PROMACTA Dailymed
Generic: eltrombopag olamine is used for the treatment of Anemia, Aplastic Purpura, Thrombocytopenic, Idiopathic

All Imprints
promacta (eltrombopag olamine) 25 mg - gs nx3 25 round orange
promacta (eltrombopag olamine) 50 mg - gs ufu 50 round blue
Boxed Warning
Warning: Risk For Hepatic Decompensation In Patients With Chronic Hepatitis C And Risk Of Hepatotoxicity

Go PRO for all pill images
Warning: Risk For Hepatic Decompensation In Patients With Chronic Hepatitis C And Risk Of Hepatotoxicity
In patients with chronic hepatitis C, PROMACTA in combination with interferon and ribavirin may increase the risk of hepatic decompensation [see Warnings and Precautions (5.1)].
PROMACTA may increase the risk of severe and potentially life-threatening hepatotoxicity. Monitor hepatic function and discontinue dosing as recommended [see Warnings and Precautions (5.2)].
WARNING: RISK FOR HEPATIC DECOMPENSATION IN PATIENTS WITH CHRONIC HEPATITIS C and RISK OF HEPATOTOXICITYSee full prescribing information for complete boxed warning.
In patients with chronic hepatitis C, PROMACTA in combination with interferon and ribavirin may increase the risk of hepatic decompensation. (5.1 )
PROMACTA may increase the risk of severe and potentially life-threatening hepatotoxicity. Monitor hepatic function and discontinue dosing as recommended. (5.2 )
1 Indications And Usage
PROMACTA is a thrombopoietin receptor agonist indicated:
- for the treatment of thrombocytopenia in adult and pediatric patients 1 year and older with persistent or chronic immune thrombocytopenia (ITP) who have had an insufficient response to corticosteroids, immunoglobulins, or splenectomy. PROMACTA should be used only in patients with ITP whose degree of thrombocytopenia and clinical condition increase the risk for bleeding. (
1.1 )- for the treatment of thrombocytopenia in patients with chronic hepatitis C to allow the initiation and maintenance of interferon-based therapy. PROMACTA should be used only in patients with chronic hepatitis C whose degree of thrombocytopenia prevents the initiation of interferon-based therapy or limits the ability to maintain interferon-based therapy. (
1.2 )- in combination with standard immunosuppressive therapy for the first-line treatment of adult and pediatric patients 2 years and older with severe aplastic anemia. (
1.3 )- for the treatment of patients with severe aplastic anemia who have had an insufficient response to immunosuppressive therapy. (
1.3 )
Limitations of Use:
- PROMACTA is not indicated for the treatment of patients with myelodysplastic syndrome (MDS). (
1.4 )- Safety and efficacy have not been established in combination with direct-acting antiviral agents used without interferon for treatment of chronic hepatitis C infection. (
1.4 )1.1 Treatment of Thrombocytopenia in Patients With Persistent or Chronic Immune Thrombocytopenia
PROMACTA is indicated for the treatment of thrombocytopenia in adult and pediatric patients 1 year and older with persistent or chronic immune thrombocytopenia (ITP) who have had an insufficient response to corticosteroids, immunoglobulins, or splenectomy. PROMACTA should be used only in patients with ITP whose degree of thrombocytopenia and clinical condition increase the risk for bleeding.
1.2 Treatment of Thrombocytopenia in Patients With Hepatitis C Infection
PROMACTA is indicated for the treatment of thrombocytopenia in patients with chronic hepatitis C to allow the initiation and maintenance of interferon-based therapy. PROMACTA should be used only in patients with chronic hepatitis C whose degree of thrombocytopenia prevents the initiation of interferon-based therapy or limits the ability to maintain interferon-based therapy.
1.3 Treatment of Severe Aplastic Anemia
- PROMACTA is indicated in combination with standard immunosuppressive therapy (IST) for the first-line treatment of adult and pediatric patients 2 years and older with severe aplastic anemia.
- PROMACTA is indicated for the treatment of patients with severe aplastic anemia who have had an insufficient response to immunosuppressive therapy.
1.4 Limitations of Use
- PROMACTA is not indicated for the treatment of patients with myelodysplastic syndromes (MDS) [see Warnings and Precautions (5.3)].
- Safety and efficacy have not been established in combination with direct-acting antiviral agents used without interferon for treatment of chronic hepatitis C infection.
2 Dosage And Administration
- Take PROMACTA without a meal or with a meal low in calcium (≤ 50 mg). Take PROMACTA at least 2 hours before or 4 hours after any medications or products containing polyvalent cations, such as antacids, calcium-rich foods, and mineral supplements. (
2.4 ,7.1 ,12.3 )- Persistent or Chronic ITP: Initiate PROMACTA at 50Â mg once daily for most adult and pediatric patients 6Â years and older, and at 25Â mg once daily for pediatric patients aged 1 to 5Â years. Dose reductions are needed for patients with hepatic impairment and some patients of East-/Southeast-Asian ancestry. Adjust to maintain platelet count greater than or equal to 50 x 109/L. Do not exceed 75Â mg per day. (
2.1 ,8.6 ,8.7 )- Chronic Hepatitis C-associated Thrombocytopenia: Initiate PROMACTA at 25Â mg once daily for all patients. Adjust to achieve target platelet count required to initiate antiviral therapy. Do not exceed a daily dose of 100Â mg. (
2.2 )- First-line Severe Aplastic Anemia: Initiate PROMACTA once daily at 2.5 mg/kg (in pediatric patients aged 2 to 5 years old), 75 mg (pediatric patients aged 6 to 11 years old), or 150 mg for patients aged 12 years and older concurrently with standard immunosuppressive therapy. Reduce initial dose in patients of East-/Southeast-Asian ancestry. Modify dosage for toxicity or elevated platelet counts. (
2.3 ,8.7 )- Refractory Severe Aplastic Anemia: Initiate PROMACTA at 50Â mg once daily. Reduce initial dose in patients with hepatic impairment or patients of East-/Southeast-Asian ancestry. Adjust to maintain platelet count greater than 50 x 109/L. Do not exceed 150Â mg per day. (
2.3 ,8.6 ,8.7 )2.1 Persistent or Chronic Immune Thrombocytopenia
Use the lowest dose of PROMACTA to achieve and maintain a platelet count greater than or equal to 50 x 109/L as necessary to reduce the risk for bleeding. Dose adjustments are based upon the platelet count response. Do not use PROMACTA to normalize platelet counts [see Warnings and Precautions (5.4)]. In clinical trials, platelet counts generally increased within 1 to 2Â weeks after starting PROMACTA and decreased within 1 to 2Â weeks after discontinuing PROMACTA [see Clinical Studies (14.1)].
Initial Dose Regimen:
Adult and Pediatric Patients 6Â Years and Older with ITP: Initiate PROMACTA at a dose of 50 mg once daily, except in patients who are of East-/Southeast-Asian ancestry or who have mild to severe hepatic impairment (Child-Pugh class A, B, C).
For patients of East-/Southeast-Asian ancestry with ITP, initiate PROMACTA at a reduced dose of 25Â mg once daily [see Use in Specific Populations (8.7), Clinical Pharmacology (12.3)].
For patients with ITP and mild, moderate, or severe hepatic impairment (Child-Pugh class A, B, C), initiate PROMACTA at a reduced dose of 25Â mg once daily [see Use in Specific Populations (8.6), Clinical Pharmacology (12.3)].
For patients of East-/Southeast-Asian ancestry with ITP and hepatic impairment (Child-Pugh class A, B, C), consider initiating PROMACTA at a reduced dose of 12.5Â mg once daily [see Clinical Pharmacology (12.3)].
Pediatric Patients with ITP Aged 1 to 5Â Years: Initiate PROMACTA at a dose of 25Â mg once daily [see Use in Specific Populations (8.7), Clinical Pharmacology (12.3)].
Monitoring and Dose Adjustment: After initiating PROMACTA, adjust the dose to achieve and maintain a platelet count greater than or equal to 50 x 109/L as necessary to reduce the risk for bleeding. Do not exceed a dose of 75 mg daily. Monitor clinical hematology and liver tests regularly throughout therapy with PROMACTA and modify the dosage regimen of PROMACTA based on platelet counts as outlined in Table 1. During therapy with PROMACTA, assess complete blood counts (CBCs) with differentials, including platelet counts, weekly until a stable platelet count has been achieved. Obtain CBCs with differentials, including platelet counts, monthly thereafter.
When switching between the oral suspension and tablet, assess platelet counts weekly for 2Â weeks, and then follow standard monthly monitoring.
Table 1. Dose Adjustments of PROMACTA in Patients With Persistent or Chronic Immune Thrombocytopenia
Platelet count result
Dose adjustment or response
< 50 x 109/L following at least 2Â weeks of PROMACTA
Increase daily dose by 25Â mg to a maximum of 75Â mg/day.
For patients taking 12.5 mg once daily, increase the dose to 25 mg daily before increasing the dose amount by 25 mg.
≥ 200 x 109/L to ≤ 400 x 109/L at any time
Decrease the daily dose by 25Â mg. Wait 2Â weeks to assess the effects of this and any subsequent dose adjustments.
For patients taking 25Â mg once daily, decrease the dose to 12.5Â mg once daily.
> 400 x 109/L
Stop PROMACTA; increase the frequency of platelet monitoring to twice weekly.
Once the platelet count is < 150 x 109/L, reinitiate therapy at a daily dose reduced by 25 mg.
For patients taking 25 mg once daily, reinitiate therapy at a daily dose of 12.5Â mg.
> 400 x 109/L after 2Â weeks of therapy at lowest dose of PROMACTA
Discontinue PROMACTA.
In patients with ITP and hepatic impairment (Child-Pugh class A, B, C), after initiating PROMACTA or after any subsequent dosing increase, wait 3Â weeks before increasing the dose.
Modify the dosage regimen of concomitant ITP medications, as medically appropriate, to avoid excessive increases in platelet counts during therapy with PROMACTA. Do not administer more than one dose of PROMACTA within any 24-hour period.
Discontinuation: Discontinue PROMACTA if the platelet count does not increase to a level sufficient to avoid clinically important bleeding after 4Â weeks of therapy with PROMACTA at the maximum daily dose of 75Â mg. Excessive platelet count responses, as outlined in Table 1, or important liver test abnormalities also necessitate discontinuation of PROMACTA [see Warnings and Precautions (5.2)]. Obtain CBCs with differentials, including platelet counts, weekly for at least 4Â weeks following discontinuation of PROMACTA.
2.2 Chronic Hepatitis C-Associated Thrombocytopenia
Use the lowest dose of PROMACTA to achieve and maintain a platelet count necessary to initiate and maintain antiviral therapy with pegylated interferon and ribavirin. Dose adjustments are based upon the platelet count response. Do not use PROMACTA to normalize platelet counts [see Warnings and Precautions (5.4)]. In clinical trials, platelet counts generally began to rise within the first week of treatment with PROMACTA [see Clinical Studies (14.2)].
Initial Dose Regimen: Initiate PROMACTA at a dose of 25Â mg once daily.
Monitoring and Dose Adjustment: Adjust the dose of PROMACTA in 25-mg increments every 2 weeks as necessary to achieve the target platelet count required to initiate antiviral therapy. Monitor platelet counts every week prior to starting antiviral therapy.
During antiviral therapy, adjust the dose of PROMACTA to avoid dose reductions of peginterferon. Monitor CBCs with differentials, including platelet counts, weekly during antiviral therapy until a stable platelet count is achieved. Monitor platelet counts monthly thereafter. Do not exceed a dose of 100 mg daily. Monitor clinical hematology and liver tests regularly throughout therapy with PROMACTA.
For specific dosage instructions for peginterferon or ribavirin, refer to their respective prescribing information.
Table 2. Dose Adjustments of PROMACTA in Adults With Thrombocytopenia Due to Chronic Hepatitis C
Platelet count result
Dose adjustment or response
< 50 x 109/L following at least 2Â weeks of PROMACTA
Increase daily dose by 25Â mg to a maximum of 100Â mg/day.
≥ 200 x 109/L to ≤ 400 x 109/L at any time
Decrease the daily dose by 25Â mg.
Wait 2Â weeks to assess the effects of this and any subsequent dose adjustments.
> 400 x 109/L
Stop PROMACTA; increase the frequency of platelet monitoring to twice weekly.
Once the platelet count is < 150 x 109/L, reinitiate therapy at a daily dose reduced by 25 mg.
For patients taking 25 mg once daily, reinitiate therapy at a daily dose of 12.5Â mg.
> 400 x 109/L after 2Â weeks of therapy at lowest dose of PROMACTA
Discontinue PROMACTA.
Discontinuation: The prescribing information for pegylated interferon and ribavirin include recommendations for antiviral treatment discontinuation for treatment futility. Refer to pegylated interferon and ribavirin prescribing information for discontinuation recommendations for antiviral treatment futility.
PROMACTA should be discontinued when antiviral therapy is discontinued. Excessive platelet count responses, as outlined in Table 2, or important liver test abnormalities also necessitate discontinuation of PROMACTA [see Warnings and Precautions (5.2)].
2.3 Severe Aplastic Anemia
First-Line Severe Aplastic Anemia
Initiate PROMACTA concurrently with standard immunosuppressive therapy [see Clinical Studies (14.3)].
Initial Dose Regimen
The recommended initial dose regimen is uled in Table 3. Do not exceed the initial dose of PROMACTA.
Table 3. Recommended Initial PROMACTA Dose Regimen in the First-Line Treatment of Severe Aplastic Anemia
Age
Dose regimen
Patients 12 years and older
150 mg once daily for 6 months
Pediatric patients 6 to 11 years
75 mg once daily for 6 months
Pediatric patients 2 to 5 years
2.5 mg/kg once daily for 6 months
For patients with severe aplastic anemia of East-/Southeast-Asian ancestry or those with mild, moderate, or severe hepatic impairment (Child-Pugh class A, B, C), decrease the initial PROMACTA dose by 50% as uled in Table 4 [see Use in Specific Populations (8.6, 8.7), Clinical Pharmacology (12.3)].
If baseline alanine aminotransferase (ALT) or aspartate aminotransferase (AST) levels are > 6 x upper limit of normal (ULN), do not initiate PROMACTA until transaminase levels are < 5 x ULN. Determine the initial dose for these patients based on Table 3 or Table 4.
Table 4. Recommended Initial PROMACTA Dose Regimen for Patients of East-/Southeast-Asian Ancestry or Those With Mild, Moderate, or Severe Hepatic Impairment (Child-Pugh class A, B, C) in the First-Line Treatment of Severe Aplastic Anemia
Age
Dose regimen
Patients 12 years and older
75 mg once daily for 6 months
Pediatric patients 6 to 11 years
37.5 mg once daily for 6 months
Pediatric patients 2 to 5 years
1.25 mg/kg once daily for 6 months
Monitoring and Dose Adjustment for PROMACTA: Perform clinical hematology and liver tests regularly throughout therapy with PROMACTA [see Warnings and Precautions (5.2)].
Modify the dosage regimen of PROMACTA based on platelet counts as outlined in Table 5.
