everolimus (everolimus 7.5 mg) Dailymed
Generic: everolimus is used for the treatment of Graft vs Host Disease Pregnancy

IMPRINT: M EVR7
SHAPE: oval
COLOR: white
All Imprints
everolimus tablet - m evr5 oval white
everolimus 10 mg - m evr10 oval white
everolimus tablet - m evr7 oval white
everolimus tablet - m evr2 oval white

Go PRO for all pill images
1 Indications And Usage
Everolimus tablets are a kinase inhibitor indicated for the treatment of:
• Postmenopausal women with advanced hormone receptor-positive, HER2-negative breast cancer in combination with exemestane after failure of treatment with letrozole or anastrozole. (1.1 )• Adults with progressive, well-differentiated, non-functional neuroendocrine tumors (NET) of gastrointestinal (GI) or lung origin that are unresectable, locally advanced or metastatic. Limitations of Use: Everolimus tablets are not indicated for the treatment of patients with functional carcinoid tumors. (1.2 )• Adults with renal angiomyolipoma and tuberous sclerosis complex (TSC), not requiring immediate surgery. (1.4 )
Everolimus tablets are a kinase inhibitor indicated for the treatment of adult and pediatric patients aged 1 year and older with TSC who have subependymal giant cell astrocytoma (SEGA) that requires therapeutic intervention but cannot be curatively resected. (1.5 )
1.1Hormone Receptor-Positive, HER2-Negative Breast Cancer
Everolimus tablets are indicated for the treatment of postmenopausal women with advanced hormone receptor-positive, HER2-negative breast cancer in combination with exemestane, after failure of treatment with letrozole or anastrozole.
1.2Neuroendocrine Tumors (NET)
Everolimus tablets are indicated for the treatment of adult patients with progressive, well-differentiated, non-functional NET of gastrointestinal (GI) or lung origin with unresectable, locally advanced or metastatic disease.
Limitations of Use: Everolimus tablets are not indicated for the treatment of patients with functional carcinoid tumors [see Clinical Studies (14.2)].
1.4Tuberous Sclerosis Complex (TSC)-Associated Renal Angiomyolipoma
Everolimus tablets are indicated for the treatment of adult patients with renal angiomyolipoma and TSC, not requiring immediate surgery.
1.5Tuberous Sclerosis Complex (TSC)-Associated Subependymal Giant Cell Astrocytoma (SEGA)
Everolimus tablets are indicated in adult and pediatric patients aged 1 year and older with TSC for the treatment of SEGA that requires therapeutic intervention but cannot be curatively resected.
2 Dosage And Administration
Do not combine everolimus tablets and AFINITOR DISPERZ® to achieve the total daily dose. (2.1 )
Modify the dose for patients with hepatic impairment or for patients taking drugs that inhibit or induce P-glycoprotein (P-gp) and CYP3A4. (2.1 )
Â
Breast Cancer:
• 10 mg orally once daily. (2.2 )
NET:
• 10 mg orally once daily. (2.3 )
TSC-Associated Renal Angiomyolipoma:
• 10 mg orally once daily. (2.5 )
TSC-Associated SEGA:
• 4.5 mg/m2 orally once daily; adjust dose to attain trough concentrations of 5-15 ng/mL. (2.6 ,2.8 )2.1Important Dosage Information
• Everolimus tablets and AFINITOR DISPERZ are two different dosage forms. Select the recommended dosage form based on the indication [see Indications and Usage (1)]. Do not combine everolimus tablets and AFINITOR DISPERZ to achieve the total dose.• Modify the dosage for patients with hepatic impairment or for patients taking drugs that inhibit or induce P-glycoprotein (P-gp) and CYP3A4 [see Dosage and Administration (2.10, 2.11, 2.12)].2.2Recommended Dosage for Hormone Receptor-Positive, HER2-Negative Breast Cancer
The recommended dosage of everolimus tablets is 10 mg orally once daily until disease progression or unacceptable toxicity.
2.3Recommended Dosage for Neuroendocrine Tumors (NET)
2.5Recommended Dosage for Tuberous Sclerosis Complex (TSC)-Associated Renal Angiomyolipoma
2.6Recommended Dosage for Tuberous Sclerosis Complex (TSC)-Associated Subependymal Giant Cell Astrocytoma (SEGA)
The recommended starting dosage of everolimus is 4.5 mg/m2 orally once daily until disease progression or unacceptable toxicity [see Dosage and Administration (2.8)].
2.8Therapeutic Drug Monitoring (TDM) and Dose Titration for Tuberous Sclerosis Complex (TSC)-Associated Subependymal Giant Cell Astrocytoma (SEGA)
• Monitor everolimus whole blood trough concentrations at time points recommended in Table 1.• Titrate the dose to attain trough concentrations of 5 ng/mL to 15 ng/mL.• Adjust the dose using the following equation: New dose* = current dose x (target concentration divided by current concentration) *The maximum dose increment at any titration must not exceed 5 mg. Multiple dose titrations may be required to attain the target trough concentration.• When possible, use the same assay and laboratory for TDM throughout treatment.
Table 1: Recommended Timing of Therapeutic Drug Monitoring Abbreviation: P-gp, P-glycoprotein.
Event
When to Assess Trough Concentrations After Event
Initiation of everolimus
1 to 2 weeks
Modification of everolimus dose
1 to 2 weeks
Switch between everolimus tablets and AFINITOR DISPERZ
1 to 2 weeks
Initiation or discontinuation of P-gp and moderate CYP3A4 inhibitor
2 weeks
Initiation or discontinuation of P-gp and strong CYP3A4 inducer
2 weeks
Change in hepatic function
2 weeks
Stable dose with changing body surface area (BSA)
Every 3 to 6 months
Stable dose with stable BSA
Every 6 to 12 months
2.9Dosage Modifications for Adverse Reactions
Table 2 summarizes recommendations for dosage modifications of everolimus for the management of adverse reactions.
Table 2: Recommended Dosage Modifications for Everolimus for Adverse Reactions
Adverse Reaction
Severity
Dosage Modification
Non-infectious
pneumonitis
[see Warnings and Precautions (5.1)]
Grade 2
Withhold until improvement to Grade 0 or 1. Resume at 50% of previous dose; change to every other day dosing if the reduced dose is lower than the lowest available strength.
Â
Permanently discontinue if toxicity does not resolve or improve to Grade 1 within 4 weeks.
Grade 3
Withhold until improvement to Grade 0 or 1. Resume at 50% of previous dose; change to every other day dosing if the reduced dose is lower than the lowest available strength.
Â
If toxicity recurs at Grade 3, permanently discontinue.
Grade 4
Permanently discontinue.
Stomatitis
[see Warnings and Precautions (5.5)]
Grade 2
Withhold until improvement to Grade 0 or 1. Resume at same dose.
Â
If recurs at Grade 2, withhold until improvement to Grade 0 or 1. Resume at 50% of previous dose; change to every other day dosing if the reduced dose is lower than the lowest available strength.
Grade 3
Withhold until improvement to Grade 0 or 1. Resume at 50% of previous dose; change to every other day dosing if the reduced dose is lower than the lowest available strength.
Grade 4
Permanently discontinue.
Metabolic events
(e.g., hyperglycemia,
dyslipidemia)
[see Warnings and Precautions (5.9)]
Grade 3
Withhold until improvement to Grade 0, 1, or 2. Resume at 50% of previous dose; change to every other day dosing if the reduced dose is lower than the lowest available strength.
Grade 4
Permanently discontinue.
Other non-hematologic toxicities
Grade 2
If toxicity becomes intolerable, withhold until improvement to Grade 0 or 1. Resume at same dose.
Â
If toxicity recurs at Grade 2, withhold until improvement to Grade 0 or 1. Resume at 50% of previous dose; change to every other day dosing if the reduced dose is lower than the lowest available strength.
Grade 3
Withhold until improvement to Grade 0 or 1. Consider resuming at 50% of previous dose; change to every other day dosing if the reduced dose is lower than the lowest available strength.
Â
If recurs at Grade 3, permanently discontinue.
Grade 4
Permanently discontinue.
Thrombocytopenia
[see Warnings and Precautions (5.10)]
Grade 2
Withhold until improvement to Grade 0 or 1. Resume at same dose.
Grade 3
OR
Grade 4
Withhold until improvement to Grade 0 or 1. Resume at 50% of previous dose; change to every other day dosing if the reduced dose is lower than the lowest available strength.
Neutropenia
[see Warnings and Precautions (5.10)]
Grade 3
Withhold until improvement to Grade 0, 1, or 2. Resume at same dose.
Grade 4
Withhold until improvement to Grade 0, 1, or 2. Resume at 50% of previous dose; change to every other day dosing if the reduced dose is lower than the lowest available strength.
Febrile neutropenia
[see Warnings and Precautions (5.10)]
Grade 3
Withhold until improvement to Grade 0, 1, or 2, and no fever. Resume at 50% of previous dose; change to every other day dosing if the reduced dose is lower than the lowest available strength.
Grade 4
Permanently discontinue.
