-amphetamine Following Oral Administration of MYDAYIS 37.5 mg vs MAS-ER 25 mg Followed by Immediate-Release MAS-IR 12.5 mg 8 Hours Later in Adults
Effect of Food
High fat meal does not affect the extent of absorption of d- and l-amphetamine when taken with MYDAYIS. Tmax is prolonged by 5 hours (from 7.0 hours at fasted state to 12.0 hours after a high-fat meal) for d-amphetamine and 4.5 hours (from 7.5 hours at fasted state to 12 hours after a high-fat meal) for l-amphetamine after administration of MYDAYIS 50 mg with high fat meal. Opening the capsule and sprinkling the contents on applesauce results in comparable absorption and exposure to the intact capsule taken in the fasted state [see Dosage and Administration (2.3)].
Effect of Alcohol
The in vitro testing showed increases in amphetamine release rate from MYDAYIS capsules in the presence of 20% and, more noticeably, 40% alcohol. There is no in vivo study conducted for the effect of alcohol on drug exposure.
Elimination
Metabolism
Amphetamine is reported to be oxidized at the 4 position of the benzene ring to form 4-hydroxyamphetamine, or on the side chain α or β carbons to form alpha-hydroxy-amphetamine or norephedrine, respectively. Norephedrine and 4-hydroxy-amphetamine are both active and each is subsequently oxidized to form 4-hydroxy-norephedrine. Alpha-hydroxy-amphetamine undergoes deamination to form phenylacetone, which ultimately forms benzoic acid and its glucuronide and the glycine conjugate hippuric acid. Although the enzymes involved in amphetamine metabolism have not yet been clearly defined, CYP2D6 is known to be involved with formation of 4-hydroxy-aphetamine. Since CYP2D6 is genetically polymorphic, population variations in amphetamine metabolism are a possibility.
Amphetamine is known to inhibit monoamine oxidase. Amphetamines are not an in vitro inhibitor of the major human CYP450 isoforms (CYP1A2, CYP2C9, CYP2C19, CYP2D6, and CYP3A4), nor was it an in vitro inducer of CYP1A2, CYP2B6 or CYP3A4/5. Amphetamines are not an in vitro substrate for P-gp.
Excretion
The renal excretion is the primary route for elimination of d- and l-amphetamine and its metabolites after administration of MYDAYIS.
At normal urine pHs, approximately half of an administered dose of amphetamine is recoverable in urine as derivatives of alpha-hydroxy-amphetamine and approximately another 30 to 40% of the dose is recoverable in urine as amphetamine itself. Urinary recovery of amphetamine is highly dependent on pH and urine flow rates. Alkaline urine pHs result in less ionization and reduced renal elimination, and acidic pHs and high flow rates result in increased renal elimination. Urinary recovery of amphetamine has been reported to range from 1% to 75%, and the fraction of a dose hepatically metabolized is dependent on urine pH. Consequently, both hepatic and renal dysfunctions have the potential to alter the elimination of amphetamine and could result in prolonged exposures [see Drug Interactions (7.1)].
Specific Populations
Age
Comparison of the pharmacokinetics of d- and l-amphetamine after oral administration of MYDAYIS in pediatric patients with ADHD 13 to 17 years old and healthy adult subjects (19 to 52 years) indicates that body weight is the primary determinant of apparent differences in the pharmacokinetics of d- and l-amphetamine across the age range.
PK data from patients age 13 to 17 years (n=14) who received a single 25 mg MYDAYIS capsule was scaled (based on PK proportionality) and compared with PK data from adult patients 19 to 51 years (n=20) who received 37.5 mg. Based on dose proportionality, a single-dose MYDAYIS capsule administered to pediatric patients age 13 to 17 years (n=14) would produce about 21% to 31% higher Cmax for d- and l-amphetamine and 21% to 31% higher AUC for d- and l-amphetamine, compared to the same dose of MYDAYIS capsule administered to adults (age 19 to 51 years).
Male and Female Patients
In pharmacokinetic studies, systemic exposure to d- and l-amphetamine was similar in women (N=41) and in men (N=61).