Table 5. Dose Adjustments of PROMACTA for Elevated Platelet Counts in the First-Line Treatment of Severe Aplastic Anemia
Platelet count result
Dose adjustment or response
> 200 x 109/L to ≤ 400 x 109/L
Decrease the daily dose by 25 mg every 2 weeks to lowest dose that maintains platelet count ≥ 50 x 109/L.In pediatric patients under 12 years of age, decrease the dose by 12.5 mg.
> 400 x 109/L
Discontinue PROMACTA for one week. Once the platelet count is < 200 x 109/L, reinitiate PROMACTA at a daily dose reduced by 25 mg (or 12.5 mg in pediatric patients under 12 years of age).
Table 6 summarizes the recommendations for dose interruption, reduction, or discontinuation of PROMACTA in the management of elevated liver transaminase levels and thromboembolic events.
Table 6. Recommended Dose Modifications for PROMACTA for ALT or AST Elevations and Thromboembolic Events Abbreviations: ALT, alanine aminotransferase; AST, aspartate aminotransferase; ULN, upper limit of normal.
Event
Recommendation
ALT or AST elevations
Increase in ALT or AST > 6 x ULN Discontinue PROMACTA. Once ALT or AST is < 5 x ULN, reinitiate PROMACTA at the same dose.
Increase in ALT or AST > 6 x ULN after reinitiating PROMACTA Discontinue PROMACTA and monitor ALT or AST at least every 3 to 4 days. Once ALT or AST is < 5 x ULN, reinitiate PROMACTA at a daily dose reduced by 25 mg compared to the previous dose.
If ALT or AST returns to > 6 x ULN on the reduced dose Reduce the daily dose of PROMACTA by 25 mg until ALT or AST is < 5 x ULN.
In pediatric patients under 12 years of age, reduce the daily dose by at least 15% to the nearest dose that can be administered.
Thromboembolic events (e.g., deep vein thrombosis, pulmonary embolus, stroke, myocardial infarction)
Discontinue PROMACTA but remain on horse antithymocyte globulin (h-ATG) and cyclosporine.
The total duration of PROMACTA treatment is 6 months.
Refractory Severe Aplastic Anemia
Use the lowest dose of PROMACTA to achieve and maintain a hematologic response. Dose adjustments are based upon the platelet count. Hematologic response requires dose titration, generally up to 150Â mg, and may take up to 16Â weeks after starting PROMACTA [see Clinical Studies (14.3)].
Initial Dose Regimen: Initiate PROMACTA at a dose of 50Â mg once daily.
For patients with severe aplastic anemia of East-/Southeast-Asian ancestry or those with mild, moderate, or severe hepatic impairment (Child-Pugh class A, B, C), initiate PROMACTA at a reduced dose of 25Â mg once daily [see Use in Specific Populations (8.6, 8.7), Clinical Pharmacology (12.3)].
Monitoring and Dose Adjustment: Adjust the dose of PROMACTA in 50-mg increments every 2 weeks as necessary to achieve the target platelet count greater than or equal to 50 x 109/L as necessary. Do not exceed a dose of 150 mg daily. Monitor clinical hematology and liver tests regularly throughout therapy with PROMACTA and modify the dosage regimen of PROMACTA based on platelet counts as outlined in Table 7.
Table 7. Dose Adjustments of PROMACTA in Patients With Refractory Severe Aplastic Anemia
Platelet count result
Dose adjustment or response
< 50 x 109/L following at least 2Â weeks of PROMACTA
Increase daily dose by 50Â mg to a maximum of 150Â mg/day.
For patients taking 25Â mg once daily, increase the dose to 50Â mg daily before increasing the dose amount by 50Â mg.
≥ 200 x 109/L to ≤ 400 x 109/L at any time
Decrease the daily dose by 50Â mg. Wait 2Â weeks to assess the effects of this and any subsequent dose adjustments.
> 400 x 109/L
Stop PROMACTA for 1Â week.
Once the platelet count is < 150 x 109/L, reinitiate therapy at a dose reduced by 50 mg.
> 400 x 109/L after 2Â weeks of therapy at lowest dose of PROMACTA
Discontinue PROMACTA.
For patients who achieve tri-lineage response, including transfusion independence, lasting at least 8 weeks: the dose of PROMACTA may be reduced by 50% [see Clinical Studies (14.3)]. If counts remain stable after 8 weeks at the reduced dose, then discontinue PROMACTA and monitor blood counts. If platelet counts drop to less than 30 x 109/L, hemoglobin to less than 9 g/dL, or absolute neutrophil count (ANC) to less than 0.5 x 109/L, PROMACTA may be reinitiated at the previous effective dose.
Discontinuation: If no hematologic response has occurred after 16 weeks of therapy with PROMACTA, discontinue therapy. If new cytogenetic abnormalities are observed, consider discontinuation of PROMACTA [see Adverse Reactions (6.1)]. Excessive platelet count responses (as outlined in Table 7) or important liver test abnormalities also necessitate discontinuation of PROMACTA [see Warnings and Precautions (5.2)].
2.4 Administration
Administration of Tablets and Oral Suspension: Take PROMACTA without a meal or with a meal low in calcium (≤ 50 mg). Take PROMACTA at least 2 hours before or 4 hours after other medications (e.g., antacids), calcium-rich foods (containing > 50 mg calcium e.g., dairy products, calcium-fortified juices, and certain fruits and vegetables), or supplements containing polyvalent cations, such as iron, calcium, aluminum, magnesium, selenium, and zinc [see Drug Interactions (7.1), Clinical Pharmacology (12.3)].
Do not split, chew, or crush tablets and mix with food or liquids.
Preparation of the Oral Suspension: Prior to use of the oral suspension, ensure patients or caregivers receive training on proper dosing, preparation, and administration of PROMACTA for oral suspension.
Administer the oral suspension immediately after preparation. Discard any suspension not administered within 30Â minutes after preparation.
Prepare the suspension with water only. NOTE: Do not use hot water to prepare the suspension.
For details on preparation and administration of the suspension, including the recommended duration of use of each oral dosing syringe, [see Instructions for Use].
3 Dosage Forms And Strengths
Tablets
- 12.5-mg tablets —– round, biconvex, white, film-coated tablets debossed with “GS MZ1” and 12.5 on one side. Each tablet, for oral administration, contains eltrombopag olamine, equivalent to 12.5 mg of eltrombopag free acid.
- 25-mg tablets —– round, biconvex, orange, film-coated tablets debossed with “GS NX3” and 25 on one side. Each tablet, for oral administration, contains eltrombopag olamine, equivalent to 25 mg of eltrombopag free acid.
- 50-mg tablets —– round, biconvex, blue, film-coated tablets debossed with “GS UFU” and 50 on one side. Each tablet, for oral administration, contains eltrombopag olamine, equivalent to 50 mg of eltrombopag free acid.
- 75-mg tablets —– round, biconvex, pink, film-coated tablets debossed with “GS FFS” and 75 on one side. Each tablet, for oral administration, contains eltrombopag olamine, equivalent to 75 mg of eltrombopag free acid.
For Oral Suspension
- 12.5-mg packet —– contains a reddish-brown to yellow powder for reconstitution.
- 25-mg packet —– contains a reddish-brown to yellow powder for reconstitution.
- Tablets: 12.5 mg, 25 mg, 50 mg, and 75 mg (
3 )- For oral suspension: 12.5 mg and 25 mg (
3 )
4 Contraindications
None.
None. (4 )
5 Warnings And Precautions
- Hepatotoxicity: Monitor liver function before and during therapy. (
5.2 )- Increased Risk of Death and Progression of Myelodysplastic Syndromes to Acute Myeloid Leukemia. (
5.3 )- Thrombotic/Thromboembolic Complications: Portal vein thrombosis has been reported in patients with chronic liver disease receiving PROMACTA. Monitor platelet counts regularly. (
5.4 )5.1 Hepatic Decompensation in Patients With Chronic Hepatitis C
In patients with chronic hepatitis C, PROMACTA in combination with interferon and ribavirin may increase the risk of hepatic decompensation. In two controlled clinical trials in patients with chronic hepatitis C and thrombocytopenia, ascites and encephalopathy occurred more frequently on the arm receiving treatment with PROMACTA plus antivirals (7%) than the placebo plus antivirals arm (4%). Patients with low albumin levels (less than 3.5Â g/dL) or Model for End-Stage Liver Disease (MELD) score greater than or equal to 10 at baseline had a greater risk for hepatic decompensation on the arm receiving treatment with PROMACTA plus antivirals. Discontinue PROMACTA if antiviral therapy is discontinued.
5.2 Hepatotoxicity
PROMACTA may increase the risk of severe and potentially life-threatening hepatotoxicity [see Adverse Reactions (6.1)]. One patient (< 1%) with ITP treated with PROMACTA in clinical trials experienced drug-induced liver injury. Eleven patients (1%) with chronic hepatitis C treated with PROMACTA in clinical trials experienced drug-induced liver injury.
Treatment of ITP, Chronic Hepatitis C-associated Thrombocytopenia, and Refractory Severe Aplastic Anemia
Measure serum ALT, AST, and bilirubin prior to initiation of PROMACTA, every 2Â weeks during the dose adjustment phase, and monthly following establishment of a stable dose. PROMACTA inhibits UDP-glucuronosyltransferase (UGT)1A1 and organic anion-transporting polypeptide (OATP)1B1, which may lead to indirect hyperbilirubinemia. If bilirubin is elevated, perform fractionation. Evaluate abnormal serum liver tests with repeat testing within 3 to 5 days. If the abnormalities are confirmed, monitor serum liver tests weekly until resolved or stabilized. Discontinue PROMACTA if ALT levels increase to greater than or equal to 3Â x ULN in patients with normal liver function or greater than or equal to 3Â x baseline (or greater than 5 x ULN, whichever is the lower) in patients with pre-treatment elevations in transaminases and are:
- progressively increasing, or
- persistent for greater than or equal to 4Â weeks, or
- accompanied by increased direct bilirubin, or
- accompanied by clinical symptoms of liver injury or evidence for hepatic decompensation.
If the potential benefit for reinitiating treatment with PROMACTA is considered to outweigh the risk for hepatotoxicity, then consider cautiously reintroducing PROMACTA and measure serum liver tests weekly during the dose adjustment phase. Hepatotoxicity may reoccur if PROMACTA is reinitiated. If liver test abnormalities persist, worsen, or recur, then permanently discontinue PROMACTA.
First-Line Treatment of Severe Aplastic Anemia
Measure ALT, AST, and bilirubin prior to initiation of PROMACTA, every other day while hospitalized for h-ATG therapy, and then every 2 weeks during treatment. During treatment, manage increases in ALT or AST levels as recommended in Table 6.
5.3 Increased Risk of Death and Progression of Myelodysplastic Syndromes to Acute Myeloid Leukemia
A randomized, double-blind, placebo-controlled, multicenter trial in patients with International Prognostic Scoring System (IPSS) intermediate-1, intermediate-2 or high risk MDS with thrombocytopenia, receiving azacitidine in combination with either PROMACTA (n = 179) or placebo (n = 177) was terminated due to lack of efficacy and safety reasons, including increased progression to acute myeloid leukemia (AML). Patients received PROMACTA or placebo at a starting dose of 200 mg once daily, up to a maximum of 300 mg once daily, in combination with azacitidine for at least six cycles. The incidence of death (overall survival) was 32% (57/179) in the PROMACTA arm versus 29% (51/177) in the placebo arm (HR [95% CI] = 1.42 [0.97, 2.08], showing an increased relative risk of death in this trial by 42% in the PROMACTA arm). The incidence of progression to AML was 12% (21/179) in the PROMACTA arm versus 6% (10/177) in the placebo arm (HR [95% CI] = 2.66 [1.31, 5.41], showing an increased relative risk of progression to AML in this trial by 166% in the PROMACTA arm).
5.4 Thrombotic/Thromboembolic Complications
Thrombotic/thromboembolic complications may result from increases in platelet counts with PROMACTA. Reported thrombotic/thromboembolic complications included both venous and arterial events and were observed at low and at normal platelet counts.
Consider the potential for an increased risk of thromboembolism when administering PROMACTA to patients with known risk factors for thromboembolism (e.g., Factor V Leiden, ATIII deficiency, antiphospholipid syndrome, chronic liver disease). To minimize the risk for thrombotic/thromboembolic complications, do not use PROMACTA in an attempt to normalize platelet counts. Follow the dose adjustment guidelines to achieve and maintain target platelet counts [see Dosage and Administration (2.1, 2.2, 2.3)].
In two controlled clinical trials in patients with chronic hepatitis C and thrombocytopenia, 3% (31/955) treated with PROMACTA experienced a thrombotic event compared with 1% (5/484) on placebo. The majority of events were of the portal venous system (1% in patients treated with PROMACTA versus less than 1% for placebo).
In a controlled trial in patients with chronic liver disease and thrombocytopenia not related to ITP undergoing elective invasive procedures (NÂ =Â 292), the risk of thrombotic events was increased in patients treated with 75Â mg of PROMACTA once daily. Seven thrombotic complications (six patients) were reported in the group that received PROMACTA and three thrombotic complications were reported in the placebo group (two patients). All of the thrombotic complications reported in the group that received PROMACTA were portal vein thrombosis (PVT). Symptoms of PVT included abdominal pain, nausea, vomiting, and diarrhea. Five of the six patients in the group that received PROMACTA experienced a thrombotic complication within 30 days of completing treatment with PROMACTA and at a platelet count above 200 x 109/L. The risk of portal venous thrombosis was increased in thrombocytopenic patients with chronic liver disease treated with 75Â mg of PROMACTA once daily for 2Â weeks in preparation for invasive procedures.
5.5 Cataracts
In the three controlled clinical trials in adults with persistent or chronic ITP, cataracts developed or worsened in 15 (7%) patients who received 50 mg of PROMACTA daily and 8 (7%) placebo-group patients. In the extension trial, cataracts developed or worsened in 11% of patients who underwent ocular examination prior to therapy with PROMACTA. In the two controlled clinical trials in patients with chronic hepatitis C and thrombocytopenia, cataracts developed or worsened in 8% of patients treated with PROMACTA and 5% of patients treated with placebo.
Cataracts were observed in toxicology studies of eltrombopag in rodents [see Nonclinical Toxicology (13.2)]. Perform a baseline ocular examination prior to administration of PROMACTA and, during therapy with PROMACTA, regularly monitor patients for signs and symptoms of cataracts.
6 Adverse Reactions
The following clinically significant adverse reactions associated with PROMACTA are described in other sections.
- Hepatic Decompensation in Patients with Chronic Hepatitis C [see Warnings and Precautions (5.1)]
- Hepatotoxicity [see Warnings and Precautions (5.2)]
- Increased Risk of Death and Progression of Myelodysplastic Syndromes to Acute Myeloid Leukemia [see Warnings and Precautions (5.3)]
- Thrombotic/Thromboembolic Complications [see Warnings and Precautions (5.4)]
- Cataracts [see Warnings and Precautions (5.5)]
Across all indications, the most common adverse reactions (≥ 20% in any indication) were: anemia, nausea, pyrexia, alanine aminotransferase increased, cough, fatigue, headache, and diarrhea. (6.1 )
To report SUSPECTED ADVERSE REACTIONS, contact Novartis Pharmaceuticals Corporation at 1-888-669-6682 or FDA at 1-800-FDA-1088 orwww.fda.gov/medwatch .