2.10Dosage Modifications for Hepatic Impairment
The recommended dosages of everolimus for patients with hepatic impairment are described in Table 3 [see Use in Specific Populations (8.6)]:
Table 3: Recommended Dosage Modifications for Patients With Hepatic Impairment Abbreviations: NET, Neuroendocrine Tumors; SEGA, Subependymal Giant Cell Astrocytoma; TSC, Tuberous Sclerosis Complex.
Indication
Dose Modification for Everolimus
Breast Cancer, NET, and TSC-Associated Renal Angiomyolipoma
• Mild hepatic impairment (Child-Pugh class A) – 7.5 mg orally once daily; decrease the dose to 5 mg orally once daily if a dose of 7.5 mg once daily is not tolerated.• Moderate hepatic impairment (Child-Pugh class B) – 5 mg orally once daily; decrease the dose to 2.5 mg orally once daily if a dose of 5 mg once daily is not tolerated.• Severe hepatic impairment (Child-Pugh class C) – 2.5 mg orally once daily if the desired benefit outweighs the risk; do not exceed a dose of 2.5 mg once daily.
TSC-Associated SEGA
• Severe hepatic impairment (Child-Pugh class C) – 2.5 mg/m2 orally once daily.• Adjust dose based on everolimus trough concentrations as recommended [see Dosage and Administration (2.8)].2.11Dosage Modifications for P-gp and CYP3A4 Inhibitors
• Avoid the concomitant use of P-gp and strong CYP3A4 inhibitors [see Drug Interactions (7.1)].• Avoid ingesting grapefruit and grapefruit juice.• Reduce the dose for patients taking everolimus with a P-gp and moderate CYP3A4 inhibitor as recommended in Table 4 [see Drug Interactions (7.1), Clinical Pharmacology (12.3)].
Table 4: Recommended Dosage Modifications for Concurrent Use of Everolimus With a P-gp and Moderate CYP3A4 Inhibitor
Indication
Dose Modification for Everolimus
Breast Cancer, NET, and TSC-Associated Renal Angiomyolipoma
• Reduce dose to 2.5 mg once daily.• May increase dose to 5 mg once daily if tolerated.• Resume dose administered prior to inhibitor initiation, once the inhibitor is discontinued for 3 days.
TSC-Associated SEGA
• Reduce the daily dose by 50%.• Change to every other day dosing if the reduced dose is lower than the lowest available strength.• Resume dose administered prior to inhibitor initiation, once the inhibitor is discontinued for 3 days.• Assess trough concentrations when initiating and discontinuing the inhibitor [see Dosage and Administration (2.8)].2.12Dosage Modifications for P-gp and CYP3A4 Inducers
• Avoid concomitant use of St. John’s Wort (Hypericum perforatum).• Increase the dose for patients taking everolimus with a P-gp and strong CYP3A4 inducer as recommended in Table 5 [see Drug Interactions (7.1), Clinical Pharmacology (12.3)].
Table 5: Recommended Dosage Modifications for Concurrent Use of Everolimus With P-gp and Strong CYP3A4 Inducers
Indication
Dose Modification for Everolimus
Breast Cancer, NET, and TSC-Associated Renal Angiomyolipoma
• Avoid coadministration where alternatives exist.• If coadministration cannot be avoided, double the daily dose using increments of 5 mg or less. Multiple increments may be required.• Resume the dose administered prior to inducer initiation, once an inducer is discontinued for 5 days.
TSC-Associated SEGA
• Double the daily dose using increments of 5 mg or less. Multiple increments may be required.• Addition of another strong CYP3A4 inducer in a patient already receiving treatment with a strong CYP3A4 inducer may not require additional dosage modification.• Assess trough concentrations when initiating and discontinuing the inducer [see Dosage and Administration (2.8)].• Resume the dose administered before starting any inducer, once all inducers are discontinued for 5 days.2.13Administration and Preparation
• Administer everolimus at the same time each day.• Administer everolimus consistently either with or without food [see Clinical Pharmacology (12.3)].• If a dose of everolimus is missed, it can be administered up to 6 hours after the time it is normally administered. After more than 6 hours, the dose should be skipped for that day. The next day, everolimus should be administered at its usual time. Double doses should not be administered to make up for the dose that was missed.
Everolimus tablets should be swallowed whole with a glass of water. Do not break or crush tablets.
3 Dosage Forms And Strengths
Everolimus Tablets are available containing 2.5 mg, 5 mg, 7.5 mg or 10 mg of everolimus.
• The 2.5 mg tablets are white to slightly yellow, capsule shaped, unscored tablets debossed with M on one side of the tablet and EVR2 on the other side.• The 5 mg tablets are white to slightly yellow, capsule shaped, unscored tablets debossed with M on one side of the tablet and EVR5 on the other side.• The 7.5 mg tablets are white to slightly yellow, capsule shaped, unscored tablets debossed with M on one side of the tablet and EVR7 on the other side.• The 10 mg tablets are white to slightly yellow, capsule shaped, unscored tablets debossed with M on one side of the tablet and EVR10 on the other side.
Everolimus tablets: 2.5 mg, 5 mg, 7.5 mg, and 10 mg tablets (3 )
4 Contraindications
Everolimus is contraindicated in patients with clinically significant hypersensitivity to everolimus or to other rapamycin derivatives [see Warnings and Precautions (5.3)].
Clinically significant hypersensitivity to everolimus or to other rapamycin derivatives. (4 )
5 Warnings And Precautions
• Non-Infectious Pneumonitis: Monitor for clinical symptoms or radiological changes. Withhold or permanently discontinue based on severity. (2.9 ,5.1 )• Infections: Monitor for signs and symptoms of infection. Withhold or permanently discontinue based on severity. (2.9 ,5.2 )• Severe Hypersensitivity Reactions: Permanently discontinue for clinically significant hypersensitivity. (5.3 )• Angioedema: Patients taking concomitant angiotensin-converting-enzyme (ACE) inhibitors may be at increased risk for angioedema. Permanently discontinue for angioedema. (5.4 ,7.2 )• Stomatitis: Initiate dexamethasone alcohol-free mouthwash when starting treatment. (5.5 ,6.1 )• Renal Failure: Monitor renal function prior to treatment and periodically thereafter. (5.6 )• Risk of Impaired Wound Healing: Withhold for at least 1 week prior to elective surgery. Do not administer for at least 2 weeks following major surgery and until adequate wound healing. The safety of resumption of treatment after resolution of wound healing complications has not been established. (5.7 )• Geriatric Patients: Monitor and adjust dose for adverse reactions. (5.8 )• Metabolic Disorders: Monitor serum glucose and lipids prior to treatment and periodically thereafter. Withhold or permanently discontinue based on severity. (2.9 ,5.9 )• Myelosuppression: Monitor hematologic parameters prior to treatment and periodically thereafter. Withhold or permanently discontinue based on severity. (2.9 ,5.10 )• Risk of Infection or Reduced Immune Response with Vaccination: Avoid live vaccines and close contact with those who have received live vaccines. Complete recommended childhood vaccinations prior to starting treatment. (5.11 )• Radiation Sensitization and Radiation Recall: Severe radiation reactions may occur. (5.12 ,6.2 )• Embryo-Fetal Toxicity: Can cause fetal harm. Advise patients of reproductive potential of the potential risk to a fetus and to use effective contraception. (5.13 ,8.1 ,8.3 )5.1Non-infectious Pneumonitis
Non-infectious pneumonitis is a class effect of rapamycin derivatives. Non-infectious pneumonitis was reported in up to 19% of patients treated with everolimus in clinical trials, some cases were reported with pulmonary hypertension (including pulmonary arterial hypertension) as a secondary event. The incidence of Grade 3 and 4 non-infectious pneumonitis was up to 4% and up to 0.2%, respectively [see Adverse Reactions (6.1)]. Fatal outcomes have been observed.
Consider a diagnosis of non-infectious pneumonitis in patients presenting with non-specific respiratory signs and symptoms. Consider opportunistic infections, such as pneumocystis jiroveci pneumonia (PJP) in the differential diagnosis. Advise patients to report promptly any new or worsening respiratory symptoms.
Continue everolimus without dose alteration in patients who develop radiological changes suggestive of non-infectious pneumonitis and have few or no symptoms. Imaging appears to overestimate the incidence of clinical pneumonitis.
For Grade 2 to 4 non-infectious pneumonitis, withhold or permanently discontinue everolimus based on severity [see Dosage and Administration (2.9)]. Corticosteroids may be indicated until clinical symptoms resolve. Administer prophylaxis for PJP when concomitant use of corticosteroids or other immunosuppressive agents are required. The development of pneumonitis has been reported even at a reduced dose.
5.2Infections
Everolimus has immunosuppressive properties and may predispose patients to bacterial, fungal, viral, or protozoal infections, including infections with opportunistic pathogens [see Adverse Reactions (6.1)]. Localized and systemic infections, including pneumonia, mycobacterial infections, other bacterial infections, invasive fungal infections (e.g., aspergillosis, candidiasis, or PJP), and viral infections (e.g., reactivation of hepatitis B virus) have occurred. Some of these infections have been severe (e.g., sepsis, septic shock, or resulting in multisystem organ failure) or fatal. The incidence of Grade 3 and 4 infections was up to 10% and up to 3%, respectively. The incidence of serious infections was reported at a higher frequency in patients < 6 years of age [see Use in Specific Populations (8.4)].