Racial Groups
Formal pharmacokinetic studies for race have not been conducted. However, amphetamine pharmacokinetics appeared to be comparable among Whites (N=41), Blacks (N=27), and Hispanics (N=34).
Patients with Renal impairment
The effect of renal impairment on d- and l-amphetamine after administration of MYDAYIS has not been studied.
In a pharmacokinetic study of lisdexamfetamine in adult subjects with normal and impaired renal function mean d-amphetamine clearance was reduced from 0.7 L/hr/kg in normal subjects to 0.4 L/hr/kg in subjects with severe renal impairment (GFR 15 to <30 mL/min/1.73 m2) patients. Dialysis did not significantly affect the clearance of d-amphetamine. The impact of renal impairment on the disposition of amphetamine would be expected to be similar between oral administration of lisdexamfetamine and MYDAYIS [see Use in Specific Populations (8.6)].
13 Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenesis
No evidence of carcinogenicity was found in studies in which d, l-amphetamine (enantiomer ratio of 1:1) was administered to mice and rats in the diet for 2 years at doses of up to 30 mg/kg/day in male mice, 19 mg/kg/day in female mice, and 5 mg/kg/day in male and female rats. These doses are approximately 3, 2, and 1 times, respectively, the maximum recommended human dose of 50 mg/day on a mg/m2 body surface area basis in adults.
Mutagenesis
Amphetamine, in the enantiomer ratio present, d- to l- ratio of 3:1, was not clastogenic in the mouse bone marrow micronucleus test in vivo and was negative when tested in the E. coli component of the Ames test in vitro. d, l-Amphetamine (1:1 enantiomer ratio) has been reported to produce a positive response in the mouse bone marrow micronucleus test, an equivocal response in the Ames test, and negative responses in the in vitro sister chromatid exchange and chromosomal aberration assays.
Impairment of Fertility
Amphetamines, in the enantiomer ratio, d- to l- ratio of 3:1, did not adversely affect fertility or early embryonic development in the rat at doses of up to 20 mg/kg/day (approximately 6 times the maximum recommended human dose of 25 mg/day given to adolescents on a mg/m2 body surface area basis).
13.2 Animal Toxicology and/or Pharmacology
Acute administration of high doses of amphetamine (d- or d, l-) has been shown to produce long-lasting neurotoxic effects, including irreversible nerve fiber damage in rodents. The significance of these findings to humans is unknown.
14 Clinical Studies
Efficacy of MYDAYIS in the treatment of ADHD was established in the following trials:
- Three short-term trials in adults (18 to 55 years, Studies 1, 2, and 3)
- Two short-term trials in pediatric patients (13 to 17 years, Studies 4 and 5)
Adult Patients (18 to 55 years) with ADHD
The approved adult doses, 12.5 mg, 25 mg, and 37.5 mg are based on Studies 1 and 3 and the 50 mg dose efficacy is based on Study 2. Doses up to 75 mg per day (1.5 times the maximum recommended adult dosage) were evaluated, but demonstrated no additional clinical benefit.
A 4 week, randomized, double-blind, multicenter, placebo-controlled, forced-dose titration, safety and efficacy study (Study 1) was conducted in adults aged 18 to 55 years (N=275) who met DSM-5 criteria for ADHD. Patients were randomized in a 1:1:1 ratio, to two MYDAYIS treatment groups and a placebo group. Group 1 received a dose of 12.5 mg/day throughout the study. Group 2 were titrated on a weekly basis from the initial dose 12.5 mg until target dose of 37.5 mg/day was reached by Week 3 and were maintained at 37.5 mg throughout the study. Group 3 received placebo.
The primary efficacy endpoint was defined as the change from baseline of the adult ADHD-Rating Scale (RS) with prompts total score at Week 4. Baseline adult ADHD-RS with prompts total score was defined as the last valid adult ADHD-RS with prompts total score assessment prior to taking the first dose of double-blind investigational product, usually at Visit 2. The primary comparison of interest was at Week 4 for each MYDAYIS dose compared with placebo. MYDAYIS demonstrated a statistically significant treatment effect compared with placebo on change of ADHD-RS total score from baseline at visit 6 (Week 4), for both 12.5 mg and 37.5 mg doses respectively (Study 1 in Table 4). Patients on MYDAYIS also showed statistically significantly greater improvement on the Clinical Global Impression of Improvement (CGI-I) score compared with placebo treatment.