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared with rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Persistent or Chronic Immune Thrombocytopenia
Adults: In clinical trials, hemorrhage was the most common serious adverse reaction and most hemorrhagic reactions followed discontinuation of PROMACTA. Other serious adverse reactions included thrombotic/thromboembolic complications [see Warnings and Precautions (5.4)]. The data described below reflect exposure of PROMACTA to patients with persistent or chronic ITP aged 18 to 85 years, of whom 66% were female, in three placebo-controlled trials and one open-label extension trial [see Clinical Studies (14.1)]. PROMACTA was administered to 330 patients for at least 6Â months and 218 patients for at least 1Â year.
Table 8 presents the most common adverse drug reactions (experienced by greater than or equal to 3% of patients receiving PROMACTA) from the three placebo-controlled trials, with a higher incidence in PROMACTA versus placebo.
Table 8. Adverse Reactions (≥ 3%) From Three Placebo-controlled Trials in Adults With Persistent or Chronic Immune Thrombocytopenia Abbreviations: ALT, alanine aminotransferase; AST, aspartate aminotransferase. aIncludes PTs of urinary tract infection, cystitis, urinary tract infection bacterial, and bacteriuria.
Adverse reaction
PROMACTA 50 mg
n = 241
(%)
Placebo
n = 128
(%)
Nausea
9
3
Diarrhea
9
7
Upper respiratory tract infection
7
6
Vomiting
6
< 1
Urinary tract infectiona
5
4
Increased ALT
5
3
Myalgia
5
2
Oropharyngeal pain
4
3
Increased AST
4
2
Pharyngitis
4
2
Back pain
3
2
Influenza
3
2
Paresthesia
3
2
Rash
3
2
In the three controlled clinical persistent or chronic ITP trials, alopecia, musculoskeletal pain, blood alkaline phosphatase increased, and dry mouth were the adverse reactions reported in 2% of patients treated with PROMACTA and in no patients who received placebo.
Among 302 patients with persistent or chronic ITP who received PROMACTA in the single-arm extension trial, the adverse reactions occurred in a pattern similar to that seen in the placebo-controlled trials. Table 9 presents the most common treatment-related adverse reactions (experienced by greater than or equal to 3% of patients receiving PROMACTA) from the extension trial.
Table 9. Treatment-related Adverse Reactions (≥ 3%) From Extension Trial in Adults With Persistent or Chronic Immune Thrombocytopenia Abbreviations: ALT, alanine aminotransferase; AST, aspartate aminotransferase.
Adverse reaction
PROMACTA 50 mg
n = 302
(%)
Headache
10
ALT increased
5
AST increased
5
Cataract
5
Fatigue
5
Blood bilirubin increased
4
Nausea
4
Hyperbilirubinemia
3
Diarrhea
3
In the three controlled persistent or chronic ITP trials, serum liver test abnormalities (predominantly Grade 2 or less in severity) were reported in 11% and 7% of patients for PROMACTA and placebo, respectively. Four patients (1%) treated with PROMACTA and three patients in the placebo group (2%) discontinued treatment due to hepatobiliary laboratory abnormalities. Seventeen of the patients treated with PROMACTA in the controlled trials with hepatobiliary laboratory abnormalities were re-exposed to PROMACTA in the extension trial. Eight of these patients again experienced liver test abnormalities (less than or equal to Grade 3) resulting in discontinuation of PROMACTA in one patient. In the extension persistent or chronic ITP trial, six additional patients had PROMACTA discontinued due to liver test abnormalities (less than or equal to Grade 3).
In the three controlled persistent or chronic ITP trials, cataracts developed or worsened in 7% of patients treated with PROMACTA and 7% of patients in the placebo group. All patients had documented, preexisting risk factors for cataractogenesis, including corticosteroid use. In the extension trial, cataracts developed or worsened in 11% of patients who underwent ocular examination prior to therapy with PROMACTA. Seventy-two percent of patients had preexisting risk factors, including corticosteroid use.
The safety of PROMACTA was also assessed in all patients treated in 7 adult persistent or chronic ITP clinical trials (N = 763 PROMACTA-treated patients and 179 placebo-treated patients). Thromboembolic events were reported in 6% of PROMACTA-treated patients versus 0% of placebo-treated patients and thrombotic microangiopathy with acute renal failure was reported in < 1% of PROMACTA-treated patients versus 0% of placebo-treated patients.
In a placebo-controlled trial of PROMACTA in patients with chronic liver disease and thrombocytopenia not related to ITP, six patients treated with PROMACTA and one patient in the placebo group developed portal vein thromboses [see Warnings and Precautions (5.4)].
Pediatric Patients: The data described below reflect median exposure to PROMACTA of 91Â days for 107Â pediatric patients (aged 1 to 17Â years) with persistent or chronic ITP, of whom 53% were female, across the randomized phase of two placebo-controlled trials.
Table 10 presents the most common adverse drug reactions (experienced by greater than or equal to 3% of pediatric patients 1 year and older receiving PROMACTA) across the two placebo-controlled trials, with a higher incidence for PROMACTA versus placebo.
Table 10. Adverse Reactions (≥ 3%) With a Higher Incidence for PROMACTA Versus Placebo From Two Placebo-controlled Trials in Pediatric Patients 1 Year and Older With Persistent or Chronic Immune Thrombocytopenia Abbreviations: ALT, alanine aminotransferase; AST, aspartate aminotransferase. aIncludes adverse reactions or laboratory abnormalities > 3 x ULN.
PROMACTA
Placebo
n = 107
n = 50
Adverse reaction
(%)
(%)
Upper respiratory tract infection
17
6
Nasopharyngitis
12
4
Cough
9
0
Diarrhea
9
2
Pyrexia
9
8
Abdominal pain
8
4
Oropharyngeal pain
8
2
Toothache
6
0
ALT increaseda
6
0
Rash
5
2
AST increased
4
0
Rhinorrhea
4
0
In the two controlled clinical persistent or chronic ITP trials, cataracts developed or worsened in 2 (1%) patients treated with PROMACTA. Both patients had received chronic oral corticosteroids, a risk factor for cataractogenesis.
Chronic Hepatitis C-associated Thrombocytopenia: In the two placebo-controlled trials, 955 patients with chronic hepatitis C-associated thrombocytopenia received PROMACTA. Table 11 presents the most common adverse drug reactions (experienced by greater than or equal to 10% of patients receiving PROMACTA compared with placebo).
Table 11. Adverse Reactions (≥ 10% and Greater Than Placebo) From Two Placebo-controlled Trials in Adults With Chronic Hepatitis C aIncludes PTs of insomnia, initial insomnia, and poor quality sleep.
Adverse reaction
PROMACTA
+ Peginterferon/Ribavirin
n = 955
(%)
Placebo
+ Peginterferon/Ribavirin
n = 484
(%)
Anemia
40
35
Pyrexia
30
24
Fatigue
28
23
Headache
21
20
Nausea
19
14
Diarrhea
19
11
Decreased appetite
18
14
Influenza-like illness
18
16
Insomniaa
16
15
Asthenia
16
13
Cough
15
12
Pruritus
15
13
Chills
14
9
Myalgia
12
10
Alopecia
10
6
Peripheral edema
10
5
Rash was reported in 9% and 7% of patients receiving PROMACTA and placebo, respectively.
In the two controlled clinical trials in patients with chronic hepatitis C, hyperbilirubinemia was reported in 8% of patients receiving PROMACTA compared with 3% for placebo. Total bilirubin greater than or equal to 1.5Â x ULN was reported in 76% and 50% of patients receiving PROMACTA and placebo, respectively. ALT or AST greater than or equal to 3Â x ULN was reported in 34% and 38% of patients for PROMACTA and placebo, respectively.
In the two controlled clinical trials in patients with chronic hepatitis C, cataracts developed or worsened in 8% of patients treated with PROMACTA and 5% of patients treated with placebo.
The safety of PROMACTA was also assessed in all patients treated with PROMACTA in the two controlled trials, including patients who initially received PROMACTA in the pre-antiviral treatment phase of the trial and were later randomized to the placebo arm (N = 1520 PROMACTA-treated patients). Hepatic failure was reported in 0.8% of PROMACTA-treated patients and 0.4% of placebo-treated patients.
Severe Aplastic Anemia
First-Line Treatment of Severe Aplastic Anemia
The safety of PROMACTA was established based upon a single-arm trial of 153 patients with severe aplastic anemia who had not received prior definitive immunosuppressive therapy. In this trial, PROMACTA was administered in combination with horse antithymocyte globulin (h-ATG) and cyclosporine [see Clinical Studies (14.3)]. Among the 153 patients who were dosed in this trial, 92 patients were evaluable for safety of the concurrent use of PROMACTA, h-ATG, and cyclosporine at the recommended dose and schedule.
In this cohort, PROMACTA was administered at up to 150 mg once daily on Day 1 to Month 6 (D1-M6) in combination with h-ATG on Days 1 to 4 and cyclosporine for 6 months, followed by low dose of cyclosporine (maintenance dose) for an additional 18 months for patients who achieved a hematologic response at 6 months. The median duration of exposure to PROMACTA in this cohort was 183 days with 70% of patients exposed for > 24 weeks.
Table 12 presents the most common adverse reactions (experienced by greater than or equal to 5% of patients) associated with PROMACTA in the D1-M6 cohort.
Table 12. Adverse Reactions (≥ 5%) From One Open-label Trial in First-Line Treatment of Patients With Severe Aplastic Anemia Abbreviations: ALT, alanine aminotransferase; AST, aspartate aminotransferase.
Adverse reaction
PROMACTAn = 92(%)
ALT increased
29
AST increased
17
Blood bilirubin increased
17
Rash
8
Skin discoloration, including hyperpigmentation
5
In the PROMACTA D1-M6 cohort, ALT increased (29%), AST increased (17%), and blood bilirubin increased (17%) were reported more frequently than in patients with refractory severe aplastic anemia (see Table 13).
New or worsening liver function laboratory abnormalities (CTCAE Grade 3 and Grade 4) in the PROMACTA D1-M6 cohort were 15% and 2% for AST, 26% and 4% for ALT, and 12% and 1% for bilirubin, respectively.
In this single-arm open-label clinical trial, ALT or AST > 3 x ULN with total bilirubin > 1.5 x ULN and ALT or AST > 3 x ULN with total bilirubin > 2 x ULN were reported in 44% and 32% of patients, respectively, in the PROMACTA D1-M6 cohort.
Pediatric Patients
A total of 34 pediatric patients (2 patients 2 to 5 years of age, 12 patients 6 to 11 years of age, and 20 patients 12 to 16 years of age) were enrolled in this single-arm trial of which 26 pediatric patients were enrolled in the PROMACTA D1-M6 cohort. In this cohort, the most frequent serious adverse reactions (experienced by ≥ 10% of patients) were upper respiratory tract infection (12% in patients age 2 to 16 years compared to 5% in patients 17 years of age and older, respectively) and rash (12% compared to 2%). The most common adverse reactions (experienced by ≥ 10% of patients) associated with PROMACTA were ALT increased (23% in patients age 2 to 16 years compared to 32% in patients 17 years of age and older, respectively), blood bilirubin increased (12% compared to 20%), AST increased (12% compared to 20%), and rash (12% compared to 6%).
Cytogenetic Abnormalities
In this trial, patients had bone marrow aspirates evaluated for cytogenetic abnormalities. Seven patients in the PROMACTA D1-M6 cohort had a new cytogenetic abnormality reported of which 4 had the loss of chromosome 7; these 4 occurred within 6.1 months. Across all cohorts, clonal cytogenetic evolution occurred in 15 out of 153 (10%) patients. Of the 15 patients who experienced a cytogenetic abnormality: 7 patients had the loss of chromosome 7, 6 of which occurred within 6.1 months; 4 patients had chromosomal aberrations which were of unclear significance; 3 patients had a deletion of chromosome 13; and 1 patient had a follow-up bone marrow assessment at 5 years with features of dysplasia with hypercellularity concerning for potential development of MDS. It is unclear whether these findings occurred due to the underlying disease, the immunosuppressive therapy, and/or treatment with PROMACTA.
Refractory Severe Aplastic Anemia
In the single-arm, open-label trial, 43 patients with refractory severe aplastic anemia received PROMACTA. Eleven patients (26%) were treated for greater than 6Â months and 7 patients (16%) were treated for greater than 1Â year. The most common adverse reactions (greater than or equal to 20%) were nausea, fatigue, cough, diarrhea, and headache.
Table 13. Adverse Reactions (≥ 10%) From One Open-label Trial in Adults With Refractory Severe Aplastic Anemia
Adverse reaction
PROMACTA
n = 43
(%)
Nausea
33
Fatigue
28
Cough
23
Diarrhea
21
Headache
21
Pain in extremity
19
Pyrexia
14
Dizziness
14
Oropharyngeal pain
14
Abdominal pain
12
Muscle spasms
12
Transaminases increased
12
Arthralgia
12
Rhinorrhea
12
Rash and hyperbilirubinemia were reported in 7% of patients; cataract was reported in 2% of patients.
In this trial, concurrent ALT or AST greater than 3 x ULN with total bilirubin greater than 1.5 x ULN were reported in 5% of patients. Total bilirubin greater than 1.5 x ULN occurred in 14% of patients.
In this trial, patients had bone marrow aspirates evaluated for cytogenetic abnormalities. Eight patients had a new cytogenetic abnormality reported on therapy, including 5 patients who had complex changes in chromosome 7.
6.2 Postmarketing Experience
The following adverse reactions have been identified during post approval use of PROMACTA. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate the frequency or establish a causal relationship to drug exposure.
Skin and Subcutaneous Tissue Disorders: Skin discoloration, including hyperpigmentation and skin yellowing.
7 Drug Interactions
7.1 Polyvalent Cations (Chelation)
Eltrombopag chelates polyvalent cations (such as iron, calcium, aluminum, magnesium, selenium, and zinc) in foods, mineral supplements, and antacids.
Take PROMACTA at least 2 hours before or 4 hours after any medications or products containing polyvalent cations, such as antacids, dairy products, and mineral supplements to avoid significant reduction in absorption of PROMACTA due to chelation [see Dosage and Administration (2.4), Clinical Pharmacology (12.3)].
7.2 Transporters
Use caution when concomitantly administering PROMACTA and drugs that are substrates of OATP1B1 (e.g., atorvastatin, bosentan, ezetimibe, fluvastatin, glyburide, olmesartan, pitavastatin, pravastatin, rosuvastatin, repaglinide, rifampin, simvastatin acid, SN-38 [active metabolite of irinotecan], valsartan) or breast cancer resistance protein (BCRP) (e.g., imatinib, irinotecan, lapatinib, methotrexate, mitoxantrone, rosuvastatin, sulfasalazine, topotecan). Monitor patients closely for signs and symptoms of excessive exposure to the drugs that are substrates of OATP1B1 or BCRP and consider reduction of the dose of these drugs, if appropriate. In clinical trials with PROMACTA, a dose reduction of rosuvastatin by 50% was recommended.