Complete treatment of preexisting invasive fungal infections prior to starting treatment. Monitor for signs and symptoms of infection. Withhold or permanently discontinue everolimus based on severity of infection [see Dosage and Administration (2.9)].
Administer prophylaxis for PJP when concomitant use of corticosteroids or other immunosuppressive agents are required.
5.3Severe Hypersensitivity Reactions
Hypersensitivity reactions to everolimus have been observed and include anaphylaxis, dyspnea, flushing, chest pain, and angioedema (e.g., swelling of the airways or tongue, with or without respiratory impairment) [see Contraindications (4)]. The incidence of Grade 3 hypersensitivity reactions was up to 1%. Permanently discontinue everolimus for the development of clinically significant hypersensitivity.
5.4Angioedema With Concomitant Use of Angiotensin-Converting Enzyme (ACE) Inhibitors
Patients taking concomitant ACE inhibitors with everolimus may be at increased risk for angioedema (e.g., swelling of the airways or tongue, with or without respiratory impairment). In a pooled analysis of randomized double-blind oncology clinical trials, the incidence of angioedema in patients taking everolimus with an ACE inhibitor was 6.8% compared to 1.3% in the control arm with an ACE inhibitor. Permanently discontinue everolimus for angioedema.
5.5Stomatitis
Stomatitis, including mouth ulcers and oral mucositis, has occurred in patients treated with everolimus at an incidence ranging from 44% to 78% across clinical trials. Grades 3-4 stomatitis was reported in 4% to 9% of patients [see Adverse Reactions (6.1)]. Stomatitis most often occurs within the first 8 weeks of treatment. When starting everolimus, initiating dexamethasone alcohol-free oral solution as a swish and spit mouthwash reduces the incidence and severity of stomatitis [see Adverse Reactions (6.1)]. If stomatitis does occur, mouthwashes and/or other topical treatments are recommended. Avoid alcohol-, hydrogen peroxide-, iodine-, or thyme- containing products, as they may exacerbate the condition. Do not administer antifungal agents, unless fungal infection has been diagnosed.
5.6Renal Failure
Cases of renal failure (including acute renal failure), some with a fatal outcome, have occurred in patients taking everolimus. Elevations of serum creatinine and proteinuria have been reported in patients taking everolimus [see Adverse Reactions (6.1)]. The incidence of Grade 3 and 4 elevations of serum creatinine was up to 2% and up to 1%, respectively. The incidence of Grade 3 and 4 proteinuria was up to 1% and up to 0.5%, respectively. Monitor renal function prior to starting everolimus and annually thereafter. Monitor renal function at least every 6 months in patients who have additional risk factors for renal failure.
5.7Risk of Impaired Wound Healing
Impaired wound healing can occur in patients who receive drugs that inhibit the VEGF signaling pathway. Therefore, everolimus has the potential to adversely affect wound healing.
Withhold everolimus for at least 1 week prior to elective surgery. Do not administer for at least 2 weeks following major surgery and until adequate wound healing. The safety of resumption of treatment upon resolution of wound healing complications has not been established.
5.8Geriatric Patients
In the randomized hormone receptor-positive, HER2-negative breast cancer study (BOLERO-2), the incidence of deaths due to any cause within 28 days of the last everolimus dose was 6% in patients ≥ 65 years of age compared to 2% in patients < 65 years of age. Adverse reactions leading to permanent treatment discontinuation occurred in 33% of patients ≥ 65 years of age compared to 17% in patients < 65 years of age. Careful monitoring and appropriate dose adjustments for adverse reactions are recommended [see Dosage and Administration (2.9), Use in Specific Populations (8.5)].
5.9Metabolic Disorders
Hyperglycemia, hypercholesterolemia, and hypertriglyceridemia have been reported in patients taking everolimus at an incidence up to 75%, 86%, and 73%, respectively. The incidence of these Grade 3 and 4 laboratory abnormalities was up to 15% and up to 0.4%, respectively [see Adverse Reactions (6.1)]. In non-diabetic patients, monitor fasting serum glucose prior to starting everolimus and annually thereafter. In diabetic patients, monitor fasting serum glucose more frequently as clinically indicated. Monitor lipid profile prior to starting everolimus and annually thereafter. When possible, achieve optimal glucose and lipid control prior to starting everolimus. For Grade 3 to 4 metabolic events, withhold or permanently discontinue everolimus based on severity [see Dosage and Administration (2.9)].
5.10Myelosuppression
Anemia, lymphopenia, neutropenia, and thrombocytopenia have been reported in patients taking everolimus. The incidence of these Grade 3 and 4 laboratory abnormalities was up to 16% and up to 2%, respectively [see Adverse Reactions (6.1)]. Monitor complete blood count (CBC) prior to starting everolimus every 6 months for the first year of treatment and annually thereafter. Withhold or permanently discontinue everolimus based on severity [see Dosage and Administration (2.9)].
5.11Risk of Infection or Reduced Immune Response With Vaccination
The safety of immunization with live vaccines during everolimus therapy has not been studied. Due to the potential increased risk of infection, avoid the use of live vaccines and close contact with individuals who have received live vaccines during treatment with everolimus. Due to the potential increased risk of infection or reduced immune response with vaccination, complete the recommended childhood series of vaccinations according to American Council on Immunization Practices (ACIP) guidelines prior to the start of therapy. An accelerated vaccination schedule may be appropriate.
5.12 Radiation Sensitization and Radiation Recall
Radiation sensitization and recall, in some cases severe, involving cutaneous and visceral organs (including radiation esophagitis and pneumonitis) have been reported in patients treated with radiation prior to, during, or subsequent to everolimus treatment [see Adverse Reactions (6.2)].
Monitor patients closely when everolimus is administered during or sequentially with radiation treatment.
5.13Embryo-Fetal Toxicity
Based on animal studies and the mechanism of action, everolimus can cause fetal harm when administered to a pregnant woman. In animal studies, everolimus caused embryo-fetal toxicities in rats when administered during the period of organogenesis at maternal exposures that were lower than human exposures at the clinical dose of 10 mg once daily. Advise pregnant women of the potential risk to a fetus. Advise female patients of reproductive potential to avoid becoming pregnant and to use effective contraception during treatment with everolimus and for 8 weeks after the last dose. Advise male patients with female partners of reproductive potential to use effective contraception during treatment with everolimus and for 4 weeks after the last dose [see Use in Specific Populations (8.1, 8.3)].
6 Adverse Reactions
The following serious adverse reactions are described elsewhere in the labeling:
• Non-Infectious Pneumonitis [see Warnings and Precautions (5.1)]• Infections [see Warnings and Precautions (5.2)]• Severe Hypersensitivity Reactions [see Warnings and Precautions (5.3)]• Angioedema with Concomitant Use of ACE inhibitors [see Warnings and Precautions (5.4)]• Stomatitis [see Warnings and Precautions (5.5)]• Renal Failure [see Warnings and Precautions (5.6)]• Impaired Wound Healing [see Warnings and Precautions (5.7)]• Metabolic Disorders [see Warnings and Precautions (5.9)]• Myelosuppression [see Warnings and Precautions (5.10)]• Radiation Sensitization and Radiation Recall [see Warnings and Precautions (5.12) ]
• Breast cancer, NET: Most common adverse reactions (incidence ≥ 30%) include stomatitis, infections, rash, fatigue, diarrhea, edema, abdominal pain, nausea, fever, asthenia, cough, headache, and decreased appetite. (6.1 )• TSC-Associated Renal Angiomyolipoma: Most common adverse reaction (incidence ≥ 30%) is stomatitis. (6.1 )• TSC-Associated SEGA: Most common adverse reactions (incidence ≥ 30%) are stomatitis and respiratory tract infection. (6.1 )
To report SUSPECTED ADVERSE REACTIONS, contact Mylan at 1-877-446-3679 (1-877-4-INFO-RX) or FDA at 1-800-FDA-1088 orwww.fda.gov/medwatch.
6.1Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, the adverse reaction rates observed cannot be directly compared to rates in other trials and may not reflect the rates observed in clinical practice.
The safety of everolimus (10 mg orally once daily) in combination with exemestane (25 mg orally once daily) (n = 485) vs. placebo in combination with exemestane (n = 239) was evaluated in a randomized, controlled trial (BOLERO-2) in patients with advanced or metastatic hormone receptor-positive, HER2-negative breast cancer. The median age of patients was 61 years (28 to 93 years), and 75% were white. The median follow-up was approximately 13 months.
The most common adverse reactions (incidence ≥ 30%) were stomatitis, infections, rash, fatigue, diarrhea, and decreased appetite. The most common Grade 3-4 adverse reactions (incidence ≥ 2%) were stomatitis, infections, hyperglycemia, fatigue, dyspnea, pneumonitis, and diarrhea. The most common laboratory abnormalities (incidence ≥ 50%) were hypercholesterolemia, hyperglycemia, increased aspartate transaminase (AST), anemia, leukopenia, thrombocytopenia, lymphopenia, increased alanine transaminase (ALT), and hypertriglyceridemia. The most common Grade 3-4 laboratory abnormalities (incidence ≥ 3%) were lymphopenia, hyperglycemia, anemia, hypokalemia, increased AST, increased ALT, and thrombocytopenia.