Two multicenter, randomized, double-blind, placebo-controlled, crossover studies of MYDAYIS 25 mg/day (Study 3) and 50 mg/day (Study 2) were conducted in adult patients who met DSM-IV TR criteria for ADHD. The efficacy was determined using the Permanent Product Measure of Performance (PERMP), a skill-adjusted math test that measures attention in ADHD. PERMP total score results from the sum of the number of math problems attempted plus the number of math problems answered correctly. Efficacy assessments were conducted at 2, 4, 8, 12, 14, and 16 hours post-dose using the PERMP. MYDAYIS treatment, compared to placebo, reached statistical significance at either 2 hours (Study 2) or 4 hours (Study 3) post-dose to 16 hours post-dose in both studies. In a prespecified supplementary analysis for Study 2, the maximum approved dose of MYDAYIS (50 mg) demonstrated a statistically significant treatment effect compared with placebo beginning at 2 to 16 hours post-dose (Study 2 and Study 3 in Table 4).
Pediatric Patients (13 to 17 years) with ADHD
A 4 week, randomized, double-blind, multicenter, placebo-controlled, dose-optimization, safety and efficacy study (Study 4) was conducted. In Study 4, the 157 pediatric patients 13 to 17 years old who met DSM-IV TR criteria for ADHD, were randomized in a 1:1 ratio to MYDAYIS or placebo group. Subjects were titrated from a dose of 12.5 mg/day until an optimal dose was reached (up to a maximum dose of 25 mg); this dose was maintained during the dose-maintenance period (Study 4 in Table 4).
The primary efficacy endpoint was defined as the change from baseline of the ADHD-RS-IV Total Score at Week 4. The baseline ADHD-RS-IV Total Score was defined as the last valid ADHD-RS-IV Total Score assessment prior to taking the first dose of double-blind investigational product, usually at Visit 2. MYDAYIS demonstrated a statistically significant treatment effect compared with placebo on the change of ADHD RS-IV total scores from baseline at Visit 6 (Week 4). MYDAYIS also showed statistically significantly greater improvement on the Clinical Global Impression of Improvement (CGI-I) score at Visit 6 (Week 4).
A multicenter, randomized, double-blind, placebo-controlled, crossover study of MYDAYIS 25 mg/day (Study 5) was conducted in adolescent patients who met DSM-IV TR criteria for ADHD. The efficacy was determined using the Permanent Product Measure of Performance (PERMP), a skill-adjusted math test that measures attention in ADHD. PERMP total score results from the sum of the number of math problems attempted plus the number of math problems answered correctly. Efficacy assessments were conducted at 2, 4, 8, 12, 14, and 16 hours post-dose using the PERMP. MYDAYIS treatment, compared to placebo, reached statistical significance at 2 to 16 hours post-dose (Study 5 in Table 4, Figure 2).
Figure 2: LS Mean (SE) PERMP Total Score by Treatment and Time-Point for Adolescents Ages 13 to 17 with ADHD After 1 Week of Double Blind Treatment (Study 5)
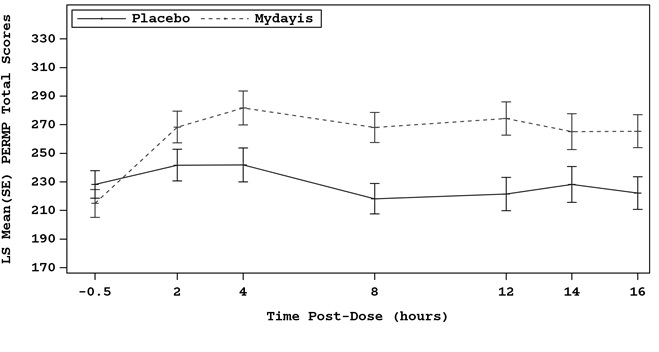
LS Mean: least-squares mean; SE: standard error
In both adults and pediatric patients, examination of a population subset based on gender or race did not reveal any differences.