7.3 Protease Inhibitors
HIV Protease Inhibitors: No dose adjustment is recommended when PROMACTA is coadministered with lopinavir/ritonavir (LPV/RTV). Drug interactions with other HIV protease inhibitors have not been evaluated.
Hepatitis C Virus Protease Inhibitors: No dose adjustments are recommended when PROMACTA is coadministered with boceprevir or telaprevir. Drug interactions with other hepatitis C virus (HCV) protease inhibitors have not been evaluated.
7.4 Peginterferon Alfa-2a/b Therapy
No dose adjustments are recommended when PROMACTA is coadministered with peginterferon alfa-2a (PEGASYS®) or -2b (PEGINTRON®).
8 Use In Specific Populations
- Lactation: Advise women not to breastfeed during treatment. (
8.2 )8.1 Pregnancy
Risk Summary
Available data from a small number of published case reports and postmarketing experience with PROMACTA use in pregnant women are insufficient to assess any drug-associated risks for major birth defects, miscarriage, or adverse maternal or fetal outcomes. In animal reproduction and developmental toxicity studies, oral administration of eltrombopag to pregnant rats during organogenesis resulted in embryolethality and reduced fetal weights at maternally toxic doses. These effects were observed at doses resulting in exposures that were six times the human clinical exposure based on area under the curve (AUC) in patients with persistent or chronic ITP at 75 mg/day, and three times the AUC in patients with chronic hepatitis C at 100 mg/day (see Data).
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and of miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
Data
Animal Data
In an early embryonic development study, female rats received oral eltrombopag at doses of 10, 20, or 60 mg/kg/day (0.8, 2, and 6 times, respectively, the human clinical exposure based on AUC in patients with ITP at 75 mg/day and 0.3, 1, and 3 times, respectively, the human clinical exposure based on AUC in patients with chronic hepatitis C at 100 mg/day). Increased pre- and post-implantation loss and reduced fetal weight were observed at the highest dose which also caused maternal toxicity.
In an embryo-fetal development study eltrombopag was administered orally to pregnant rats during the period of organogenesis at doses of 10, 20, or 60 mg/kg/day (0.8, 2, and 6 times, respectively, the human clinical exposure based on AUC in patients with ITP at 75 mg/day and 0.3, 1, and 3 times, respectively, the human clinical exposure based on AUC in patients with chronic hepatitis C at 100 mg/day). Decreased fetal weights (6% to 7%) and a slight increase in the presence of cervical ribs were observed at the highest dose which also caused maternal toxicity. However, no evidence of major structural malformations was observed.
In an embryo-fetal development study eltrombopag was administered orally to pregnant rabbits during the period of organogenesis at doses of 30, 80, or 150 mg/kg/day (0.04, 0.3, and 0.5 times, respectively, the human clinical exposure based on AUC in patients with ITP at 75 mg/day and 0.02, 0.1, and 0.3 times, respectively, the human clinical exposure based on AUC in patients with chronic hepatitis C at 100 mg/day). No evidence of fetotoxicity, embryolethality, or teratogenicity was observed.
In a pre- and post-natal developmental toxicity study in pregnant rats (F0), oral eltrombopag was administered from gestation Day 6 through lactation Day 20. No adverse effects on maternal reproductive function or on the development of the offspring (F1) were observed at doses up to 20 mg/kg/day (2 times the human clinical exposure based on AUC in patients with ITP at 75 mg/day and similar to the human clinical exposure based on AUC in patients with chronic hepatitis C at 100 mg/day). Eltrombopag was detected in the plasma of offspring (F1). The plasma concentrations in pups increased with dose following administration of drug to the F0 dams.
8.2 Lactation
Risk Summary
There are no data regarding the presence of eltrombopag or its metabolites in human milk, the effects on the breastfed child, or the effects on milk production. However, eltrombopag was detected in the pups of lactating rats 10 days postpartum suggesting the potential for transfer during lactation. Due to the potential for serious adverse reactions in a breastfed child from PROMACTA, breastfeeding is not recommended during treatment.
8.3 Females and Males of Reproductive Potential
Contraception
Based on animal reproduction studies, PROMACTA can cause fetal harm when administered to a pregnant woman. Sexually-active females of reproductive potential should use effective contraception (methods that result in less than 1% pregnancy rates) when using PROMACTA during treatment and for at least 7 days after stopping treatment with PROMACTA.
8.4 Pediatric Use
The safety and efficacy of PROMACTA have been established in pediatric patients 1 year and older with persistent or chronic ITP and in pediatric patients 2 years and older with IST-naĂŻve severe aplastic anemia (in combination with h-ATG and cyclosporine). Safety and efficacy in pediatric patients below the age of 1 year with ITP have not been established. Safety and efficacy in pediatric patients with thrombocytopenia associated with chronic hepatitis C and refractory severe aplastic anemia have not been established.
The safety and efficacy of PROMACTA in pediatric patients 1Â year and older with persistent or chronic ITP were evaluated in two double-blind, placebo-controlled trials [see Adverse Reactions (6.1), Clinical Studies (14.1)]. The pharmacokinetics of eltrombopag have been evaluated in 168 pediatric patients 1 year and older with ITP dosed once daily [see Clinical Pharmacology (12.3)]. See Dosage and Administration (2.1) for dosing recommendations for pediatric patients 1Â year and older.
The safety and efficacy of PROMACTA in combination with h-ATG and cyclosporine for the first-line treatment of severe aplastic anemia in pediatric patients 2 years and older were evaluated in a single-arm, open-label trial [see Adverse Reactions (6.1), Clinical Studies (14.3)]. A total of 26 pediatric patients (ages 2 to < 17 years) were evaluated; 12 children (aged 2 to < 12 years) and 14 adolescents (aged 12 to < 17). See Dosage and Administration (2.3) for dosing recommendations for pediatric patients 2 years and older. The safety and efficacy of PROMACTA in combination with h-ATG and cyclosporine in pediatric patients younger than 2 years for the first-line treatment of severe aplastic anemia have not yet been established. In patients 2 to 16 years of age, 69% of patients experienced serious adverse events compared to 42% in patients 17 years and older. Among the 12 patients who were 2 to 11 years of age in the PROMACTA D1-M6 cohort and reached the 6-month assessment or withdrew earlier, the complete response rate at Month 6 was 8% versus 46% in patients age 12 to 16 years and 50% in patients 17 years of age and older.
8.5 Geriatric Use
Of the 106 patients in two randomized clinical trials of PROMACTA 50 mg in persistent or chronic ITP, 22% were 65 years of age and over, while 9% were 75 years of age and over. Of the 1439 patients in two randomized clinical trials of PROMACTA in patients with chronic hepatitis C and thrombocytopenia, 7% were 65 years of age and over, while < 1% were 75 years of age and over. Of the 196 patients who received PROMACTA for the treatment of severe aplastic anemia, 18% were 65 years of age and over, while 3% were 75 years of age and over. No overall differences in safety or effectiveness were observed between these patients and younger patients.
8.6 Hepatic Impairment
Patients With Persistent or Chronic ITP and Severe Aplastic Anemia
Reduce the initial dose of PROMACTA in patients with persistent or chronic ITP (adult and pediatric patients 6 years and older only) or refractory severe aplastic anemia who also have hepatic impairment (Child-Pugh class A, B, C) [see Dosage and Administration (2.1, 2.3), Warnings and Precautions (5.2), Clinical Pharmacology (12.3)].
In a clinical trial in patients with severe aplastic anemia who had not received prior definitive immunosuppressive therapy, patients with baseline ALT or AST > 5 x ULN were ineligible to participate. If a patient with hepatic impairment (Child-Pugh class A, B, C) initiates therapy with PROMACTA for the first-line treatment of severe aplastic anemia, reduce the initial dose [see Dosage and Administration (2.3), Warnings and Precautions (5.2), Clinical Pharmacology (12.3)].
Patients With Chronic Hepatitis C
No dosage adjustment is recommended in patients with chronic hepatitis C and hepatic impairment [see Clinical Pharmacology (12.3)].
8.7 Ethnicity
Reduce the initial dose of PROMACTA for patients of East-/Southeast-Asian ancestry with ITP (adult and pediatric patients 6 years and older only) or severe aplastic anemia [see Dosage and Administration (2.1, 2.3), Clinical Pharmacology (12.3)]. No reduction in the initial dose of PROMACTA is recommended in patients of East-/Southeast-Asian ancestry with chronic hepatitis C [see Clinical Pharmacology (12.3)].
10 Overdosage
In the event of overdose, platelet counts may increase excessively and result in thrombotic/thromboembolic complications.
In one report, a subject who ingested 5000 mg of PROMACTA had a platelet count increase to a maximum of 929 x 109/L at 13 days following the ingestion. The patient also experienced rash, bradycardia, ALT/AST elevations, and fatigue. The patient was treated with gastric lavage, oral lactulose, intravenous fluids, omeprazole, atropine, furosemide, calcium, dexamethasone, and plasmapheresis; however, the abnormal platelet count and liver test abnormalities persisted for 3 weeks. After 2 months’ follow-up, all events had resolved without sequelae.
In case of an overdose, consider oral administration of a metal cation-containing preparation, such as calcium, aluminum, or magnesium preparations to chelate eltrombopag and thus limit absorption. Closely monitor platelet counts. Reinitiate treatment with PROMACTA in accordance with dosing and administration recommendations [see Dosage and Administration (2.1, 2.2)].
11 Description
PROMACTA (eltrombopag) tablets contain eltrombopag olamine, a small molecule thrombopoietin (TPO) receptor agonist for oral administration.
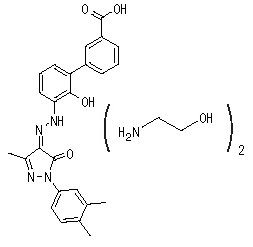
Eltrombopag olamine is a biphenyl hydrazone. The chemical name for eltrombopag olamine is 3'-{(2Z)-2-[1-(3,4-dimethylphenyl)-3-methyl-5-oxo-1,5-dihydro-4H-pyrazol-4-ylidene]hydrazino}-2'-hydroxy-3-biphenylcarboxylic acid - 2-aminoethanol (1:2). It has the molecular formula C25H22N4O4 • 2(C2H7NO). The molecular weight is 564.65 g/mol for eltrombopag olamine and 442.5 g/mol for eltrombopag free acid. Eltrombopag olamine has the following structural formula:
Eltrombopag olamine is practically insoluble in aqueous buffer across a pH range of 1 to 7.4, and is sparingly soluble in water.
PROMACTA (eltrombopag) tablets contain eltrombopag olamine in the amount equivalent to 12.5 mg, 25 mg, 50 mg, or 75 mg of eltrombopag free acid. The inactive ingredients of PROMACTA tablets are:
Tablet Core: magnesium stearate, mannitol, microcrystalline cellulose, povidone, and sodium starch glycolate. Coating: FD&C Blue No. 2 aluminum lake (50-mg tablet), FD&C Yellow No. 6 aluminum lake (25-mg tablet), hypromellose, Iron Oxide Black and Iron Oxide Red (75-mg tablet), polyethylene glycol 400, polysorbate 80 (12.5-mg tablet), or titanium dioxide.
PROMACTA (eltrombopag) for oral suspension packets contain a reddish-brown to yellow powder which produces a reddish-brown suspension when reconstituted with water. Each packet delivers eltrombopag olamine equivalent to 12.5 mg or 25Â mg of eltrombopag free acid. The inactive ingredients of PROMACTA for oral suspension are mannitol, sucralose, and xanthan gum.
12 Clinical Pharmacology
12.1 Mechanism of Action
Eltrombopag is a TPO-receptor agonist that interacts with the transmembrane domain of the human TPO-receptor (also known as cMpl) and initiates signaling cascades that induce proliferation and differentiation of megakaryocytes leading to increased platelet production.
12.2 Pharmacodynamics
In clinical trials, treatment with PROMACTA resulted in dose-dependent increases in platelet counts following repeated (daily) dosing. The increase in platelet counts reached a maximum approximately two weeks after the initiation of dosing, and returned to baseline within approximately two weeks after the last dose of PROMACTA.
Cardiac Electrophysiology
At doses up to 150 mg (the maximum recommended dose) daily for 5 days, PROMACTA did not prolong the QT/QTc interval to any relevant extent.
12.3 Pharmacokinetics
Eltrombopag demonstrated a dose-proportional increase in exposure between doses of 50 to 150 mg/day in healthy adult subjects. Eltrombopag AUC was approximately 1.7-fold higher in patients with persistent or chronic ITP and approximately 2.8-fold higher in patients with HCV compared to healthy subjects. Steady-state was achieved after approximately 1 week of once daily treatment, with geometric mean accumulation ratio of 1.56 (90% confidence interval 1.20, 1.63) at 75 mg/day. Eltrombopag AUC was approximately 3.2-fold higher in patients with definitive immunosuppressive therapy-naĂŻve severe aplastic anemia compared to healthy subjects suggesting higher relative exposure compared to healthy subjects or patients with ITP and similar exposure compared to patients with chronic hepatitis C. Eltrombopag for oral suspension delivered 22% higher plasma AUC0-INF than the tablet formulation.
Absorption
Eltrombopag is absorbed with a peak concentration occurring 2 to 6 hours after oral administration. Oral absorption of drug-related material following administration of a single 75-mg solution dose was estimated to be at least 52%.
Effect of Food
A standard high-fat breakfast (876 calories, 52 g fat, 71 g carbohydrate, 34 g protein, and 427 mg calcium) significantly decreased plasma eltrombopag AUC0-INF by approximately 59% and Cmax by 65% and delayed Tmax by 1 hour. The decrease in exposure is primarily due to the high calcium content.
A meal low in calcium (≤ 50 mg calcium) did not significantly impact plasma eltrombopag exposure, regardless of calorie and fat content.
The effect of administration of a single 25-mg dose of eltrombopag for oral suspension with a high-calcium, moderate-fat, moderate calorie meal on AUC0-INF and Cmax in healthy adult subjects is presented in Table 14.
Table 14. Effect on Plasma Eltrombopag Pharmacokinetic Parameters After Administration of a Single 25-mg Dose of Eltrombopag for Oral Suspension With a High Calcium Meala in Healthy Adult Subjects a372 calories, 9 g fat, and 448 mg calcium. Timing of eltrombopag for oral suspension dose Mean (90% CI) reduction in plasma eltrombopag AUC0-INF Mean (90% CI) reduction in plasma eltrombopag Cmax With a high-calcium, moderate-fat, moderate-calorie meal 75% (71%, 88%) 79% (76%, 82%) 2 hours after the high-calcium, moderate-fat, moderate-calorie meal 47% (40%, 53%) 48% (40%, 54%) 2 hours before the high-calcium, moderate-fat, moderate-calorie meal 20% (9%, 29%) 14% (2%, 25%)
Distribution
The concentration of eltrombopag in blood cells is approximately 50% to 79% of plasma concentrations based on a radiolabel study. In vitro studies suggest that eltrombopag is highly bound to human plasma proteins (greater than 99%). Eltrombopag is a substrate of BCRP, but is not a substrate for P-glycoprotein (P-gp) or OATP1B1.