Fatal adverse reactions occurred in 2% of patients who received everolimus. The rate of adverse reactions resulting in permanent discontinuation was 24% for the everolimus arm. Dose adjustments (interruptions or reductions) occurred in 63% of patients in the everolimus arm.
Adverse reactions reported with an incidence of ≥ 10% for patients receiving everolimus vs. placebo are presented in Table 6. Laboratory abnormalities are presented in Table 7. The median duration of treatment with everolimus was 23.9 weeks; 33% were exposed to everolimus for a period of ≥ 32 weeks.
Table 6: Adverse Reactions Reported in ≥ 10% of Patients With Hormone Receptor-Positive Breast Cancer in BOLERO-2 Grading according to NCI CTCAE Version 3.0.
Everolimus
with Exemestane
N = 482
Placebo
with Exemestane
N = 238
All Grades
%
Grade 3-4
%
All Grades
%
Grade 3-4
%
Gastrointestinal
  StomatitisIncludes stomatitis, mouth ulceration, aphthous stomatitis, glossodynia, gingival pain, glossitis, and lip ulceration.
67
8No Grade 4 adverse reactions were reported.
11
0.8
  Diarrhea
33
2
18
0.8
  Nausea
29
0.4
28
1
  Vomiting
17
1
12
0.8
  Constipation
14
0.4
13
0.4
  Dry mouth
11
0
7
0
General
  Fatigue
36
4
27
1
  Edema peripheral
19
1
6
0.4
  Pyrexia
15
0.2
7
0.4
  Asthenia
13
2
4
0
Infections
  InfectionsIncludes all reported infections, including but not limited to, urinary tract infections, respiratory tract (upper and lower) infections, skin infections, and gastrointestinal tract infections.
50
6
25
2
Investigations
  Weight loss
25
1
6
0
Metabolism and nutrition
  Decreased appetite
30
1
12
0.4
  Hyperglycemia
14
5
2
0.4
Musculoskeletal and connective tissue
  Arthralgia
20
0.8
17
0
  Back pain
14
0.2
10
0.8
  Pain in extremity
9
0.4
11
2
Nervous system
  Dysgeusia
22
0.2
6
0
  Headache
21
0.4
14
0
Psychiatric
  Insomnia
13
0.2
8
0
Respiratory, thoracic and mediastinal
  Cough
24
0.6
12
0
  Dyspnea
21
4
11
1
  Epistaxis
17
0
1
0
  PneumonitisIncludes pneumonitis, interstitial lung disease, lung infiltration, and pulmonary fibrosis.
19
4
0.4
0
Skin and subcutaneous tissue
  Rash
39
1
6
0
  Pruritus
13
0.2
5
0
  Alopecia
10
0
5
0
Vascular
  Hot flush
6
0
14
0
Table 7: Selected Laboratory Abnormalities Reported in ≥ 10% of Patients With Hormone Receptor-Positive Breast Cancer in BOLERO-2 Grading according to NCI CTCAE Version 3.0.
Laboratory Parameter
Everolimus
with Exemestane
N = 482
Placebo
with Exemestane
N = 238
All Grades
%
Grade 3-4
%
All Grades
%
Grade 3-4
%
HematologyReflects corresponding adverse drug reaction reports of anemia, leukopenia, lymphopenia, neutropenia, and thrombocytopenia (collectively as pancytopenia), which occurred at lower frequency.
  Anemia
68
6
40
1
  Leukopenia
58
2No Grade 4 laboratory abnormalities were reported.
28
6
  Thrombocytopenia
54
3
5
0.4
  Lymphopenia
54
12
37
6
  Neutropenia
31
2
11
2
Chemistry
  Hypercholesterolemia
70
1
38
2
  Hyperglycemia
69
9
44
1
  Increased AST
69
4
45
3
  Increased ALT
51
4
29
5
  Hypertriglyceridemia
50
0.8
26
0
  Hypoalbuminemia
33
0.8
16
0.8
  Hypokalemia
29
4
7
1
  Increased creatinine
24
2
13
0
In a single arm study (SWISH; N = 92) in postmenopausal women with hormone receptor-positive, HER2-negative breast cancer beginning everolimus (10 mg orally once daily) in combination with exemestane (25 mg orally once daily), patients started dexamethasone 0.5 mg/5 mL alcohol-free mouthwash (10 mL swished for 2 minutes and spat, 4 times daily for 8 weeks) concurrently with everolimus and exemestane. No food or drink was to be consumed for at least 1 hour after swishing and spitting the dexamethasone mouthwash. The primary objective of this study was to assess the incidence of Grade 2 to 4 stomatitis within 8 weeks. The incidence of Grade 2 to 4 stomatitis within 8 weeks was 2%, which was lower than the 33% reported in the BOLERO-2 trial. The incidence of Grade 1 stomatitis was 19%. No cases of Grade 3 or 4 stomatitis were reported. Oral candidiasis was reported in 2% of patients in this study compared to 0.2% in the BOLERO-2 trial.
Coadministration of everolimus and dexamethasone alcohol-free oral solution has not been studied in pediatric patients.
In a randomized, controlled trial (RADIANT-4) of everolimus (n = 202 treated) vs. placebo (n = 98 treated) in patients with advanced non-functional NET of GI or lung origin, the median age of patients was 63 years (22-86 years), 76% were white, and 53% were female. The median duration of exposure to everolimus was 9.3 months; 64% of patients were treated for ≥ 6 months and 39% were treated for ≥ 12 months. Everolimus was discontinued for adverse reactions in 29% of patients, dose reduction or delay was required in 70% of everolimus-treated patients.
Serious adverse reactions occurred in 42% of everolimus-treated patients and included 3 fatal events (cardiac failure, respiratory failure, and septic shock). Adverse reactions occurring at an incidence of ≥ 10% and at ≥ 5% absolute incidence over placebo (all Grades) or ≥ 2% higher incidence over placebo (Grade 3 and 4) are presented in Table 10. Laboratory abnormalities are presented in Table 11.
Table 10: Adverse Reactions in ≥ 10% of Everolimus-Treated Patients With Non-Functional NET of GI or Lung Origin in RADIANT-4 Grading according to NCI CTCAE Version 4.03.
Everolimus
N = 202
Placebo
N = 98
All Grades
%
Grade 3-4
%
All Grades
%
Grade 3-4
%
Gastrointestinal
    StomatitisIncludes stomatitis, mouth ulceration, aphthous stomatitis, gingival pain, glossitis, tongue ulceration, and mucosal inflammation.
63
9No Grade 4 adverse reactions were reported.
22
0
    Diarrhea
41
9
31
2
    Nausea
26
3
17
1
    Vomiting
15
4
12
2
General
    Peripheral edema
39
3
6
1
    Fatigue
37
5
36
1
    Asthenia
23
3
8
0
    Pyrexia
23
2
8
0
Infections
    InfectionsUrinary tract infection, nasopharyngitis, upper respiratory tract infection, lower respiratory tract infection (pneumonia, bronchitis), abscess, pyelonephritis, septic shock and viral myocarditis.
58
11
29
2
Investigations
    Weight loss
22
2
11
1
Metabolism and nutrition
    Decreased appetite
22
1
17
1
Nervous system
    Dysgeusia
18
1
4
0
Respiratory, thoracic and mediastinal
    Cough
27
0
20
0
    Dyspnea
20
3
11
2
    PneumonitisIncludes pneumonitis and interstitial lung disease.
16
2
2
0
    Epistaxis
13
1
3
0
Skin and subcutaneous
    Rash
30
1
9
0
    Pruritis
17
1
9
0
Â
Table 11: Selected Laboratory Abnormalities in ≥ 10% of Everolimus-Treated Patients With Non-Functional NET of GI or Lung Origin in RADIANT-4 Grading according to NCI CTCAE Version 4.03.
Everolimus
N = 202
Placebo
N = 98
All Grades
%
Grade 3-4
%
All Grades
%
Grade 3-4
%
Hematology
    Anemia
81
5No Grade 4 laboratory abnormalities were reported.
41
2
    Lymphopenia
66
16
32
2
    Leukopenia
49
2
17
0
    Thrombocytopenia
33
2
11
0
    Neutropenia
32
2
15
3
Chemistry
    Hypercholesterolemia
71
0
37
0
    Increased AST
57
2
34
2
    Hyperglycemia (fasting)
55
6
36
1
    Increased ALT
46
5
39
1
    Hypophosphatemia
43
4
15
2
    Hypertriglyceridemia
30
3
8
1
    Hypokalemia
27
6
12
3
    Hypoalbuminemia
18
0
8
0
Â
The data described below are based on a randomized (2:1), double-blind, placebo-controlled trial (EXIST-2) of everolimus tablets in 118 patients with renal angiomyolipoma as a feature of TSC (n = 113) or sporadic lymphangioleiomyomatosis (n = 5). The median age of patients was 31 years (18 to 61 years), 89% were white, and 34% were male. The median duration of blinded study treatment was 48 weeks (2 to 115 weeks) for patients receiving everolimus tablets.