Table 4: Summary of Primary Efficacy Results from Short-Term Studies of MYDAYIS in Adult and Pediatric Patients with ADHD Study Number (Age range) Primary Endpoint Treatment Group Mean Baseline Score (SD) LS Mean Change from Baseline Placebo-subtracted Difference Difference (drug minus placebo) in least-squares mean change from baseline. (95% CI)SD: standard deviation; LS Mean: least-squares mean; CI: confidence interval. Adult Studies Study 1(18 to 55 years) ADHD-RS MYDAYIS (12.5 mg/day) Doses statistically significantly superior to placebo. 39.8 (6.38) -18.5 -8.1 (-11.7, -4.4) MYDAYIS (37.5 mg/day) 39.9 (7.07) -23.8 -13.4 (-17.1, -9.7) Placebo 40.5 (6.52) -10.4 Study 2(18 to 55 years) Average PERMP MYDAYIS (50 mg/day) 239.2 (75.6) Pre-dose PERMP total score. 293.23 LS Mean for PERMP is post-dose average score over all sessions of the treatment day, rather than change from baseline. 18.38 (11.28, 25.47) Placebo 249.6 (76.7) 274.85 Study 3(18 to 55 years) Average PERMP MYDAYIS (25 mg/day) 217.5 (59.6) 267.96 19.29 (10.95, 27.63) Placebo 226.9 (61.7) 248.67 Pediatric Studies Study 4(13 to 17 years) Results represent subgroup of Study 4 and not the total population. ADHD-RS-IV MYDAYIS (12.5-25 mg/day) 36.7 (6.15) -20.3 -8.7 (-12.6, -4.8) Placebo 38.3 (6.67) -11.6 Study 5(13 to 17 years) Average PERMP MYDAYIS (25 mg/day) 214.5 (87.8) 272.67 41.26 (32.24, 50.29) Placebo 228.7 (101) 231.41
16 How Supplied/storage And Handling
How Supplied
MYDAYIS extended-release capsules are available as:
- 12.5 mg: Green body/green cap (imprinted with black SHIRE 465 and 12.5 mg), bottles of 100, NDC 54092-468-01
- 25 mg: Ivory body/green cap (imprinted with black SHIRE 465 and 25 mg), bottles of 100, NDC 54092-471-01
- 37.5 mg: Ivory body/caramel cap (imprinted with black SHIRE 465 and 37.5 mg), bottles of 100, NDC 54092-474-01
- 50 mg: Ivory body/purple cap (imprinted with black SHIRE 465 and 50 mg), bottles of 100, NDC 54092-477-01
STORAGE AND HANDLING SECTION
Storage and Handling
Dispense in a tight, light-resistant container as defined in the USP.
Store at room temperature, 20ÂşC to 25ÂşC (68ÂşF to 77ÂşF). Excursions permitted between 15ÂşC to 30ÂşC (59ÂşF to 86ÂşF) [see USP Controlled Room Temperature].
17 Patient Counseling Information
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
Abuse, Misuse, and Addiction
Educate patients and their families about the risks of abuse, misuse, and addiction of MYDAYIS, which can lead to overdose and death, and proper disposal of any unused drug [see Warnings and Precautions (5.1), Drug Abuse and Dependence (9.2), Overdosage (10)]. Advise patients to store MYDAYIS in a safe place, preferably locked, and instruct patients to not give MYDAYIS to anyone else.
Risks to Patients with Serious Cardiac Disease
Advise patients that there are potential risks to patients with serious cardiac disease, including sudden death with MYDAYIS use. Instruct patients to contact a healthcare provider immediately if they develop symptoms such as exertional chest pain, unexplained syncope, or other symptoms suggestive of cardiac disease [see Warnings and Precautions (5.2)].
Increased Blood Pressure and Heart Rate
Instruct patients that MYDAYIS can cause elevations of their blood pressure and pulse rate and they should be monitored for such effects [see Warnings and Precautions (5.3)].