Elimination
The plasma elimination half-life of eltrombopag is approximately 21 to 32 hours in healthy subjects and 26 to 35 hours in patients with ITP.
Metabolism: Absorbed eltrombopag is extensively metabolized, predominantly through pathways, including cleavage, oxidation, and conjugation with glucuronic acid, glutathione, or cysteine. In vitro studies suggest that CYP1A2 and CYP2C8 are responsible for the oxidative metabolism of eltrombopag. UGT1A1 and UGT1A3 are responsible for the glucuronidation of eltrombopag.
Excretion: The predominant route of eltrombopag excretion is via feces (59%), and 31% of the dose is found in the urine. Unchanged eltrombopag in feces accounts for approximately 20% of the dose; unchanged eltrombopag is not detectable in urine.
Specific Populations
Ethnicity
Eltrombopag concentrations in East-/Southeast-Asian ancestry patients with ITP or chronic hepatitis C, were 50% to 55% higher compared with non-Asian subjects [see Dosage and Administration (2.1, 2.3)].
Eltrombopag exposure in healthy African-American subjects was approximately 40% higher than that observed in Caucasian subjects in one clinical pharmacology trial and similar in three other clinical pharmacology trials. The effect of African-American ethnicity on exposure and related safety and efficacy of eltrombopag has not been established.
Hepatic Impairment
Following a single dose of PROMACTA (50 mg), plasma eltrombopag AUC0-INF was 41% higher in patients with mild hepatic impairment (Child-Pugh class A) compared with subjects with normal hepatic function. Plasma eltrombopag AUC0-INF was approximately 2-fold higher in patients with moderate (Child-Pugh class B) and severe hepatic impairment (Child-Pugh class C) compared with subjects with normal hepatic function. The half-life of eltrombopag was prolonged 2-fold in these patients. This clinical trial did not evaluate protein-binding effects.
Chronic Liver Disease
Following repeat doses of eltrombopag in patients with thrombocytopenia and with chronic liver disease, mild hepatic impairment resulted in an 87% to 110% higher plasma eltrombopag AUC(0-Ď„) and moderate hepatic impairment resulted in approximately 141% to 240% higher plasma eltrombopag AUC(0-Ď„) values compared with patients with normal hepatic function. The half-life of eltrombopag was prolonged 3-fold in patients with mild hepatic impairment and 4-fold in patients with moderate hepatic impairment. This clinical trial did not evaluate protein-binding effects.
Chronic Hepatitis C
Patients with chronic hepatitis C treated with PROMACTA had higher plasma AUC(0-Ď„) values as compared with healthy subjects, and AUC(0-Ď„) increased with increasing Child-Pugh score. Patients with chronic hepatitis C and mild hepatic impairment had approximately 100% to 144% higher plasma AUC(0-Ď„) compared with healthy subjects. This clinical trial did not evaluate protein-binding effects.
Renal Impairment
Following a single dose of PROMACTA (50 mg), the average total plasma eltrombopag AUC0-INF was 32% to 36% lower in subjects with mild (estimated creatinine clearance (CLCr) by Cockcroft-Gault equation: 50 to 80 mL/min), to moderate (CLCr of 30 to 49 mL/min) renal impairment and 60% lower in subjects with severe (CLCr less than 30 mL/min) renal impairment compared with healthy subjects. The effect of renal impairment on unbound (active) eltrombopag exposure has not been assessed.
Pediatric Patients
The pharmacokinetics of eltrombopag have been evaluated in 168 pediatric patients 1 year and older with ITP dosed once daily in two trials. Plasma eltrombopag apparent clearance following oral administration (CL/F) increased with increasing body weight. East-/Southeast-Asian pediatric patients with ITP had approximately 43% higher plasma eltrombopag AUC(0-Ď„) values as compared with non-Asian patients.
Plasma eltrombopag AUC(0-Ď„) and Cmax in pediatric patients aged 12 to 17Â years was similar to that observed in adults. The pharmacokinetic parameters of eltrombopag in pediatric patients with ITP are shown in Table 15.
Table 15. Geometric Mean (95% CI) Steady-state Plasma Eltrombopag Pharmacokinetic Parametersa in Patients With ITP (Normalized to a Once-daily 50-mg Dose) aPK parameters presented as geometric mean (95% CI). bBased on population PK post-hoc estimates.
Cmax b
AUC(0-Ď„) b
Age
(mcg/mL)
(mcg·hr/mL)
Adults (n = 108)
7.03
101
(6.44, 7.68)
(91.4, 113)
12 to 17 years (n = 62)
6.80
103
(6.17, 7.50)
(91.1, 116)
6 to 11 years (n = 68)
10.3
153
(9.42, 11.2)
(137, 170)
1 to 5 years (n = 38)
11.6
162
(10.4, 12.9)
(139, 187)
Drug Interaction Studies
Clinical Studies
Effect of Drugs on Eltrombopag
Effect of Polyvalent Cation-containing Antacids on Eltrombopag:
The coadministration of a single dose of PROMACTA (75 mg) with a polyvalent cation-containing antacid (1,524 mg aluminum hydroxide, 1,425 mg magnesium carbonate, and sodium alginate) decreased plasma eltrombopag AUC0-INF and Cmax by approximately 70%. The contribution of sodium alginate to this interaction is not known.
Effect of HIV Protease Inhibitors on Eltrombopag:
The coadministration of repeat-dose lopinavir 400 mg/ritonavir 100 mg (twice daily) with a single dose of PROMACTA (100 mg) decreased plasma eltrombopag AUC0-INF by 17%.
Effect of HCV Protease Inhibitors on Eltrombopag:
The coadministration of repeat-dose telaprevir (750 mg every 8 hours) or boceprevir (800 mg every 8 hours) with a single dose of PROMACTA (200 mg) to healthy adult subjects in a clinical trial did not alter plasma eltrombopag AUC0-INF or Cmax to a significant extent.
Effect of Cyclosporine on Eltrombopag:
The coadministration of a single dose of PROMACTA (50 mg) with a single dose of an OATP and BCRP inhibitor cyclosporine (200 mg or 600 mg) decreased plasma eltrombopag AUC0-INF by 18% to 24% and Cmax by 25% to 39%.
Effect of Pegylated Interferon alfa-2a + Ribavirin and Pegylated Interferon alfa-2b + Ribavirin on Eltrombopag:
The presence of pegylated interferon alfa + ribavirin therapy did not significantly affect the clearance of eltrombopag.
Effect of Eltrombopag on Other Drugs
Effect of Eltrombopag on Cytochrome P450 Enzymes Substrates:
The coadministration of multiple doses of PROMACTA (75 mg once daily for 7 days) did not result in the inhibition or induction of the metabolism of a combination of probe substrates for CYP1A2 (caffeine), CYP2C19 (omeprazole), CYP2C9 (flurbiprofen), or CYP3A4 (midazolam) in humans.
Effect of Eltrombopag on Rosuvastatin:
The coadministration of multiple doses of PROMACTA (75 mg once daily for 5 days) with a single dose of rosuvastatin (OATP1B1 and BCRP substrate; 10 mg) increased plasma rosuvastatin AUC0-INF by 55% and Cmax by 103%.
Effect of Eltrombopag on HCV Protease Inhibitors:
The coadministration of repeat-dose telaprevir (750 mg every 8 hours) or boceprevir (800 mg every 8 hours) with a single dose of PROMACTA (200 mg) to healthy adult subjects in a clinical trial did not alter plasma telaprevir or boceprevir AUC0-INF or Cmax to a significant extent.
In vitro Studies
Eltrombopag Effect on Metabolic Enzymes
Eltrombopag has demonstrated the potential to inhibit CYP2C8, CYP2C9, UGT1A1, UGT1A3, UGT1A4, UGT1A6, UGT1A9, UGT2B7, and UGT2B15.
Eltrombopag Effect on Transporters
Eltrombopag has demonstrated the potential to inhibit OATP1B1 and BCRP.
13 Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Eltrombopag does not stimulate platelet production in rats, mice, or dogs because of unique TPO receptor specificity. Data from these animals do not fully model effects in humans.
Eltrombopag was not carcinogenic in mice at doses up to 75Â mg/kg/day or in rats at doses up to 40Â mg/kg/day (exposures up to 4 times the human clinical exposure based on AUC in patients with ITP at 75 mg/day and 2 times the human clinical exposure based on AUC in patients with chronic hepatitis C at 100 mg/day).
Eltrombopag was not mutagenic or clastogenic in a bacterial mutation assay or in two in vivo assays in rats (micronucleus and unscheduled DNA synthesis, 10 times the human clinical exposure based on Cmax in patients with ITP at 75 mg/day and 7 times the human clinical exposure based on Cmax in patients with chronic hepatitis C at 100Â mg/day). In the in vitro mouse lymphoma assay, eltrombopag was marginally positive (less than 3-fold increase in mutation frequency).
Eltrombopag did not affect female fertility in rats at doses up to 20Â mg/kg/day (2 times the human clinical exposure based on AUC in patients with ITP at 75Â mg/day and similar to the human clinical exposure based on AUC in patients with chronic hepatitis C at 100 mg/day). Eltrombopag did not affect male fertility in rats at doses up to 40Â mg/kg/day, the highest dose tested (3 times the human clinical exposure based on AUC in patients with ITP at 75 mg/day and 2 times the human clinical exposure based on AUC in patients with chronic hepatitis C at 100Â mg/day).
13.2 Animal Pharmacology and/or Toxicology
Treatment-related cataracts were detected in rodents in a dose- and time-dependent manner. At greater than or equal to 6 times the human clinical exposure based on AUC in patients with ITP at 75Â mg/day and 3 times the human clinical exposure based on AUC in patients with chronic hepatitis C at 100Â mg/day, cataracts were observed in mice after 6 weeks and in rats after 28Â weeks of dosing. At greater than or equal to 4Â times the human clinical exposure based on AUC in patients with ITP at 75Â mg/day and 2 times the human clinical exposure based on AUC in patients with chronic hepatitis C at 100Â mg/day, cataracts were observed in mice after 13Â weeks and in rats after 39Â weeks of dosing [see Warnings and Precautions (5.5)].
Renal tubular toxicity was observed in studies up to 14 days in duration in mice and rats at exposures that were generally associated with morbidity and mortality. Tubular toxicity was also observed in a 2-year oral carcinogenicity study in mice at doses of 25, 75, and 150Â mg/kg/day. The exposure at the lowest dose was 1.2 times the human clinical exposure based on AUC in patients with ITP at 75 mg/day and 0.6 times the human clinical exposure based on AUC in patients with chronic hepatitis C at 100Â mg/day. No similar effects were observed in mice after 13 weeks at exposures greater than those associated with renal changes in the 2-year study, suggesting that this effect is both dose- and time-dependent.
14 Clinical Studies
14.1 Persistent or Chronic ITP
Adults: The efficacy and safety of PROMACTA in adult patients with persistent or chronic ITP were evaluated in three randomized, double-blind, placebo-controlled trials and in an open-label extension trial.
In Study TRA100773B and Study TRA100773A (referred to as Study 773B and Study 773A, respectively [NCT00102739]), patients who had completed at least one prior ITP therapy and who had a platelet count less than 30 x 109/L were randomized to receive either PROMACTA or placebo daily for up to 6Â weeks, followed by 6Â weeks off therapy. During the trials, PROMACTA or placebo was discontinued if the platelet count exceeded 200 x 109/L.
The median age of the patients was 50Â years and 60% were female. Approximately 70% of the patients had received at least 2 prior ITP therapies (predominantly corticosteroids, immunoglobulins, rituximab, cytotoxic therapies, danazol, and azathioprine) and 40% of the patients had undergone splenectomy. The median baseline platelet counts (approximately 18 x 109/L) were similar among all treatment groups.
Study 773B randomized 114 patients (2:1) to PROMACTA 50 mg or placebo. Of 60 patients with documented time since diagnosis, approximately 17% met the definition of persistent ITP with time since diagnosis of 3-12 months. Study 773A randomized 117 patients (1:1:1:1) among placebo or 1 of 3 dose regimens of PROMACTA, 30 mg, 50 mg, or 75 mg each administered daily. Of 51 patients with documented time since diagnosis, approximately 14% met the definition of persistent ITP.
The efficacy of PROMACTA in this trial was evaluated by response rate, defined as a shift from a baseline platelet count of less than 30 x 109/L to greater than or equal to 50 x 109/L at any time during the treatment period (Table 16).
Table 16. Studies 773B and 773A: Platelet Count Response (≥ 50 x 109/L) Rates in Adults With Persistent or Chronic Immune Thrombocytopenia a p-value < 0.001 for PROMACTA versus placebo.
Study
PROMACTA
50 mg Daily
Placebo
773B
43/73 (59%)a
6/37 (16%)
773A
19/27 (70%)a
3/27 (11%)
The platelet count response to PROMACTA was similar among patients who had or had not undergone splenectomy. In general, increases in platelet counts were detected 1 week following initiation of PROMACTA and the maximum response was observed after 2 weeks of therapy. In the placebo and 50-mg–dose groups of PROMACTA, the trial drug was discontinued due to an increase in platelet counts to greater than 200 x 109/L in 3% and 27% of the patients, respectively. The median duration of treatment with the 50-mg dose of PROMACTA was 43 days in Study 773B and 42 days in Study 773A.
Of 7 patients who underwent hemostatic challenges, additional ITP medications were required in 3 of 3 placebo group patients and 0 of 4 patients treated with PROMACTA. Surgical procedures accounted for most of the hemostatic challenges. Hemorrhage requiring transfusion occurred in one placebo group patient and no patients treated with PROMACTA.
In the RAISE study (NCT00370331), 197 patients were randomized (2:1) to receive either PROMACTA 50 mg once daily (n = 135) or placebo (n = 62) for 6 months, during which time the dose of PROMACTA could be adjusted based on individual platelet counts. Of 145 patients with documented time since diagnosis, 19% met the definition of persistent ITP. Patients were allowed to taper or discontinue concomitant ITP medications after being treated with PROMACTA for 6 weeks. Patients were permitted to receive rescue treatments at any time during the trial as clinically indicated.
The median ages of the patients treated with PROMACTA and placebo were 47Â years and 52.5Â years, respectively. Approximately half of the patients treated with PROMACTA and placebo (47% and 50%, respectively) were receiving concomitant ITP medication (predominantly corticosteroids) at randomization and had baseline platelet counts less than or equal to 15 x 109/L (50% and 48%, respectively). A similar percentage of patients treated with PROMACTA and placebo (37% and 34%, respectively) had a prior splenectomy.