The most common adverse reaction reported for everolimus tablets (incidence ≥ 30%) was stomatitis. The most common Grade 3-4 adverse reactions (incidence ≥ 2%) were stomatitis and amenorrhea. The most common laboratory abnormalities (incidence ≥ 50%) were hypercholesterolemia, hypertriglyceridemia, and anemia. The most common Grade 3-4 laboratory abnormality (incidence ≥ 3%) was hypophosphatemia.
The rate of adverse reactions resulting in permanent discontinuation was 3.8% in the everolimus tablets-treated patients. Adverse reactions leading to permanent discontinuation in the everolimus tablets arm were hypersensitivity/angioedema/bronchospasm, convulsion, and hypophosphatemia. Dose adjustments (interruptions or reductions) due to adverse reactions occurred in 52% of everolimus tablets-treated patients. The most common adverse reaction leading to everolimus tablets dose adjustment was stomatitis.
Adverse reactions reported with an incidence of ≥ 10% for patients receiving everolimus tablets and occurring more frequently with everolimus tablets than with placebo are presented in Table 14. Laboratory abnormalities are presented in Table 15.
Table 14: Adverse Reactions Reported in ≥ 10% of Everolimus Tablets-Treated Patients With TSC-Associated Renal Angiomyolipoma in EXIST-2 Grading according to NCI CTCAE Version 3.0.
Everolimus Tablets
N = 79
Placebo
N = 39
All Grades
%
Grade 3-4
%
All Grades
%
Grade 3-4
%
Gastrointestinal
   StomatitisIncludes stomatitis, aphthous stomatitis, mouth ulceration, gingival pain, glossitis, and glossodynia.
78
6No Grade 4 adverse reactions were reported.
23
0
   Vomiting
15
0
5
0
   Diarrhea
14
0
5
0
General
   Peripheral edema
13
0
8
0
Infections
   Upper respiratory tract infection
11
0
5
0
Musculoskeletal and connective tissue
   Arthralgia
13
0
5
0
Respiratory, thoracic and mediastinal
   Cough
20
0
13
0
Skin and subcutaneous tissue
   Acne
22
0
5
0
Amenorrhea occurred in 15% of everolimus tablets-treated females (8 of 52). Other adverse reactions involving the female reproductive system were menorrhagia (10%), menstrual irregularities (10%), and vaginal hemorrhage (8%).
The following additional adverse reactions occurred in less than 10% of everolimus tablets-treated patients: epistaxis (9%), decreased appetite (6%), otitis media (6%), depression (5%), abnormal taste (5%), increased blood luteinizing hormone (LH) levels (4%), increased blood follicle stimulating hormone (FSH) levels (3%), hypersensitivity (3%), ovarian cyst (3%), pneumonitis (1%), and angioedema (1%).
Table 15: Selected Laboratory Abnormalities Reported in Everolimus Tablets-Treated Patients With TSC-Associated Renal Angiomyolipoma in EXIST-2 Grading according to NCI CTCAE Version 3.0.
Everolimus Tablets
N = 79
Placebo
N = 39
All Grades
%
Grade 3-4
%
All Grades
%
Grade 3-4
%
Hematology
   Anemia
61
0
49
0
   Leukopenia
37
0
21
0
   Neutropenia
25
1
26
0
   Lymphopenia
20
1No Grade 4 laboratory abnormalities were reported.
8
0
   Thrombocytopenia
19
0
3
0
Chemistry
   Hypercholesterolemia
85
1
46
0
   Hypertriglyceridemia
52
0
10
0
   Hypophosphatemia
49
5
15
0
   Increased alkaline phosphatase
32
1
10
0
   Increased AST
23
1
8
0
   Increased ALT
20
1
15
0
   Hyperglycemia (fasting)
14
0
8
0
Updated safety information from 112 patients treated with everolimus tablets for a median duration of 3.9 years identified the following additional adverse reactions and selected laboratory abnormalities: increased partial thromboplastin time (63%), increased prothrombin time (40%), decreased fibrinogen (38%), urinary tract infection (31%), proteinuria (18%), abdominal pain (16%), pruritus (12%), gastroenteritis (12%), myalgia (11%), and pneumonia (10%).
The data described below are based on a randomized (2:1), double-blind, placebo-controlled trial (EXIST-1) of everolimus in 117 patients with SEGA and TSC. The median age of patients was 9.5 years (0.8 to 26 years), 93% were white, and 57% were male. The median duration of blinded study treatment was 52 weeks (24 to 89 weeks) for patients receiving everolimus.
The most common adverse reactions reported for everolimus (incidence ≥ 30%) were stomatitis and respiratory tract infection. The most common Grade 3-4 adverse reactions (incidence ≥ 2%) were stomatitis, pyrexia, pneumonia, gastroenteritis, aggression, agitation, and amenorrhea. The most common laboratory abnormalities (incidence ≥ 50%) were hypercholesterolemia and elevated partial thromboplastin time. The most common Grade 3-4 laboratory abnormality (incidence ≥ 3%) was neutropenia.
There were no adverse reactions resulting in permanent discontinuation. Dose adjustments (interruptions or reductions) due to adverse reactions occurred in 55% of everolimus-treated patients. The most common adverse reaction leading to everolimus dose adjustment was stomatitis.
Adverse reactions reported with an incidence of ≥ 10% for patients receiving everolimus and occurring more frequently with everolimus than with placebo are reported in Table 16. Laboratory abnormalities are presented in Table 17.
Table 16: Adverse Reactions Reported in ≥ 10% of Everolimus-Treated Patients With TSC-Associated SEGA in EXIST-1 Grading according to NCI CTCAE Version 3.0.
Everolimus
N = 78
Placebo
N = 39
All
Grades
%
Grade 3-4
%
All
Grades
%
Grade 3-4
%
Gastrointestinal
  StomatitisIncludes mouth ulceration, stomatitis, and lip ulceration.
62
9No Grade 4 adverse reactions were reported.
26
3
  Vomiting
22
1
13
0
  Diarrhea
17
0
5
0
  Constipation
10
0
3
0
Infections
  Respiratory tract infectionIncludes respiratory tract infection, upper respiratory tract infection, and respiratory tract infection viral.
31
3
23
0
  GastroenteritisIncludes gastroenteritis, gastroenteritis viral, and gastrointestinal infection.
10
5
3
0
  Pharyngitis streptococcal
10
0
3
0
General
  Pyrexia
23
6
18
3
  Fatigue
14
0
3
0
Psychiatric
  Anxiety, aggression or other behavioral disturbanceIncludes agitation, anxiety, panic attack, aggression, abnormal behavior, and obsessive compulsive disorder.
21
5
3
0
Skin and subcutaneous tissue
  RashIncludes rash, rash generalized, rash macular, rash maculo-papular, rash papular, dermatitis allergic, and urticaria.
21
0
8
0
  Acne
10
0
5
0
Amenorrhea occurred in 17% of everolimus-treated females aged 10 to 55 years (3 of 18). For this same group of everolimus-treated females, the following menstrual abnormalities were reported: dysmenorrhea (6%), menorrhagia (6%), metrorrhagia (6%), and unspecified menstrual irregularity (6%).
The following additional adverse reactions occurred in less than 10% of everolimus-treated patients: nausea (8%), pain in extremity (8%), insomnia (6%), pneumonia (6%), epistaxis (5%), hypersensitivity (3%), increased blood luteinizing hormone (LH) levels (1%), and pneumonitis (1%).
Table 17: Selected Laboratory Abnormalities Reported in Everolimus-Treated Patients With TSC-Associated SEGA in EXIST-1 Grading according to NCI CTCAE Version 3.0.
Everolimus
N = 78
Placebo
N = 39
All Grades
%
Grade 3-4
%
All Grades
%
Grade 3-4
%
Hematology
  Elevated partial thromboplastin time
72
3No Grade 4 laboratory abnormalities were reported.
44
5
  Neutropenia
46
9
41
3
  Anemia
41
0
21
0
Chemistry
  Hypercholesterolemia
81
0
39
0
  Elevated AST
33
0
0
0
  Hypertriglyceridemia
27
0
15
0
  Elevated ALT
18
0
3
0
  Hypophosphatemia
9
1
3
0
Updated safety information from 111 patients treated with everolimus for a median duration of 47 months identified the following additional notable adverse reactions and selected laboratory abnormalities: decreased appetite (14%), hyperglycemia (13%), hypertension (11%), urinary tract infection (9%), decreased fibrinogen (8%), cellulitis (6%), abdominal pain (5%), decreased weight (5%), elevated creatinine (5%), and azoospermia (1%).
6.2Postmarketing Experience
The following adverse reactions have been identified during postapproval use of everolimus. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate frequency or establish a causal relationship to drug exposure:
• Blood and Lymphatic Disorders: Thrombotic microangiopathy• Cardiac: Cardiac failure with some cases reported with pulmonary hypertension (including pulmonary arterial hypertension) as a secondary event• Gastrointestinal:  Acute pancreatitis• Hepatobiliary:  Cholecystitis and cholelithiasis• Infections: Sepsis and septic shock• Nervous System: Reflex sympathetic dystrophy• Vascular: Arterial thrombotic events, lymphedema• Injury, Poisoning and Procedural Complications:  Radiation Sensitization and Radiation Recall
7 Drug Interactions
• P-gp and strong CYP3A4 inhibitors: Avoid concomitant use. (2.11 ,7.1 )• P-gp and moderate CYP3A4 inhibitors: Reduce the dose as recommended. (2.11 ,7.1 )• P-gp and strong CYP3A4 inducers: Increase the dose as recommended. (2.12 ,7.1 )7.1Effect of Other Drugs on Everolimus
Avoid the concomitant use of P-gp and strong CYP3A4 inhibitors [see Dosage and Administration (2.11), Clinical Pharmacology (12.3)].