Psychiatric Adverse Reactions
Advise patients that MYDAYIS, at recommended doses, may cause psychotic or manic symptoms even in patients without prior history of psychotic symptoms or mania [see Warnings and Precautions (5.4)].
Long-Term Suppression of Growth in Pediatric Patients
Advise patients, family members, and caregivers that MYDAYIS may cause slowing of growth including weight loss [see Warnings and Precautions (5.5)].
Circulation Problems in Fingers and Toes [Peripheral Vasculopathy, Including Raynaud's Phenomenon]
Instruct patients beginning treatment with MYDAYIS about the risk of peripheral vasculopathy, including Raynaud's phenomenon, and associated signs and symptoms: fingers or toes may feel numb, cool, painful, and/or may change from pale, to blue, to red. Instruct patients to report to their physician any new numbness, pain, skin color change, or sensitivity to temperature in fingers or toes. Instruct patients to call their physician immediately with any signs of unexplained wounds appearing on fingers or toes while taking MYDAYIS. Further clinical evaluation (e.g., rheumatology referral) may be appropriate for certain patients [see Warnings and Precautions (5.6)].
Seizures
Caution patient that MYDAYIS may lower the convulsive threshold. Advise patients to contact their healthcare provider immediately and to discontinue MYDAYIS if a seizure occurs [see Warnings and Precautions (5.7)].
Serotonin Syndrome
Caution patients about the risk of serotonin syndrome with concomitant use of MYDAYIS and other serotonergic drugs including SSRIs, SNRIs, triptans, tricyclic antidepressants, fentanyl, lithium, tramadol, tryptophan, buspirone, St. John's Wort, and with drugs that impair metabolism of serotonin (in particular MAOIs, both those intended to treat psychiatric disorders and also others such as linezolid [see Contraindications (4), Warnings and Precautions (5.8), Drug Interactions (7.1)]. Advise patients to contact their healthcare provider or report to the emergency room if they experience signs or symptoms of serotonin syndrome.
Motor and Verbal Tics, and Worsening of Tourette's Syndrome
Advise patients that motor and verbal tics and worsening of Tourette’s syndrome may occur during treatment with MYDAYIS. Instruct patients to notify their healthcare provider if emergence of new tics or worsening of tics or Tourette’s syndrome occurs [see Warnings and Precautions (5.10)].
Concomitant Medications
Advise patients to notify their physicians if they are taking, or plan to take, any prescription or over-the-counter drugs because there is a potential for interactions [see Drug Interactions (7.1)].
Pregnancy Registry
Advise patients that there is a pregnancy exposure registry that monitors pregnancy outcomes in women exposed to MYDAYIS during pregnancy [see Use in Specific Populations (8.1)].
Pregnancy
Advise patients of the potential fetal effects from the use of MYDAYIS during pregnancy. Advise patients to notify their healthcare provider if they become pregnant or intend to become pregnant during treatment with MYDAYIS [see Use in Specific Populations (8.1)].
Lactation
Advise women not to breastfeed if they are taking MYDAYIS [see Use in Specific Populations (8.2)].
Alcohol
Advise patients to avoid alcohol while taking MYDAYIS. Consumption of alcohol while taking MYDAYIS may result in a more rapid release of the dose of mixed amphetamine salts [see Clinical Pharmacology (12.3)].
Distributed by: Takeda Pharmaceuticals America, Inc. Lexington, MA 02421
Made in USA.
For more information call 1-800-828-2088
MYDAYIS® and the MYDAYIS Logo® are registered trademarks of Takeda Pharmaceuticals U.S.A., Inc.
© 2023 Takeda Pharmaceuticals U.S.A., Inc. All rights reserved.
Patented: see www.takeda.com/en-us/patents.
MYD365
Spl Medguide Section
MEDICATION GUIDEMYDAYIS (my-DAY-is)(mixed salts of a single-entity amphetamine product)extended-release capsules, CII Â Â This Medication Guide has been approved by the U.S. Food and Drug Administration MYD365 Revised: 10/2023 What is the most important information I should know about MYDAYIS?MYDAYIS may cause serious side effects, including:
- Abuse, misuse, and addiction. MYDAYIS has a high chance for abuse and misuse and may lead to substance use problems, including addiction. Misuse and abuse of MYDAYIS, other amphetamine containing medicines, and methylphenidate containing medicines, can lead to overdose and death. The risk of overdose and death is increased with higher doses of MYDAYIS or when it is used in ways that are not approved, such as snorting or injection.