The efficacy of PROMACTA in this trial was evaluated by the odds of achieving a platelet count greater than or equal to 50 x 109/L and less than or equal to 400 x 109/L for patients receiving PROMACTA relative to placebo and was based on patient response profiles throughout the 6-month treatment period. In 134 patients who completed 26 weeks of treatment, a sustained platelet response (platelet count greater than or equal to 50 x 109/L and less than or equal to 400 x 109/L for 6 out of the last 8 weeks of the 26-week treatment period in the absence of rescue medication at any time) was achieved by 60% of patients treated with PROMACTA, compared with 10% of patients treated with placebo (splenectomized patients: PROMACTA 51%, placebo 8%; non-splenectomized patients: PROMACTA 66%, placebo 11%). The proportion of responders in the group of patients treated with PROMACTA was between 37% and 56% compared with 7% and 19% in the placebo treatment group for all on-therapy visits. Patients treated with PROMACTA were significantly more likely to achieve a platelet count between 50 x 109/L and 400 x 109/L during the entire 6-month treatment period compared with those patients treated with placebo.
Outcomes of treatment are presented in Table 17 for all patients enrolled in the trial.
Table 17. RAISE: Outcomes of Treatment in Adults With Persistent or Chronic Immune Thrombocytopenia
Outcome
PROMACTA
n = 135
Placebo
n = 62
Mean number of weeks with platelet counts ≥ 50 x 109/L
11.3
2.4
Requiring rescue therapy, n (%)
24 (18)
25 (40)
Among 94 patients receiving other ITP therapy at baseline, 37 (59%) of 63 patients treated with PROMACTA and 10 (32%) of 31 patients in the placebo group discontinued concomitant therapy at some time during the trial.
In the EXTEND study (NCT00351468), patients who completed any prior clinical trial with PROMACTA were enrolled in an open-label, single-arm trial in which attempts were made to decrease the dose or eliminate the need for any concomitant ITP medications. PROMACTA was administered to 302 patients in EXTEND; 218 patients completed 1 year, 180 patients completed 2 years, 107 patients completed 3 years, 75 patients completed 4 years, 34 patients completed 5 years, and 18 patients completed 6 years of therapy. The median baseline platelet count was 19 x 109/L prior to administration of PROMACTA. Median platelet counts at 1, 2, 3, 4, 5, 6, and 7 years on study were 85 x 109/L, 85 x 109/L, 105 x 109/L, 64 x 109/L, 75 x 109/L, 119 x 109/L, and 76 x 109/L, respectively.
Pediatric Patients: The efficacy and safety of PROMACTA in pediatric patients 1 year and older with persistent or chronic ITP were evaluated in two double-blind, placebo-controlled trials. The trials differed in time since ITP diagnosis: at least 6 months versus at least 12 months. During the trials, doses could be increased every 2 weeks to a maximum of 75 mg once daily. The dose of PROMACTA was reduced if the platelet count exceeded 200 x 109/L and interrupted and reduced if it exceeded 400 x 109/L.
In the PETIT2 study (NCT01520909), patients refractory or relapsed to at least one prior ITP therapy with a platelet count less than 30 x 109/L (n = 92) were stratified by age and randomized (2:1) to PROMACTA (n = 63) or placebo (n = 29). The starting dose for patients aged 6 to 17 years was 50 mg once daily for those at least 27 kg and 37.5 mg once daily for those less than 27 kg, administered as oral tablets. A reduced dose of 25 mg once daily was used for East-/Southeast-Asian patients aged 6 to 17 years regardless of weight. The starting dose for patients aged 1 to 5 years was 1.2 mg/kg once daily (0.8 mg/kg once daily for East-/Southeast-Asian patients) administered as oral suspension.
The 13-week, randomized, double-blind period was followed by a 24-week, open-label period where patients from both arms were eligible to receive PROMACTA.
The median age of the patients was 9 years and 48% were female. Approximately 62% of patients had a baseline platelet count less than or equal to 15 x 109/L, a characteristic that was similar between treatment arms. The percentage of patients with at least 2 prior ITP therapies (predominantly corticosteroids and immunoglobulins) was 73% in the group treated with PROMACTA and 90% in the group treated with placebo. Four patients in the group treated with PROMACTA had undergone splenectomy.
The efficacy of PROMACTA in this trial was evaluated by the proportion of subjects on PROMACTA achieving platelet counts ≥ 50 x 109/L (in the absence of rescue therapy) for at least 6 out of 8 weeks between Weeks 5 to 12 of the randomized, double-blind period (Table 18).
Table 18. PETIT2: Platelet Count Response (≥ 50 x 109/L Without Rescue) for 6 out of 8 Weeks (between Weeks 5 to 12) Overall and by Age Cohort in Pediatric Patients 1 Year and Older With Chronic Immune Thrombocytopenia a p-value = < 0.001 for PROMACTA versus placebo.
Age cohort
PROMACTA
Placebo
Overall
26/63 (41%)a
1/29 (3%)
    12 to 17 years
10/24 (42%)
1/10 (10%)
    6 to 11 years
11/25 (44%)
0/13 (0%)
    1 to 5 years
5/14 (36%)
0/6 (0%)
More pediatric patients treated with PROMACTA (75%) compared with placebo (21%) had at least one platelet count greater than or equal to 50 x 109/L during the first 12 weeks of randomized treatment in absence of rescue therapy. Fewer pediatric patients treated with PROMACTA required rescue treatment during the randomized, double-blind period compared with placebo-treated patients (19% [12/63] versus 24% [7/29]). In the patients who achieved a platelet response (≥ 50 x 109/L without rescue) for 6 out of 8 weeks (between weeks 5 to 12), 62% (16/26) had an initial response in the first 2 weeks after starting PROMACTA.
Patients were permitted to reduce or discontinue baseline ITP therapy only during the open-label phase of the trial. Among 15 patients receiving other ITP therapy at baseline, 53% (8/15) reduced (n = 1) or discontinued (n = 7) concomitant therapy, mainly corticosteroids, without needing rescue therapy.
In the PETIT study (NCT00908037), patients refractory or relapsed to at least one prior ITP therapy with a platelet count less than 30 x 109/L (n = 67) were stratified by age and randomized (2:1) to PROMACTA (n = 45) or placebo (n = 22). Approximately 15% of patients met the definition of persistent ITP. The starting dose for patients aged 12 to 17 years was 37.5 mg once daily regardless of weight or race. The starting dose for patients aged 6 to 11 years was 50 mg once daily for those greater than or equal to 27 kg and 25 mg once daily for those less than 27 kg, administered as oral tablets. Reduced doses of 25 mg (for those greater than or equal to 27 kg) and 12.5 mg (for those less than 27 kg), each once daily, were used for East-/Southeast-Asian patients in this age range. The starting dose for patients aged 1 to 5 years was 1.5 mg/kg once daily (0.8 mg/kg once daily for East-/Southeast-Asian patients) administered as oral suspension.
The 7-week, randomized, double-blind period was followed by an open-label period of up to 24 weeks where patients from both arms were eligible to receive PROMACTA.
The median age of the patients was 10 years and 60% were female. Approximately 51% of patients had a baseline platelet count less than or equal to 15 x 109/L. The percentage of patients with at least 2 prior ITP therapies (predominantly corticosteroids and immunoglobulins) was 84% in the group treated with PROMACTA and 86% in the group treated with placebo. Five patients in the group treated with PROMACTA had undergone splenectomy.
The efficacy of PROMACTA in this trial was evaluated by the proportion of patients achieving platelet counts greater than or equal to 50 x 109/L (in absence of rescue therapy) at least once between Weeks 1 and 6 of the randomized, double-blind period (Table 19). Platelet response to PROMACTA was consistent across the age cohorts.
Table 19. PETIT: Platelet Count Response (≥ 50 x 109/L Without Rescue) Rates in Pediatric Patients 1 Year and Older With Persistent or Chronic Immune Thrombocytopenia a p-value = 0.011 for PROMACTA versus placebo.
Age cohort
PROMACTA
Placebo
Overall
28/45 (62%)a
7/22 (32%)
    12 to 17 years
10/16 (62%)
0/8 (0%)
    6 to 11 years
12/19 (63%)
3/9 (33%)
    1 to 5 years
6/10 (60%)
4/5 (80%)
Fewer pediatric patients treated with PROMACTA required rescue treatment during the randomized, double-blind period compared with placebo-treated patients (13% [6/45] versus 50% [11/22]).
Patients were permitted to reduce or discontinue baseline ITP therapy only during the open-label phase of the trial. Among 13 patients receiving other ITP therapy at baseline, 46% (6/13) reduced (n = 3) or discontinued (n = 3) concomitant therapy, mainly corticosteroids, without needing rescue therapy.
14.2 Chronic Hepatitis C-Associated Thrombocytopenia
The efficacy and safety of PROMACTA for the treatment of thrombocytopenia in adult patients with chronic hepatitis C were evaluated in two randomized, double-blind, placebo-controlled trials. The ENABLE1 study (NCT00516321) utilized peginterferon alfa-2a (PEGASYS®) plus ribavirin for antiviral treatment and the ENABLE2 study (NCT00529568) utilized peginterferon alfa-2b (PEGINTRON®) plus ribavirin. In both trials, patients with a platelet count of less than 75 x 109/L were enrolled and stratified by platelet count, screening HCV RNA, and HCV genotype. Patients were excluded if they had evidence of decompensated liver disease with Child-Pugh score greater than 6 (class B and C), history of ascites, or hepatic encephalopathy. The median age of the patients in both trials was 52 years, 63% were male, and 74% were Caucasian. Sixty-nine percent of patients had HCV genotypes 1, 4, 6, with the remainder genotypes 2 and 3. Approximately 30% of patients had been previously treated with interferon and ribavirin. The majority of patients (90%) had bridging fibrosis and cirrhosis, as indicated by noninvasive testing. A similar proportion (95%) of patients in both treatment groups had Child-Pugh class A (score 5 to 6) at baseline. A similar proportion of patients (2%) in both treatment groups had baseline international normalized ratio (INR) greater than 1.7. Median baseline platelet counts (approximately 60 x 109/L) were similar in both treatment groups. The trials consisted of 2 phases – a pre-antiviral treatment phase and an antiviral treatment phase. In the pre-antiviral treatment phase, patients received open-label PROMACTA to increase the platelet count to a threshold of greater than or equal to 90 x 109/L for ENABLE1 and greater than or equal to 100 x 109/L for ENABLE2. PROMACTA was administered at an initial dose of 25 mg once daily for 2 weeks and increased in 25-mg increments over 2- to 3-week periods to achieve the optimal platelet count to initiate antiviral therapy. The maximal time patients could receive open-label PROMACTA was 9 weeks. If threshold platelet counts were achieved, patients were randomized (2:1) to the same dose of PROMACTA at the end of the pre-treatment phase or to placebo. PROMACTA was administered in combination with pegylated interferon and ribavirin per their respective prescribing information for up to 48 weeks.
The efficacy of PROMACTA for both trials was evaluated by sustained virologic response (SVR) defined as the percentage of patients with undetectable HCV-RNA at 24Â weeks after completion of antiviral treatment. The median time to achieve the target platelet count greater than or equal to 90 x 109/L was approximately 2Â weeks. Ninety-five percent of patients were able to initiate antiviral therapy.
In both trials, a significantly greater proportion of patients treated with PROMACTA achieved SVR (see Table 20). The improvement in the proportion of patients who achieved SVR was consistent across subgroups based on baseline platelet count (less than 50 x 109/L versus greater than or equal to 50 x 109/L). In patients with high baseline viral loads (greater than or equal to 800,000), the SVR rate was 18% (82/452) for PROMACTA versus 8% (20/239) for placebo.
Table 20. ENABLE1 and ENABLE2: Sustained Virologic Response (SVR) in Adults With Chronic Hepatitis C Abbreviation: HCV, hepatitis C virus. aPROMACTA given in combination with peginterferon alfa-2a (180 mcg once weekly for 48 weeks for genotypes 1/4/6; 24 weeks for genotype 2 or 3) plus ribavirin (800 to 1,200 mg daily in 2 divided doses orally). bPROMACTA given in combination with peginterferon alfa-2b (1.5 mcg/kg once weekly for 48 weeks for genotypes 1/4/6; 24 weeks for genotype 2 or 3) plus ribavirin (800 to 1,400 mg daily in 2 divided doses orally). cTarget platelet count was ≥ 90 x 109/L for ENABLE1 and ≥ 100 x 109/L for ENABLE2. d p-value < 0.05 for PROMACTA versus placebo.
ENABLE1a
ENABLE2b
Pre-antiviral treatment phase
n = 715
n = 805
% Patients who achieved target platelet counts and initiated antiviral therapyc
95%
94%
Antiviral treatment phase
PROMACTA
n = 450
%
Placebo
n = 232
%
PROMACTA
n = 506
%
Placebo
n = 253
%
Overall SVRd
23
14
19
13
    HCV genotype 2,3
35
24
34
25
    HCV genotype 1, 4, 6
18
10
13
7
The majority of patients treated with PROMACTA (76%) maintained a platelet count greater than or equal to 50Â x 109/L compared with 19% for placebo. A greater proportion of patients on PROMACTA did not require any antiviral dose reduction as compared with placebo (45% versus 27%).
14.3 Severe Aplastic Anemia
First-Line Treatment of Severe Aplastic Anemia
PROMACTA in combination with h-ATG and cyclosporine was investigated in a single-arm, single-center, open-label sequential cohort trial (Study ETB115AUS01T, referred to as Study US01T [NCT01623167]) in patients with severe aplastic anemia who had not received prior immunosuppressive therapy (IST) with any ATG, alemtuzumab, or high dose cyclophosphamide. A total of 153 patients received PROMACTA in Study US01T in three sequential cohorts and an extension of the third cohort. The multiple cohorts received the same PROMACTA starting dose but differed by treatment start day and duration. The starting dose of PROMACTA for patients 12 years and older was 150 mg once daily (a reduced dose of 75 mg was administered for East-/Southeast-Asians), 75 mg once daily for pediatric patients aged 6 to 11 years (a reduced dose of 37.5 mg was administered for East-/Southeast-Asians), and 2.5 mg/kg once daily for pediatric patients aged 2 to 5 years (a reduced dose of 1.25 mg/kg was administered for East-/Southeast-Asians).
- Cohort 1 (n = 30): PROMACTA on Day 14 to Month 6 (D14-M6) plus h-ATG and cyclosporine
- Cohort 2 (n = 31): PROMACTA on Day 14 to Month 3 (D14-M3) plus h-ATG and cyclosporine
- Cohort 3 + Extension cohort [PROMACTA D1-M6 cohort] (n = 92): PROMACTA on Day 1 to Month 6 (D1-M6) plus h-ATG and cyclosporine (with all patients eligible to receive low dose of cyclosporine (maintenance dose) if they achieved a hematologic response at 6 months)
PROMACTA dose reductions were conducted for elevated platelet counts and hepatic impairment. Table 21 includes the dosages of h-ATG and cyclosporine administered in combination with PROMACTA in Study US01T.
Data from the Cohort 3 + Extension cohort support the efficacy of PROMACTA for the first-line treatment of patients with severe aplastic anemia (Table 22). The results presented in this section represent the findings from the Cohort 3 and Extension cohort (n = 92).
Table 21. Dosages of Immunosuppressive Therapy Administered With PROMACTA in Study US01T aDose of cyclosporine was adjusted to achieve the above recommended target trough levels; refer to the appropriate cyclosporine prescribing information. bCalculated as the midpoint between the ideal body weight and actual body weight.