Reduce the dose for patients taking everolimus with a P-gp and moderate CYP3A4 inhibitor as recommended [see Dosage and Administration (2.11), Clinical Pharmacology (12.3)].
Increase the dose for patients taking everolimus with a P-gp and strong CYP3A4 inducer as recommended [see Dosage and Administration (2.12), Clinical Pharmacology (12.3)].
7.2Effects of Combination Use of Angiotensin-Converting Enzyme (ACE) Inhibitors
Patients taking concomitant ACE inhibitors with everolimus may be at increased risk for angioedema. Avoid the concomitant use of ACE inhibitors with everolimus [see Warnings and Precautions (5.4)].
8 Use In Specific Populations
Â
• For breast cancer, NET, or TSC-associated renal angiomyolipoma patients with hepatic impairment, reduce the dose. (2.10 ,8.6 )• For patients with TSC-associated SEGA and severe hepatic impairment, reduce the starting dose and adjust dose to attain target trough concentrations. (2.8 ,2.10 ,8.6 )8.1 Pregnancy
Based on animal studies and the mechanism of action [see Clinical Pharmacology (12.1)], everolimus can cause fetal harm when administered to a pregnant woman. There are limited case reports of everolimus use in pregnant women; however, these reports are not sufficient to inform about risks of birth defects or miscarriage. In animal studies, everolimus caused embryo-fetal toxicities in rats when administered during the period of organogenesis at maternal exposures that were lower than human exposures at the recommended dose of everolimus 10 mg orally once daily (see Data). Advise pregnant women of the potential risk to the fetus.
In the U.S. general population, the estimated background risk of major birth defects and miscarriage is 2% to 4% and 15% to 20% of clinically recognized pregnancies, respectively.
Animal Data
In animal reproductive studies, oral administration of everolimus to female rats before mating and through organogenesis induced embryo-fetal toxicities, including increased resorption, pre-implantation and post-implantation loss, decreased numbers of live fetuses, malformation (e.g., sternal cleft), and retarded skeletal development. These effects occurred in the absence of maternal toxicities. Embryo-fetal toxicities in rats occurred at doses ≥ 0.1 mg/kg (0.6 mg/m2) with resulting exposures of approximately 4% of the human exposure at the recommended dose of everolimus 10 mg orally once daily based on area under the curve (AUC). In rabbits, embryo-toxicity evident as an increase in resorptions occurred at an oral dose of 0.8 mg/kg (9.6 mg/m2), approximately 1.6 times the recommended dose of everolimus 10 mg orally once daily or the median dose administered to patients with tuberous sclerosis complex (TSC)-associated subependymal giant cell astrocytoma (SEGA). The effect in rabbits occurred in the presence of maternal toxicities.
In a pre- and post-natal development study in rats, animals were dosed from implantation through lactation. At the dose of 0.1 mg/kg (0.6 mg/m2), there were no adverse effects on delivery and lactation or signs of maternal toxicity; however, there were reductions in body weight (up to 9% reduction from the control) and in survival of offspring (~5% died or missing). There were no drug-related effects on the developmental parameters (morphological development, motor activity, learning, or fertility assessment) in the offspring.
8.2 Lactation
There are no data on the presence of everolimus or its metabolites in human milk, the effects of everolimus on the breastfed infant or on milk production. Everolimus and its metabolites passed into the milk of lactating rats at a concentration 3.5 times higher than in maternal serum. Because of the potential for serious adverse reactions in breastfed infants from everolimus, advise women not to breastfeed during treatment with everolimus and for 2 weeks after the last dose.
8.3 Females and Males of Reproductive Potential
Verify the pregnancy status of females of reproductive potential prior to starting everolimus [see Use in Specific Populations (8.1) ].
Everolimus can cause fetal harm when administered to pregnant women [see Use in Specific Populations (8.1)].
Females
Advise female patients of reproductive potential to use effective contraception during treatment with everolimus and for 8 weeks after the last dose.
Males
Advise male patients with female partners of reproductive potential to use effective contraception during treatment with everolimus and for 4 weeks after the last dose.
Females
Menstrual irregularities, secondary amenorrhea, and increases in luteinizing hormone (LH) and follicle stimulating hormone (FSH) occurred in female patients taking everolimus. Based on these findings, everolimus may impair fertility in female patients [see Adverse Reactions (6.1), Nonclinical Toxicology (13.1)].
Males
Cases of reversible azoospermia have been reported in male patients taking everolimus. In male rats, sperm motility, sperm count, plasma testosterone levels and fertility were diminished at AUC similar to those of the clinical dose of everolimus 10 mg orally once daily. Based on these findings, everolimus may impair fertility in male patients [see Nonclinical Toxicology (13.1)].
8.4 Pediatric Use
The safety and effectiveness of everolimus have been established in pediatric patients age 1 year and older with TSC-associated SEGA that requires therapeutic intervention but cannot be curatively resected. Use of everolimus for this indication is supported by evidence from a randomized, double-blind, placebo-controlled trial in adult and pediatric patients (EXIST-1); an open-label, single-arm trial in adult and pediatric patients (Study 2485); and additional pharmacokinetic data in pediatric patients [see Adverse Reactions (6.1), Clinical Pharmacology (12.3), Clinical Studies (14.5)]. The safety and effectiveness of everolimus have not been established in pediatric patients less than 1 year of age with TSC-associated SEGA.
In EXIST-1, the incidence of infections and serious infections were reported at a higher frequency in patients < 6 years of age. Ninety-six percent of 23 everolimus-treated patients < 6 years had at least one infection compared to 67% of 55 everolimus-treated patients ≥ 6 years. Thirty-five percent of 23 everolimus-treated patients < 6 years of age had at least 1 serious infection compared to 7% of 55 everolimus-treated patients ≥ 6 years.
Although a conclusive determination cannot be made due to the limited number of patients and lack of a comparator arm in the open label follow-up periods of EXIST-1 and Study 2485, everolimus did not appear to adversely impact growth and pubertal development in the 115 pediatric patients treated with everolimus for a median duration of 4.1 years.
The safety and effectiveness of everolimus in pediatric patients have not been established in:
• Hormone receptor-positive, HER2-negative breast cancer• Neuroendocrine tumors (NET)• TSC-associated renal angiomyolipoma8.5 Geriatric Use
In BOLERO-2, 40% of patients with breast cancer treated with everolimus were ≥ 65 years of age, while 15% were ≥ 75 years of age. No overall differences in effectiveness were observed between elderly and younger patients. The incidence of deaths due to any cause within 28 days of the last everolimus dose was 6% in patients ≥ 65 years of age compared to 2% in patients < 65 years of age. Adverse reactions leading to permanent treatment discontinuation occurred in 33% of patients ≥ 65 years of age compared to 17% in patients < 65 years of age.
8.6Hepatic Impairment
Everolimus exposure may increase in patients with hepatic impairment [see Clinical Pharmacology (12.3)].
For patients with breast cancer, NET, and TSC-associated renal angiomyolipoma who have hepatic impairment, reduce the everolimus dose as recommended [see Dosage and Administration (2.10)].
For patients with TSC-associated SEGA who have severe hepatic impairment (Child-Pugh class C), reduce the starting dose of everolimus as recommended and adjust the dose based on everolimus trough concentrations [see Dosage and Administration (2.8, 2.10)].
11 Description
Everolimus tablets are a kinase inhibitor.
The chemical name of everolimus is (3S,6R,7E,9R,10R,12R,14S,15E,17E,19E,21S,23S,26R,27R, 34aS)-9,10,12,13,14,21,22,23,24,25,26,27,32,33,34,34a-Hexadecahydro-9,27-dihydroxy-3-[(1R)-2-[(1S,3R,4R)-4-(2-hydroxyethoxy)-3-methoxycyclohexyl]-1-methylethyl]-10,21-dimethoxy-6,8,12,14,20,26-hexamethyl-23,27-epoxy-3H-pyrido[2,1-c][1,4]oxaazacyclohentriacontine-1,5,11,28,29(4H,6H,31H)-pentone. The molecular formula is C53H83NO14 and the molecular weight is 958.2. The structural formula is:
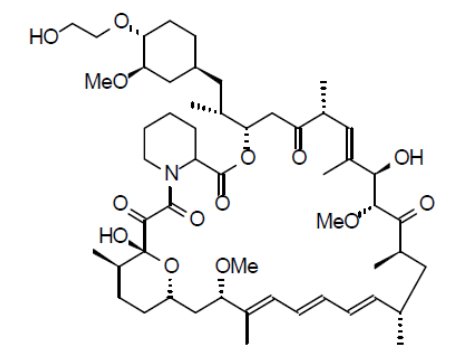
Everolimus tablets for oral administration contain 2.5 mg, 5 mg, 7.5 mg, or 10 mg of everolimus and the following inactive ingredients: anhydrous lactose, butylated hydroxytoluene, colloidal silicon dioxide, croscarmellose sodium, hypromellose and magnesium stearate.