- Your healthcare provider should check you or your child’s risk for abuse, misuse, and addiction before starting treatment with MYDAYIS and will monitor you or your child during treatment.
- MYDAYIS may lead to physical dependence after prolonged use, even if taken as directed by your healthcare provider.
- Do not give MYDAYIS to anyone else. See “ What is MYDAYIS?” for more information.
- Keep MYDAYIS in a safe place and properly dispose of any unused medicine. See “How should I store MYDAYIS?” for more information.Tell your healthcare provider if you or your child have ever abused or been dependent on alcohol, prescription medicines or street drugs.
- Risks for people with serious heart disease. Sudden death has happened in people who have heart defects or other serious heart disease.Your healthcare provider should check you or your child carefully for heart problems before starting MYDAYIS. Tell your healthcare provider if you or your child have any heart problems, heart disease, heart defects. Call your healthcare provider or go to the nearest hospital emergency room right away if you or your child have any signs of heart problems such as chest pain, shortness of breath, or fainting during treatment with MYDAYIS.
- Increased blood pressure and heart rate. Your healthcare provider should check you or your child’s blood pressure and heart rate regularly during treatment with MYDAYIS.
- Mental (psychiatric) problems, including:
- new or worse behavior and thought problems
- new or worse bipolar illness
- new psychotic symptoms (such as hearing voices, or seeing or believing things that are not real) or new manic symptomsTell your healthcare provider about any mental problems you or your child have, or about a family history of suicide, bipolar illness, or depression. Call your healthcare provider right away if you or your child have any new or worsening mental symptoms or problems while taking MYDAYIS, especially hearing voices, seeing or believing things that are not real, or new manic symptoms.
What is MYDAYIS? MYDAYIS is a central nervous system (CNS) stimulant prescription medicine used for the treatment of Attention Deficit Hyperactivity Disorder (ADHD) in people 13 years of age and older. MYDAYIS is not for use in children 12 years of age and younger. MYDAYIS is a federally controlled substance (CII) because it contains amphetamine that can be a target for people who abuse prescription medicines or street drugs. Keep MYDAYIS in a safe place to protect it from theft. Never give your MYDAYIS to anyone else, because it may cause death or harm them. Selling or giving away MYDAYIS may harm others and is against the law. Do not take MYDAYIS if you or your child are:
- allergic to amphetamine or any of the ingredients in MYDAYIS. See the end of the Medication Guide for a complete ul of ingredients in MYDAYIS.
- taking, or have taken within the past 14 days, a medicine used to treat depression called a monoamine oxidase inhibitor (MAOI).
Before taking MYDAYIS, tell your healthcare provider about all medical conditions, including if you or your child:
- have heart problems, heart disease, heart defects or high blood pressure
- have mental problems including psychosis, mania, bipolar illness or depression, or have a family history of suicide, bipolar illness, or depression
- have circulation problems in fingers and toes
- have or have had seizures
- have kidney problems
- have or had repeated movements or sounds (tics) or Tourette’s syndrome, or have a family history of tics or Tourette’s syndrome
- are pregnant or plan to become pregnant. It is not known if MYDAYIS will harm your unborn baby. Tell your healthcare provider if you become pregnant during treatment with MYDAYIS.
- There is a pregnancy registry for females who are exposed to MYDAYIS during pregnancy. The purpose of the registry is to collect information about the health of females exposed to MYDAYIS and their baby. If you or your child becomes pregnant during treatment with MYDAYIS, talk to your healthcare provider about registering with the National Pregnancy Registry for Psychiatric Medications at 1-866-961-2388 or visit online at https://womensmentalhealth.org/research/pregnancyregistry/.
- are breastfeeding or plan to breastfeed. MYDAYIS passes into breast milk. You should not breastfeed during treatment with MYDAYIS.