Agent
Dose Administered in the Pivotal Trial
Horse antithymocyte globulin (h-ATG)
40 mg/kg/day, based on actual body weight, administered intravenously on Days 1 to 4 of the 6-month treatment period
Cyclosporinea (therapeutic dose for 6 months, from Day 1 to Month 6, adjusted to obtain a target therapeutic trough level between 200 mcg/L and 400 mcg/L)
Patients 12 years and older (total daily dose of 6 mg/kg/day)
3 mg/kg, based on actual body weight, orally every 12 hours for 6 months, starting on Day 1
Patients > 20 years of age with a body mass index > 35 or patients 12 to 20 years of age with a body mass index > 95th percentile:
3 mg/kg, based on adjusted body weightb, orally every 12 hours for 6 months, starting on Day 1
Patients 2 to 11 years of age (total daily dose of 12 mg/kg/day)
6 mg/kg, based on actual body weight, orally every 12 hours for 6 months, starting on Day 1
Patients 2 to 11 years of age with a body mass index > 95th percentile:
6 mg/kg, based on adjusted body weightb, orally every 12 hours for 6 months, starting on Day 1
Cyclosporine(maintenance dose, from Month 6 to Month 24)
For patients who achieve a hematologic response at 6 months
2 mg/kg/day administered orally at a fixed dose for an additional 18 months
In the PROMACTA D1-M6 cohort, the median age was 28 years (range, 5 to 82 years) with 16% and 28% of patients ≥ 65 years of age and < 17 years of age, respectively. Forty-six percent of patients were male and the majority of patients were White (62%). Patients weighing 12 kg or less or patients with ALT or AST > 5x upper limit of normal were excluded from the trial.
The efficacy of PROMACTA in combination with h-ATG and cyclosporine was established on the basis of complete hematological response at 6 months. A complete response was defined as hematological parameters meeting all 3 of the following values on 2 consecutive serial blood count measurements at least one week apart: absolute neutrophil count (ANC) > 1000/mcL, platelet count > 100 x 109/L and hemoglobin > 10 g/dL. A partial response was defined as blood counts no longer meeting the standard criteria for severe pancytopenia in severe aplastic anemia equivalent to 2 of the following values on 2 consecutive serial blood count measurements at least one week apart: ANC > 500/mcL, platelet count > 20 x 109/L, or reticulocyte count > 60,000/mcL. Overall response rate is defined as the number of partial responses plus complete responses.
Table 22. Study US01T: Hematologic Response in First-Line Treatment of Patients With Severe Aplastic Anemia Abbreviation: NE, not estimable. aThe number of patients who reached the 6-month assessment or withdrew earlier is the denominator for percentage calculation. bNumber of responders at any time.
PROMACTA D1-M6 + h-ATG + cyclosporinen = 92
Month 6, na
Overall response, n (%) [95% CI]
Complete response, n (%) [95% CI]
87 69 (79) [69, 87] 38 (44) [33, 55]
 Â
Median duration of overall response, nb
70
Months (95% CI)
24.3 (21.4, NE)
Median duration of complete response, nb
46
Months (95% CI)
24.3 (23.0, NE)
The overall and complete hematological response rates at Year 1 (n = 78) are 56.4% and 38.5% and at Year 2 (n = 62) are 38.7% and 30.6%, respectively.
Pediatric Patients
Thirty-four patients 2 to 16 years of age were enrolled in Study US01T. In the D1-M6 cohort, 7 and 17 out of 25 pediatric patients achieved a complete and overall response, respectively, at 6 months.
Refractory Severe Aplastic Anemia
PROMACTA was studied in a single-arm, single-center, open-label trial (Study ETB115AUS28T, referred to as Study US28T [NCT00922883]) in 43 patients with severe aplastic anemia who had an insufficient response to at least one prior immunosuppressive therapy and who had a platelet count less than or equal to 30 x 109/L. PROMACTA was administered at an initial dose of 50 mg once daily for 2 weeks and increased over 2-week periods up to a maximum dose of 150 mg once daily. The efficacy of PROMACTA in the study was evaluated by the hematologic response assessed after 12 weeks of treatment. Hematologic response was defined as meeting 1 or more of the following criteria: 1) platelet count increases to 20 x 109/L above baseline, or stable platelet counts with transfusion independence for a minimum of 8 weeks; 2) hemoglobin increase by greater than 1.5 g/dL, or a reduction in greater than or equal to 4 units of red blood cell (RBC) transfusions for 8 consecutive weeks; 3) ANC increase of 100% or an ANC increase greater than 0.5 x 109/L. PROMACTA was discontinued after 16 weeks if no hematologic response was observed. Patients who responded continued therapy in an extension phase of the trial.
The treated population had median age of 45 years (range, 17 to 77 years) and 56% were male. At baseline, the median platelet count was 20 x 109/L, hemoglobin was 8.4 g/dL, ANC was 0.58 x 109/L, and absolute reticulocyte count was 24.3 x 109/L. Eighty-six percent of patients were red blood cell (RBC) transfusion dependent and 91% were platelet transfusion dependent. The majority of patients (84%) received at least 2 prior immunosuppressive therapies. Three patients had cytogenetic abnormalities at baseline.
Table 23 presents the efficacy results.
Table 23. Study US28T: Hematologic Response in Patients With Refractory Severe Aplastic Anemia aIncludes single- and multi-lineage. bNRÂ =Â not reached due to few events (relapsed).
Outcome
PROMACTA
n = 43
Response ratea, n (%)
    95% CI (%)
17 (40)
(25, 56)
Median of duration of response in months (95% CI)
NRb (3.0, NRb)
In the 17 responders, the platelet transfusion-free period ranged from 8 to 1096Â days with a median of 200Â days, and the RBC transfusion-free period ranged from 15 to 1082Â days with a median of 208Â days.
In the extension phase, 8 patients achieved a multi-lineage response; 4 of these patients subsequently tapered off treatment with PROMACTA and maintained the response (median follow up: 8.1Â months, range, 7.2 to 10.6Â months).
16 How Supplied/storage And Handling
16.1 Tablets
- The 12.5-mg tablets are round, biconvex, white, film-coated tablets debossed with “GS MZ1” and 12.5 on one side and are available in bottles of 30: NDC 0078-0684-15
- The 25-mg tablets are round, biconvex, orange, film-coated tablets debossed with “GS NX3” and 25 on one side and are available in bottles of 30: NDC 0078-0685-15
- The 50-mg tablets are round, biconvex, blue, film-coated tablets debossed with “GS UFU” and 50 on one side and are available:
- Bottles of 14Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â NDC 0078-0686-55
- Bottles of 30Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â Â NDC 0078-0686-15
- The 75-mg tablets are round, biconvex, pink, film-coated tablets debossed with “GS FFS” and 75 on one side and are available in bottles of 30: NDC 0078-0687-15
Store at room temperature between 20°C to 25°C (68°F to 77°F); excursions permitted between 15°C and 30°C (59°F and 86°F) [see USP Controlled Room Temperature]. Dispense in original bottle.
16.2 For Oral Suspension
- The 12.5-mg for oral suspension is a reddish-brown to yellow powder in unit-dose packets, copackaged in a kit with a 40-cc reconstitution vessel, a threaded closure with syringe-port capability, and 30 single-use oral dosing syringes.
Each kit (NDC 0078-0972-61) contains 30 packets: NDC 0078-0972-19
- The 25-mg for oral suspension is a reddish-brown to yellow powder in unit-dose packets, copackaged in a kit with a 40-cc reconstitution vessel, a threaded closure with syringe-port capability, and 30 single-use oral dosing syringes.
Each kit (NDC 0078-0697-61) contains 30 packets: NDC 0078-0697-19
Store at room temperature between 20°C to 25°C (68°F to 77°F); excursions permitted between 15°C and 30°C (59°F and 86°F) [see USP Controlled Room Temperature]. Following reconstitution, the product should be administered immediately but may be stored for a maximum period of 30 minutes between 20°C to 25°C (68°F to 77°F); excursions permitted between 15°C and 30°C (59°F and 86°F) [see USP Controlled Room Temperature]. Throw away (discard) the mixture if not used within 30 minutes.
17 Patient Counseling Information
Advise the patient or caregiver to read the FDA-approved patient labeling (Medication Guide and Instructions for Use).
Prior to treatment, patients should fully understand and be informed of the following risks and considerations for PROMACTA:
Risks
Hepatotoxicity
- Therapy with PROMACTA may be associated with hepatobiliary laboratory abnormalities [see Warnings and Precautions (5.2)].
- Advise patients with chronic hepatitis C and cirrhosis that they may be at risk for hepatic decompensation when receiving PROMACTA with alfa interferon therapy [see Warnings and Precautions (5.1)].
- Advise patients that they should report any of the following signs and symptoms of liver problems to their healthcare provider right away [see Warnings and Precautions (5.2)].
- yellowing of the skin or the whites of the eyes (jaundice)
- unusual darkening of the urine
- unusual tiredness
- right upper stomach area pain
- confusion
- swelling of the stomach area (abdomen)
Risk of Bleeding Upon PROMACTA Discontinuation
- Advise patients that thrombocytopenia and risk of bleeding may reoccur upon discontinuing PROMACTA, particularly if PROMACTA is discontinued while the patient is on anticoagulants or antiplatelet agents. Advise patients that during therapy with PROMACTA, they should continue to avoid situations or medications that may increase the risk for bleeding.
Thrombotic/Thromboembolic Complications
- Advise patients that too much PROMACTA may result in excessive platelet counts and a risk for thrombotic/thromboembolic complications [see Warnings and Precautions (5.4)].
Cataracts
- Advise patients to have a baseline ocular examination prior to administration of PROMACTA and be monitored for signs and symptoms of cataracts during therapy [see Warnings and Precautions (5.5)].
Drug Interactions
- Advise patients to take PROMACTA at least 2 hours before or 4 hours after calcium-rich foods, mineral supplements, and antacids which contain polyvalent cations, such as iron, calcium, aluminum, magnesium, selenium, and zinc [see Dosage and Administration (2.4), Drug Interactions (7.1)].
Lactation
- Advise women not to breastfeed during treatment with PROMACTA [see Use in Specific Populations (8.2)].
Administration of PROMACTA
- For patients with persistent or chronic ITP, therapy with PROMACTA is administered to achieve and maintain a platelet count greater than or equal to 50 x 109/L as necessary to reduce the risk for bleeding [see Indications and Usage (1.1)].
- For patients with chronic hepatitis C, therapy with PROMACTA is administered to achieve and maintain a platelet count necessary to initiate and maintain antiviral therapy with pegylated interferon and ribavirin [see Indications and Usage (1.2)].
- Advise patients to take PROMACTA without a meal or with a meal low in calcium (≤ 50 mg) and at least 2 hours before or 4 hours after other medications (e.g., antacids) and calcium-rich foods [see Dosage and Administration (2.4)].
- Prior to use of the oral suspension, ensure patients or caregivers receive training on proper dosing, preparation, and administration [see Dosage and Administration (2.4)].
- Inform patients or caregivers how many packets to administer to get the full dose [see Instructions for Use].
- Inform patients or caregivers to use a new oral dosing syringe to prepare each dose of PROMACTA for oral suspension [see Instructions for Use].
The following are registered trademarks of their respective owners: PEGASYS/Hoffmann-La Roche Inc.; PEGINTRON/Schering Corporation.
Distributed by:Novartis Pharmaceuticals CorporationEast Hanover, New Jersey 07936
© Novartis
T2023-13
Spl Medguide Section
This Medication Guide has been approved by the U.S. Food and Drug Administration Revised: March 2023 MEDICATION GUIDE PROMACTA® (pro-MAC-ta)(eltrombopag)tablets PROMACTA® (pro-MAC-ta)(eltrombopag)for oral suspension What is the most important information I should know about PROMACTA? PROMACTA can cause serious side effects, including: Liver problems:
- If you have chronic hepatitis C virus and take PROMACTA with interferon and ribavirin treatment, PROMACTA may increase your risk of liver problems. If your healthcare provider tells you to stop your treatment with interferon and ribavirin, you will also need to stop taking PROMACTA.
- PROMACTA may increase your risk of liver problems that may be severe and possibly life threatening. Your healthcare provider will do blood tests to check your liver function before you start taking PROMACTA and during your treatment. Your healthcare provider may stop your treatment with PROMACTA if you have changes in your liver function blood tests.
Tell your healthcare provider right away if you have any of these signs and symptoms of liver problems:
- yellowing of the skin or the whites of the eyes (jaundice)
- unusual darkening of the urine
- unusual tiredness
- right upper stomach area (abdomen) pain
- confusion
- swelling of the stomach area (abdomen)
See “What are the possible side effects of PROMACTA?” for other side effects of PROMACTA. What is PROMACTA? PROMACTA is a prescription medicine used to treat adults and children 1 year of age and older with low blood platelet counts due to persistent or chronic immune thrombocytopenia (ITP), when other medicines to treat ITP or surgery to remove the spleen have not worked well enough.
PROMACTA is also used to treat people with:
- low blood platelet counts due to chronic hepatitis C virus (HCV) infection before and during treatment with interferon.
- severe aplastic anemia (SAA) in combination with other medicines to treat SAA, as the first treatment for adults and children 2 years of age and older.
- severe aplastic anemia (SAA) when other medicines to treat SAA have not worked well enough.
PROMACTA is used to try to raise platelet counts in order to lower your risk for bleeding.
PROMACTA is not used to make platelet counts normal.
PROMACTA is not for use in people with a pre-cancerous condition called myelodysplastic syndrome (MDS), or in people with low platelet counts caused by certain other medical conditions or diseases.
It is not known if PROMACTA is safe and effective when used with other antiviral medicines to treat chronic hepatitis C.
It is not known if PROMACTA is safe and effective in children:
- younger than 1 year with ITP
- with low blood platelet counts due to chronic hepatitis C
- whose severe aplastic anemia (SAA) has not improved after previous treatments.
- younger than 2 years when used in combination with other medicines to treat SAA as the first treatment for SAA.
Before you take PROMACTA, tell your healthcare provider about all of your medical conditions, including if you:
- have liver problems
- have a precancerous condition called MDS or a blood cancer
- have or had a blood clot
- have a history of cataracts
- have had surgery to remove your spleen (splenectomy)
- have bleeding problems
- are of East-/Southeast-Asian ancestry. You may need a lower dose of PROMACTA.
- are pregnant or plan to become pregnant. It is not known if PROMACTA will harm an unborn baby. Tell your healthcare provider if you become pregnant or think you may be pregnant during treatment with PROMACTA.
- Females who are able to become pregnant, should use effective birth control (contraception) during treatment with PROMACTA and for at least 7 days after stopping treatment with PROMACTA. Talk to your healthcare provider about birth control methods that may be right for you during this time.
- are breastfeeding or plan to breastfeed. You should not breastfeed during your treatment with PROMACTA. Talk to your healthcare provider about the best way to feed your baby during this time.
- Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. PROMACTA may affect the way certain medicines work. Certain other medicines may affect the way PROMACTA works.