12 Clinical Pharmacology
Â
12.1 Mechanism of Action
Everolimus is an inhibitor of mammalian target of rapamycin (mTOR), a serine-threonine kinase, downstream of the PI3K/AKT pathway. The mTOR pathway is dysregulated in several human cancers and in tuberous sclerosis complex (TSC). Everolimus binds to an intracellular protein, FKBP-12, resulting in an inhibitory complex formation with mTOR complex 1 (mTORC1) and thus inhibition of mTOR kinase activity. Everolimus reduced the activity of S6 ribosomal protein kinase (S6K1) and eukaryotic initiation factor 4E-binding protein (4E-BP1), downstream effectors of mTOR, involved in protein synthesis. S6K1 is a substrate of mTORC1 and phosphorylates the activation domain 1 of the estrogen receptor which results in ligand-independent activation of the receptor. In addition, everolimus inhibited the expression of hypoxia-inducible factor (e.g., HIF-1) and reduced the expression of vascular endothelial growth factor (VEGF). Inhibition of mTOR by everolimus has been shown to reduce cell proliferation, angiogenesis, and glucose uptake in in vitro and/or in vivo studies.
Constitutive activation of the PI3K/Akt/mTOR pathway can contribute to endocrine resistance in breast cancer. In vitro studies show that estrogen-dependent and HER2+ breast cancer cells are sensitive to the inhibitory effects of everolimus, and that combination treatment with everolimus and Akt, HER2, or aromatase inhibitors enhances the anti-tumor activity of everolimus in a synergistic manner.
Two regulators of mTORC1 signaling are the oncogene suppressors tuberin-sclerosis complexes 1 and 2 (TSC1, TSC2). Loss or inactivation of either TSC1 or TSC2 leads to activation of downstream signaling. In TSC, a genetic disorder, inactivating mutations in either the TSC1 or the TSC2 gene lead to hamartoma formation throughout the body as well as seizures and epileptogenesis. Overactivation of mTOR results in neuronal dysplasia, aberrant axonogenesis and dendrite formation, increased excitatory synaptic currents, reduced myelination, and disruption of the cortical laminar structure causing abnormalities in neuronal development and function. Treatment with an mTOR inhibitor in animal models of mTOR dysregulation in the brain resulted in seizure suppression, prevention of the development of new-onset seizures, and prevention of premature death.
12.2 Pharmacodynamics
In patients with TSC-associated subependymal giant cell astrocytoma (SEGA), the magnitude of the reduction in SEGA volume was correlated with the everolimus trough concentration.
In a randomized, placebo-controlled, cross-over study, 59 healthy subjects were administered a single oral dose of everolimus (20 mg and 50 mg) and placebo. Everolimus at single doses up to 50 mg did not prolong the QT/QTc interval.
12.3 Pharmacokinetics
After administration of everolimus in patients with advanced solid tumors, peak everolimus concentrations are reached 1 to 2 hours after administration of oral doses ranging from 5 mg to 70 mg. Following single doses, Cmax is dose-proportional with daily dosing between 5 mg and 10 mg. With single doses of 20 mg and higher, the increase in Cmax is less than dose-proportional; however, AUC shows dose-proportionality over the 5 mg to 70 mg dose range. Steady-state was achieved within 2 weeks following once-daily dosing.
In patients with TSC-associated SEGA, everolimus Cmin was approximately dose-proportional within the dose range from 1.35 mg/m2 to 14.4 mg/m2.
Effect of Food
In healthy subjects, a high-fat meal (containing approximately 1000 calories and 55 grams of fat) reduced systemic exposure to everolimus 10 mg (as measured by AUC) by 22% and the peak blood concentration Cmax by 54%. Light-fat meals (containing approximately 500 calories and 20 grams of fat) reduced AUC by 32% and Cmax by 42%.
The blood-to-plasma ratio of everolimus, which is concentration-dependent over the range of 5 to 5000 ng/mL, is 17% to 73%. The amount of everolimus confined to the plasma is approximately 20% at blood concentrations observed in cancer patients given everolimus 10 mg orally once daily. Plasma protein binding is approximately 74% both in healthy subjects and in patients with moderate hepatic impairment.
The mean elimination half-life of everolimus is approximately 30 hours.
Metabolism
Everolimus is a substrate of CYP3A4. Following oral administration, everolimus is the main circulating component in human blood. Six main metabolites of everolimus have been detected in human blood, including three monohydroxylated metabolites, two hydrolytic ring-opened products, and a phosphatidylcholine conjugate of everolimus. These metabolites were also identified in animal species used in toxicity studies, and showed approximately 100-times less activity than everolimus itself.
Excretion
No specific elimination studies have been undertaken in cancer patients. Following the administration of a 3 mg single dose of radiolabeled everolimus in patients who were receiving cyclosporine, 80% of the radioactivity was recovered from the feces, while 5% was excreted in the urine. The parent substance was not detected in urine or feces.
No relationship was apparent between oral clearance and age or sex in patients with cancer.
Patients with Renal Impairment
No significant influence of creatinine clearance (25 to 178 mL/min) was detected on oral clearance (CL/F) of everolimus.
Patients with Hepatic Impairment
Compared to normal subjects, there was a 1.8-fold, 3.2-fold, and 3.6-fold increase in AUC for subjects with mild (Child-Pugh class A), moderate (Child-Pugh class B), and severe (Child-Pugh class C) hepatic impairment, respectively. In another study, the average AUC of everolimus in subjects with moderate hepatic impairment (Child-Pugh class B) was twice that found in subjects with normal hepatic function [see Dosage and Administration (2.10), Use in Specific Populations (8.6)].
Pediatric Patients
In patients with TSC-associated SEGA, the mean Cmin values normalized to mg/m2 dose in pediatric patients (< 18 years of age) were lower than those observed in adults, suggesting that everolimus clearance adjusted to BSA was higher in pediatric patients as compared to adults.
Race or Ethnicity
Based on a cross-study comparison, Japanese patients had on average exposures that were higher than non-Japanese patients receiving the same dose. Oral clearance (CL/F) is on average 20% higher in black patients than in white patients.
Effect of CYP3A4 and P-glycoprotein (P-gp) Inhibitors on Everolimus
Everolimus exposure increased when everolimus was coadministered with:
• ketoconazole (a P-gp and strong CYP3A4 inhibitor) - Cmax and AUC increased by 3.9- and 15-fold, respectively.• erythromycin (a P-gp and moderate CYP3A4 inhibitor) - Cmax and AUC increased by 2- and 4.4-fold, respectively.• verapamil (a P-gp and moderate CYP3A4 inhibitor) - Cmax and AUC increased by 2.3- and 3.5-fold, respectively.Effect of CYP3A4 and P-gp Inducers on Everolimus
The coadministration of everolimus with rifampin, a P-gp and strong inducer of CYP3A4, decreased everolimus AUC by 63% and Cmax by 58% compared to everolimus alone [see Dosage and Administration (2.12)].
Effect of Everolimus on CYP3A4 Substrates
No clinically significant pharmacokinetic interactions were observed between everolimus and the HMG-CoA reductase inhibitors atorvastatin (a CYP3A4 substrate), pravastatin (a non-CYP3A4 substrate), and simvastatin (a CYP3A4 substrate).
The coadministration of an oral dose of midazolam (sensitive CYP3A4 substrate) with everolimus resulted in a 25% increase in midazolam Cmax and a 30% increase in midazolam AUC0-inf.
The coadministration of everolimus with exemestane increased exemestane Cmin by 45% and C2h by 64%; however, the corresponding estradiol levels at steady state (4 weeks) were not different between the 2 treatment arms. No increase in adverse reactions related to exemestane was observed in patients with hormone receptor-positive, HER2-negative advanced breast cancer receiving the combination.
The coadministration of everolimus with long-acting octreotide increased octreotide Cmin by approximately 50%.
Effect of Everolimus on Antiepileptic Drugs (AEDs)
Everolimus increased pre-dose concentrations of the carbamazepine, clobazam, oxcarbazepine, and clobazam’s metabolite N-desmethylclobazam by about 10%. Everolimus had no impact on pre-dose concentrations of AEDs that are substrates of CYP3A4 (e.g., clonazepam and zonisamide) or other AEDs, including valproic acid, topiramate, phenobarbital, and phenytoin.
13 Nonclinical Toxicology
Â
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Administration of everolimus for up to 2 years did not indicate oncogenic potential in mice and rats up to the highest doses tested (0.9 mg/kg) corresponding, respectively to 3.9 and 0.2 times the estimated human exposure based on AUC at the recommended dose of everolimus 10 mg orally once daily.
Everolimus was not genotoxic in a battery of in vitro assays (Ames mutation test in Salmonella, mutation test in L5178Y mouse lymphoma cells, and chromosome aberration assay in V79 Chinese hamster cells). Everolimus was not genotoxic in an in vivo mouse bone marrow micronucleus test at doses up to 500 mg/kg/day (1500 mg/m2/day, approximately 255-fold the recommended dose of everolimus 10 mg orally once daily, and approximately 200-fold the median dose administered to patients with TSC-associated SEGA, based on the BSA), administered as 2 doses, 24 hours apart.