Tell your healthcare provider about all the medicines that you or your child take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. MYDAYIS may affect the way other medicines work and other medicines may affect how MYDAYIS works. Taking MYDAYIS with other medicines can cause serious side effects. Especially tell your healthcare provider if you or your child take:
- selective serotonin reuptake inhibitors (SSRIs)
- medicines used to treat migraine headaches called triptans
- lithium
- tramadol
- buspirone
- serotonin norepinephrine reuptake inhibitors (SNRIs)
- tricyclic antidepressants
- fentanyl
- tryptophan
- St. John’s Wort
Know the medicines that you or your child take. Keep a ul of your medicines with you to show your or your child’s healthcare provider and pharmacist when you or your child get a new medicine. Your healthcare provider will decide whether MYDAYIS can be taken with other medicines. Do not start any new medicine during treatment with MYDAYIS without talking to your or your child’s healthcare provider first. How should I take MYDAYIS? If you or your child take too much MYDAYIS, call your healthcare provider or Poison Help line at 1-800-222-1222 or go to the nearest hospital emergency room right away.
- Take MYDAYIS exactly as prescribed by your healthcare provider.
- Your healthcare provider may change the dose if needed.
- Take MYDAYIS 1 time each day in the morning right after you wake-up. MYDAYIS may last up to 16 hours and can cause difficulty sleeping.
- If you miss a dose of MYDAYIS, do not take your dose later in the day or double your dose to make up for a missed dose. Take your MYDAYIS dose the next morning at your regularly scheduled time.
- MYDAYIS can be taken with or without food but take it the same way each time.
- MYDAYIS capsules may be swallowed whole or if MYDAYIS capsules cannot be swallowed whole, the capsules may be opened and sprinkled over a spoonful of applesauce.
- swallow all of the applesauce and medicine mixture right away
- do not chew the applesauce and medicine mixture
- do not store the sprinkled applesauce
What should I avoid during treatment with MYDAYIS? You should avoid drinking alcohol during treatment with MYDAYIS. What are possible side effects of MYDAYIS?MYDAYIS may cause serious side effects, including:
- See "What is the most important information I should know about MYDAYIS?"
- Slowing of growth (height and weight) in children. Children should have their height and weight checked often during treatment with MYDAYIS. Your healthcare provider may stop your child's MYDAYIS treatment if they are not growing or gaining weight as expected.
- Circulation problems in fingers and toes (peripheral vasculopathy, including Raynaud's phenomenon). Signs and symptoms may include:
Tell your healthcare provider if you have or your child has any numbness, pain, skin color change, or sensitivity to temperature in your fingers or toes. Call your healthcare provider if you or your child have any signs of unexplained wounds appearing on fingers or toes during treatment with MYDAYIS.
- fingers or toes may feel numb, cool, painful
- fingers or toes may change color from pale, to blue, to red
- Seizures. Your healthcare provider will stop treatment with MYDAYIS if you have a seizure.
- New or worsening tics or worsening Tourette's Syndrome. Tell your healthcare provider if you or your child get any new or worsening tics or worsening Tourette’s syndrome during treatment with MYDAYIS.
- Serotonin syndrome. This problem may happen when MYDAYIS is taken with certain other medicines and may be life-threatening. Call your healthcare provider or go to the nearest hospital emergency room if you get symptoms of serotonin syndrome which may include:
- agitation, hallucinations, coma
- loss of coordination
- flushing
- fast heartbeat
- seizures
- confusion
- sweating or fever
- nausea, vomiting, or diarrhea
- dizziness
- changes in blood pressure
- muscle stiffness or tightness
- high body temperature (hyperthermia)
  The most common side effects of MYDAYIS include:
- trouble sleeping
- decreased appetite
- dry mouth
- increased heart rate
- anxiety
- nausea
- irritability
- weight loss
These are not all the possible side effects of MYDAYIS. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. How should I store MYDAYIS? Keep MYDAYIS and all medicines out of the reach of children.
- Store MYDAYIS at room temperature between 68°F to 77°F (20°C to 25°C).
- Protect MYDAYIS from light.