Especially tell your healthcare provider if you take:
- certain medicines used to treat high cholesterol, called “statins”
- a blood thinner medicine
Certain medicines may keep PROMACTA from working correctly. Take PROMACTA at least 2Â hours before or 4Â hours after taking these products:
- antacid medicine used to treat stomach ulcers or heartburn
- multivitamins or products that contain iron, calcium, aluminum, magnesium, selenium, and zinc which may be found in mineral supplements
Ask your healthcare provider if you are not sure if your medicine is one that is uled above.Know the medicines you take. Keep a ul of them and show it to your healthcare provider and pharmacist when you get a new medicine.
How should I take PROMACTA?
- Take PROMACTA exactly as your healthcare provider tells you to take it. Your healthcare provider will prescribe the dose of PROMACTA tablets or PROMACTA for oral suspension that is right for you.
- If your healthcare provider prescribes PROMACTA tablets, take PROMACTA tablets whole. Do not split, chew, or crush PROMACTA tablets and do not mix with food or liquids.
- If your healthcare provider prescribes PROMACTA for oral suspension, see the “Instructions for Use” that comes with your medicine for instructions on how to correctly mix and take a dose of PROMACTA.
- Use a new single-use oral dosing syringe to prepare each dose of PROMACTA for oral suspension. Do not re-use the oral dosing syringe.
- Do not stop taking PROMACTA without talking with your healthcare provider first. Do not change your dose or schedule for taking PROMACTA unless your healthcare provider tells you to change it.
- Take PROMACTA without a meal or with a meal low in calcium (50 mg or less) and at least 2 hours before or 4 hours after eating calcium-rich foods, such as dairy products, calcium-fortified juices, and certain fruits and vegetables.
- If you miss a dose of PROMACTA, wait and take your next scheduled dose. Do not take more than 1 dose of PROMACTA in 1 day.
- If you take too much PROMACTA, you may have a higher risk of serious side effects. Call your healthcare provider right away.
- Your healthcare provider will check your platelet count during your treatment with PROMACTA and change your dose of PROMACTA as needed.
- Tell your healthcare provider about any bruising or bleeding that happens while you take and after you stop taking PROMACTA.
- If you have SAA, your healthcare provider may do tests to monitor your bone marrow during treatment with PROMACTA.
What should I avoid while taking PROMACTA? Avoid situations and medicines that may increase your risk of bleeding. What are the possible side effects of PROMACTA? PROMACTA may cause serious side effects, including:
- See “What is the most important information I should know about PROMACTA?”
- Increased risk of worsening of a precancerous blood condition called myelodysplastic syndrome (MDS) to acute myelogenous leukemia (AML). PROMACTA is not for use in people with a precancerous condition called myelodysplastic syndromes (MDS). See “What is PROMACTA?” If you have MDS and receive PROMACTA, you have an increased risk that your MDS condition may worsen and become a blood cancer called AML. If your MDS worsens to become AML, you may have an increased risk of death from AML.
- High platelet counts and higher risk for blood clots. Your risk of getting a blood clot is increased if your platelet count is too high during treatment with PROMACTA. Your risk of getting a blood clot may also be increased during treatment with PROMACTA if you have normal or low platelet counts. You may have severe problems or die from some forms of blood clots, such as clots that travel to the lungs or that cause heart attacks or strokes. Your healthcare provider will check your blood platelet counts, and change your dose or stop PROMACTA if your platelet counts get too high. Tell your healthcare provider right away if you have signs and symptoms of a blood clot in the leg, such as swelling, pain, or tenderness in your leg. People with chronic liver disease may be at risk for a type of blood clot in the stomach area (abdomen). Tell your healthcare provider right away if you have stomach-area (abdomen) pain, nausea, vomiting, or diarrhea as these may be symptoms of this type of blood clot.
- New or worsened cataracts (a clouding of the lens in the eye). New or worsened cataracts can happen in people taking PROMACTA. Your healthcare provider will check your eyes before and during your treatment with PROMACTA. Tell your healthcare provider about any changes in your eyesight while taking PROMACTA.
The most common side effects of PROMACTA in adults and children include:
- low red blood cell count (anemia)
- nausea
- fever
- abnormal liver function tests
- cough
- tiredness
- headache
- diarrhea
Laboratory tests may show abnormal changes to the cells in your bone marrow.Tell your healthcare provider if you have any side effect that bothers you or that does not go away. These are not all of the possible side effects of PROMACTA. For more information, ask your healthcare provider or pharmacist.Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. How should I store PROMACTA tablets and PROMACTA for oral suspension? Tablets: For oral suspension:
- Store PROMACTA tablets at room temperature between 68°F to 77°F (20°C to 25°C).
- Keep PROMACTA in the bottle given to you.
Keep PROMACTA and all medicines out of the reach of children.
- Store PROMACTA for oral suspension at room temperature between 68°F to 77°F (20°C to 25°C).
- After mixing, PROMACTA should be taken right away but may be stored for no more than 30 minutes between 68°F to 77°F (20°C to 25°C). Throw away (discard) the mixture if not used within 30 minutes.
General information about the safe and effective use of PROMACTA Medicines are sometimes prescribed for purposes other than those uled in a Medication Guide. Do not use PROMACTA for a condition for which it was not prescribed. Do not give PROMACTA to other people, even if they have the same symptoms that you have. It may harm them. You can ask your healthcare provider or pharmacist for information about PROMACTA that is written for health professionals. What are the ingredients in PROMACTA? Tablets Active ingredient: eltrombopag olamine Inactive ingredients:
- Tablet Core: magnesium stearate, mannitol, microcrystalline cellulose, povidone, and sodium starch glycolate.
- Coating: FD&C Blue No. 2 aluminum lake (50-mg tablet), FD&C Yellow No. 6 aluminum lake (25-mg tablet), hypromellose, Iron Oxide Black and Iron Oxide Red (75-mg tablet), polyethylene glycol 400, polysorbate 80 (12.5-mg tablet), or titanium dioxide.
For oral suspension Active ingredient: eltrombopag olamine Inactive ingredients: mannitol, sucralose, and xanthan gum Distributed by: Novartis Pharmaceuticals Corporation, East Hanover, New Jersey 07936© Novartis For more information about PROMACTA, go to www.PROMACTA.com or call 1-888-669-6682.
T2023-14
INSTRUCTIONS FOR USE SECTION
INSTRUCTIONS FOR USE
PROMACTA® [pro-MAC-ta]
(eltrombopag)
for oral suspension
Read all the Instructions for Use and follow the steps below to mix and give a dose of PROMACTA for oral suspension.
Important information you need to know before taking PROMACTA for oral suspension:
- Do not take PROMACTA for oral suspension or give it to someone else until you have been shown how to properly mix and give a dose of PROMACTA for oral suspension. Your healthcare provider or nurse will show you how to mix and give a dose of PROMACTA for oral suspension properly.
- PROMACTA for oral suspension must be mixed with cool or cold water only. Do not use hot water to prepare the oral suspension.
- Give the dose of suspension right away after mixing with water. If the medicine is not given within 30 minutes, you will have to mix a new dose. Throw away (discard) the unused mixture into the trash. Do not pour it down the drain.
- If PROMACTA for oral suspension comes in contact with your skin, wash the skin right away with soap and water. Call your healthcare provider if you have a skin reaction or if you have any questions. If you spill any powder or liquid, follow the clean-up instructions in Step 12.
- Contact your healthcare provider or pharmacist if you have any questions about how to mix or give PROMACTA to your child, or if you damage or lose any of the supplies in your kit.
- Do not re-use the oral dosing syringe. Use a new single-use oral dosing syringe to prepare each dose of PROMACTA for oral suspension.
- After you have used all 30 packets, throw all the remaining supplies (mixing bottle, lid with cap, and oral dosing syringe) away in the trash.
Each PROMACTA for oral suspension kit contains the following supplies:
30 packets of PROMACTA for oral suspension

1 Reusable mixing bottle with lid and cap
30 Single-use 20-mL oral dosing syringes (Use a new (single-use) oral dosing syringe to prepare each dose of PROMACTA for oral suspension)
You will need the following to give a dose of PROMACTA for oral suspension.
From the kit:
- prescribed number of packets
- 1 reusable mixing bottle with lid and cap. Note: Due to its small size, the cap may pose a danger of choking to small children.
- 1 single-use 20-mL oral dosing syringe (Use a new (single-use) oral dosing syringe to prepare each dose of PROMACTA for oral suspension)
Not included in the kit:
- 1 clean glass or cup filled with drinking water
- scissors to cut packet
- paper towels or disposable cloth
- disposable gloves (optional)
This Instructions for Use has been approved by the U.S. Food and Drug Administration. Revised: April 2020
How do I prepare a dose of PROMACTA for oral suspension?
Step 1. Make sure that the mixing bottle, cap, lid and oral dosing syringe are dry before use. Remove the lid from the mixing bottle.
- Prepare a clean, flat work surface.
- Wash and dry your hands before preparing the medicine.
Step 2. Fill the oral dosing syringe with 20Â mL of drinking water from the glass or cup.
- Start with the plunger pushed all the way into the syringe.
- Place the tip of the oral dosing syringe all the way into the water and pull back on the plunger to the 20Â mL mark on the barrel of the oral dosing syringe.
Note: Use a new (single-use) oral dosing syringe to prepare each dose of PROMACTA for oral suspension.
Figure 1.
Step 3. Place the tip of the oral dosing syringe into the open mixing bottle. Empty water into open mixing bottle by slowly pushing the plunger all the way into the oral dosing syringe.
Figure 2. Step 4. Take only the prescribed number of packets for one dose out of the kit. You may need to use more than one packet to prepare the entire dose. 12.5 mg packets  Dose                        Number of 12.5 mg Packets Needed   12.5 mg dose          1 packet  25 mg dose             2 packets  50 mg dose             4 packets  75 mg dose             6 packets 25 mg packets  Dose                        Number of 25 mg Packets Needed   12.5 mg dose          1 packet (Note: See Step 9 for instructions on how to give a 12.5-mg dose using a 25-mg packet.)  25 mg dose             1 packet  50 mg dose             2 packets  75 mg dose             3 packets
Step 5. Add the prescribed number of packets to the mixing bottle.
- Tap the top of each packet to make sure the contents fall to the bottom.
- Cut off the top of the packet with scissors and empty the entire contents of the packet into the mixing bottle.
- Make sure not to spill the powder outside the mixing bottle.
Figure 3.
Step 6. Screw the lid tightly onto the mixing bottle. Make sure the cap is pushed onto the lid.
Step 7. Gently and slowly shake the mixing bottle back and forth for at least 20Â seconds to mix the water with the powder.
- To prevent the mixture from foaming, do not shake the mixing bottle hard.
Figure 4.
How should I give a dose of PROMACTA for oral suspension?
Step 8. Make sure the plunger is pushed all the way into the oral dosing syringe. Pull cap off the mixing bottle lid and insert the tip of the oral dosing syringe into the hole in the lid.
Step 9. Transfer the mixture into the oral dosing syringe. The liquid will be dark brown in color.
- Turn the mixing bottle upside down along with the oral dosing syringe.
- Pull back the plunger:
12.5-mg packet
o until all the medicine is in the oral dosing syringe (12.5-mg, 25-mg, 50-mg, or 75-mg dose)
25-mg packet
o to the 10 mL mark on the oral dosing syringe for a 12.5-mg dose only ORo until all the medicine is in the oral dosing syringe (25-mg, 50-mg, or 75-mg dose).Figure 5.
Step 10. Return the mixing bottle to the upright position and remove the oral dosing syringe from the mixing bottle.
Figure 6.
Step 11. Giving a dose of PROMACTA for oral suspension to a child.
- Place the tip of the oral dosing syringe into the inside of the child’s cheek.
- Slowly push the plunger all the way down to give the entire dose. Make sure the child has time to swallow the medicine.
Figure 7.
How should I clean up?
Step 12. Carefully clean up any spill of the powder or suspension with a damp paper towel or disposable cloth.
- To avoid possibly staining your skin, consider using disposable gloves.
- Throw away (discard) used paper towel or disposable cloth and gloves in the trash.
Step 13. Clean the mixing supplies.
- Do not reuse any of the mixture remaining in the mixing bottle.
- Throw away (discard) any mixture remaining in the mixing bottle in the trash. Do not pour down the drain.
- Throw away (discard) the used oral dosing syringe. Use a new (single-use) oral dosing syringe to prepare each dose of PROMACTA for oral suspension.
- Rinse the mixing bottle and lid under running water and air dry. The mixing bottle may become stained from the medicine. This is normal.
- Wash hands with soap and water.
How should I store PROMACTA for oral suspension?
- Store PROMACTA for oral suspension at room temperature between 68°F to 77°F (20°C to 25°C).
- After mixing, PROMACTA should be taken right away but may be stored for no more than 30 minutes between 68°F to 77°F (20°C to 25°C). Throw away (discard) the mixture if not used within 30 minutes.
Keep PROMACTA and all medicines out of the reach of children.
Distributed by:Novartis Pharmaceuticals CorporationEast Hanover, New Jersey 07936
© Novartis
T2020-61
Principal Display Panel
NDC 0078-0684-15
Rx only
Promacta® (eltrombopag) Tablets12.5 mg*
Swallow tablets whole. Do notsplit, chew, or crush tablets.
Dispense with Medication Guideattached or provided separately.
NOVARTIS
30 Tablets

Principal Display Panel
NDC 0078-0685-15
Rx only
Promacta® (eltrombopag) Tablets25 mg*
Swallow tablets whole. Do notsplit, chew, or crush tablets.
Dispense with Medication Guideattached or provided separately.
NOVARTIS
30 Tablets

Principal Display Panel
NDC 0078-0686-15
Rx only
Promacta® (eltrombopag) Tablets50 mg*
Swallow tablets whole. Do notsplit, chew, or crush tablets.
Dispense with Medication Guideattached or provided separately.
NOVARTIS
30 Tablets

Principal Display Panel
NDC 0078-0687-15
Rx only
Promacta® (eltrombopag) Tablets75 mg*
Swallow tablets whole. Do notsplit, chew, or crush tablets.
Dispense with Medication Guideattached or provided separately.
NOVARTIS
30 Tablets

Principal Display Panel
NDC 0078-0972-61
Rx only
Promacta® (eltrombopag)for Oral Suspension
12.5 mg
Dispense with Medication Guide enclosed or provided separately.
30 Packets
NOVARTIS

Principal Display Panel
NDC 0078-0697-61
Rx only
Promacta® (eltrombopag)for Oral Suspension
25 mg
Dispense with Medication Guide enclosed or provided separately.
30 Packets
NOVARTIS

DISCLAIMER:
"This tool does not provide medical advice, and is for informational and educational purposes only, and is not a substitute for professional medical advice, treatment or diagnosis. Call your doctor to receive medical advice. If you think you may have a medical emergency, please dial 911."
"Do not rely on openFDA to make decisions regarding medical care. While we make every effort to ensure that data is accurate, you should assume all results are unvalidated. We may limit or otherwise restrict your access to the API in line with our Terms of Service."
"This product uses publicly available data from the U.S. National Library of Medicine (NLM), National Institutes of Health, Department of Health and Human Services; NLM is not responsible for the product and does not endorse or recommend this or any other product."
PillSync may earn a commission via links on our site