- Store MYDAYIS in a safe place, like a locked cabinet.
- Dispose of remaining, unused, or expired MYDAYIS by a medicine take-back program at a U.S. Drug Enforcement Administration (DEA) authorized collection site. If no take-back program or DEA authorized collector is available, mix MYDAYIS with an undesirable, nontoxic substance such as dirt, cat litter, or used coffee grounds to make it less appealing to children and pets. Place the mixture in a container such as a sealed plastic bag and throw away MYDAYIS in the household trash. Visit www.fda.gov/drugdisposal for additional information on disposal of unused medicines.
General information about the safe and effective use of MYDAYIS Medicines are sometimes prescribed for purposes other than those uled in a Medication Guide. Do not use MYDAYIS for a condition for which it was not prescribed. Do not give MYDAYIS to other people, even if they have the same condition. It may harm them and it is against the law. You can ask your healthcare provider or pharmacist for information about MYDAYIS that is written for healthcare professionals. What are the ingredients in MYDAYIS? Active ingredients: dextroamphetamine sulfate and amphetamine sulfate, dextroamphetamine saccharate and amphetamine aspartate monohydrate Inactive ingredients: hard gelatin capsules, ethylcellulose, hydroxypropyl methylcellulose, methacrylic acid copolymer, methyl acrylate, methyl methacrylate, opadry beige, sugar spheres, talc, and triethyl citrate. Gelatin capsules contain gelatin, titanium dioxide, yellow iron oxide and edible inks. The 12.5 mg and 25 mg capsules also contain FD&C Blue #2. The 37.5 mg also contains red iron oxide. The 50 mg capsule also contains D&C Red #28, D&C Red #33, and FD&C Blue #1. Distributed by: Takeda Pharmaceuticals America, Inc., Lexington, MA 02421, Made in USA. MYDAYIS® and the MYDAYIS Logo® are registered trademarks of Takeda Pharmaceuticals U.S.A., Inc. © 2023 Takeda Pharmaceuticals U.S.A., Inc. All rights reserved. For more information about MYDAYIS go to www.mydayis.com or call 1-800-828-2088.
Principal Display Panel - 12.5 Mg Capsule Bottle Label
ONCE-DAILYNDC 54092-468-01
Mydayis® (Mixed Salts of A Single-EntityAmphetamine Product)
Extended-Release Capsules
12.5 mg
100 Capsules
Do not substitute for Adderall XR.
CII Â Rx only
Takeda

Principal Display Panel - 25 Mg Capsule Bottle Label
ONCE-DAILYNDC 54092-471-01
Mydayis® (Mixed Salts of A Single-EntityAmphetamine Product)
Extended-Release Capsules
25 mg
100 Capsules
Do not substitute for Adderall XR.
CII Â Rx only
Takeda

Principal Display Panel - 37.5 Mg Capsule Bottle Label
ONCE-DAILYNDC 54092-474-01
Mydayis® (Mixed Salts of A Single-EntityAmphetamine Product)
Extended-Release Capsules
37.5 mg
100 Capsules
Do not substitute for Adderall XR.
CII Â Rx only
Takeda

Principal Display Panel - 50 Mg Capsule Bottle Label
ONCE-DAILYNDC 54092-477-01
Mydayis® (Mixed Salts of A Single-EntityAmphetamine Product)
Extended-Release Capsules
50 mg
100 Capsules
Do not substitute for Adderall XR.
CII Â Rx only
Takeda

DISCLAIMER:
"This tool does not provide medical advice, and is for informational and educational purposes only, and is not a substitute for professional medical advice, treatment or diagnosis. Call your doctor to receive medical advice. If you think you may have a medical emergency, please dial 911."
"Do not rely on openFDA to make decisions regarding medical care. While we make every effort to ensure that data is accurate, you should assume all results are unvalidated. We may limit or otherwise restrict your access to the API in line with our Terms of Service."
"This product uses publicly available data from the U.S. National Library of Medicine (NLM), National Institutes of Health, Department of Health and Human Services; NLM is not responsible for the product and does not endorse or recommend this or any other product."
PillSync may earn a commission via links on our site