Based on non-clinical findings, everolimus may impair male fertility. In a 13-week male fertility study in rats, testicular morphology was affected at doses of 0.5 mg/kg and above. Sperm motility, sperm count, and plasma testosterone levels were diminished in rats treated with 5 mg/kg. The exposures at these doses (52 ng•hr/mL and 414 ng•hr/mL, respectively) were within the range of human exposure at the recommended dose of everolimus 10 mg orally once daily (560 ng•hr/mL) and resulted in infertility in the rats at 5 mg/kg. Effects on male fertility occurred at AUC0-24h values 10% to 81% lower than human exposure at the recommended dose of everolimus 10 mg orally once daily. After a 10-13 week non-treatment period, the fertility index increased from zero (infertility) to 60%.
Oral doses of everolimus in female rats at doses ≥ 0.1 mg/kg (approximately 4% the human exposure based on AUC at the recommended dose of everolimus 10 mg orally once daily) resulted in increased incidence of pre-implantation loss, suggesting that the drug may reduce female fertility.
13.2 Animal Toxicology and/or Pharmacology
In juvenile rat toxicity studies, dose-related delayed attainment of developmental landmarks, including delayed eye-opening, delayed reproductive development in males and females and increased latency time during the learning and memory phases were observed at doses as low as 0.15 mg/kg/day.
14 Clinical Studies
14.1Hormone Receptor-Positive, HER2-Negative Breast Cancer
A randomized, double-blind, multicenter study (BOLERO-2, NCT00863655) of everolimus in combination with exemestane vs. placebo in combination with exemestane was conducted in 724 postmenopausal women with estrogen receptor-positive, HER2-negative advanced breast cancer with recurrence or progression following prior therapy with letrozole or anastrozole. Randomization was stratified by documented sensitivity to prior hormonal therapy (yes vs. no) and by the presence of visceral metastasis (yes vs. no). Sensitivity to prior hormonal therapy was defined as either (1) documented clinical benefit (complete response [CR], partial response [PR], stable disease ≥ 24 weeks) to at least one prior hormonal therapy in the advanced setting or (2) at least 24 months of adjuvant hormonal therapy prior to recurrence. Patients were permitted to have received 0-1 prior lines of chemotherapy for advanced disease. The major efficacy outcome measure was progression-free survival (PFS) evaluated by RECIST (Response Evaluation Criteria in Solid Tumors), based on investigator (local radiology) assessment. Other outcome measures included overall survival (OS) and objective response rate (ORR).
Patients were randomized 2:1 to everolimus 10 mg orally once daily in combination with exemestane 25 mg once daily (n = 485) or to placebo in combination with exemestane 25 mg orally once daily (n = 239). The two treatment groups were generally balanced with respect to baseline demographics and disease characteristics. Patients were not permitted to cross over to everolimus at the time of disease progression.
The trial demonstrated a statistically significant improvement in PFS by investigator assessment (Table 20 and Figure 1). The results of the PFS analysis based on independent central radiological assessment were consistent with the investigator assessment. PFS results were also consistent across the subgroups of age, race, presence and extent of visceral metastases, and sensitivity to prior hormonal therapy.
ORR was higher in the everolimus in combination with exemestane arm vs. the placebo in combination with exemestane arm (Table 20). There were 3 complete responses (0.6%) and 58 partial responses (12%) in the everolimus arm. There were no complete responses and 4 partial responses (1.7%) in the placebo in combination with exemestane arm.
After a median follow-up of 39.3 months, there was no statistically significant difference in OS between the everolimus in combination with exemestane arm and the placebo in combination with exemestane arm [HR 0.89 (95% CI: 0.73, 1.10)].
Table 20: Efficacy Results in Hormone-Receptor Positive, HER-2 Negative Breast Cancer in BOLERO-2
Analysis
Everolimus
with Exemestane
N = 485
Placebo
with Exemestane
N = 239
Hazard Ratio
p-value
Median progression-free survival (months, 95% CI)
Investigator radiological review
7.8
(6.9, 8.5)
3.2
(2.8, 4.1)
0.45Hazard ratio is obtained from the stratified Cox proportional-hazards model by sensitivity to prior hormonal therapy and presence of visceral metastasis.
(0.38, 0.54)
< 0.0001p-value is obtained from the one-sided log-rank test stratified by sensitivity to prior hormonal therapy and presence of visceral metastasis.
Independent radiological review
11.0
(9.7, 15.0)
4.1
(2.9, 5.6)
0.38
(0.3, 0.5)
< 0.0001
Best overall response (%, 95% CI)
Objective response rate (ORR)Objective response rate = proportion of patients with CR or PR.
12.6%
(9.8, 15.9)
1.7% (0.5, 4.2)
n/aNot applicable.
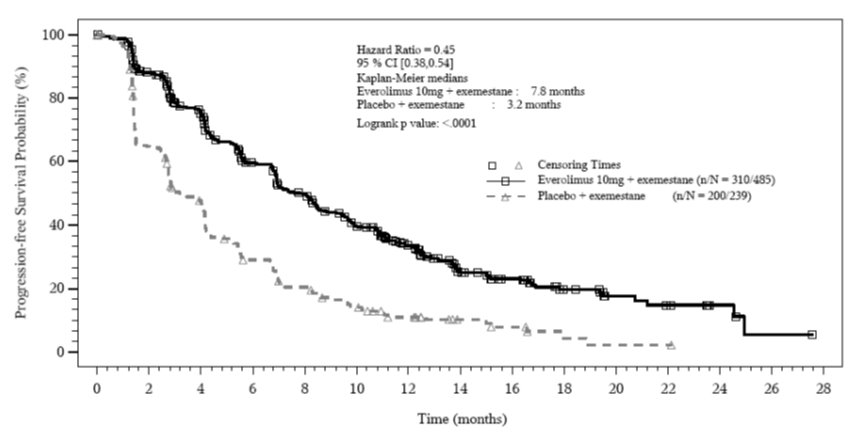
Figure 1: Kaplan-Meier Curves for Progression-Free Survival by Investigator Radiological Review in Hormone Receptor-Positive, HER-2 Negative Breast Cancer in BOLERO-2 14.2Neuroendocrine Tumors (NET)
A randomized, double-blind, multicenter study (RADIANT-4, NCT01524783) of everolimus in combination with BSC compared to placebo in combination with BSC was conducted in patients with unresectable, locally advanced or metastatic, well differentiated, non-functional NET of GI (excluding pancreatic) or lung origin. The study required that patients had well-differentiated (low or intermediate grade) histology, no prior or current history of carcinoid symptoms, and evidence of disease progression within 6 months prior to randomization. Patients were randomized 2:1 to receive either everolimus 10 mg once daily or placebo, and stratified by prior somatostatin analog use (yes vs. no), tumor origin and WHO performance status (0 vs. 1). The major efficacy outcome measure was PFS based on independent radiological assessment evaluated by RECIST. Additional efficacy outcome measures were OS and ORR.
A total of 302 patients were randomized, 205 to the everolimus arm and 97 to the placebo arm. The median age was 63 years (22 to 86 years); 47% were male; 76% were white; 74% had WHO performance status of 0 and 26% had WHO performance status of 1. The most common primary sites of tumor were lung (30%), ileum (24%), and rectum (13%).
The study demonstrated a statistically significant improvement in PFS per independent radiological review (Table 22 and Figure 3). The final OS analysis did not show a statistically significant difference between those patients who received everolimus or placebo (HR = 0.90 [95% CI: 0.66, 1.24]).
Table 22: Progression-Free Survival in Neuroendocrine Tumors of Gastrointestinal or Lung Origin in RADIANT-4
Everolimus
N = 205
Placebo
N = 97
Progression-Free Survival
Number of Events
113 (55%)
65 (67%)
Progressive Disease
104 (51%)
60 (62%)
Death
9 (4%)
5 (5%)
Median PFS in months (95% CI)
11.0 (9.2, 13.3)
3.9 (3.6, 7.4)
Hazard Ratio (95% CI)Hazard ratio is obtained from the stratified Cox model.
0.48 (0.35, 0.67)
p-valuep-value is obtained from the stratified log-rank test.
< 0.001
Overall Response Rate
2%
1%
Â
Figure 3: Kaplan-Meier Curves for Progression-Free Survival in NET of GI or Lung Origin in RADIANT-4
Â
The safety and effectiveness of everolimus in patients with locally advanced or metastatic functional carcinoid tumors have not been demonstrated. In a randomized (1:1), double-blind, multicenter trial (RADIANT-2, NCT00412061) in 429 patients with carcinoid tumors, everolimus in combination with long-acting octreotide (Sandostatin LAR®) was compared to placebo in combination with long-acting octreotide. After documented radiological progression, patients on the placebo arm could receive everolimus; of those randomized to placebo, 67% received open-label everolimus in combination with long-acting octreotide. The study did not meet its major efficacy outcome measure of a statistically significant improvement in PFS and the final analysis of OS favored the placebo in combination with long-acting octreotide arm.
14.4Tuberous Sclerosis Complex (TSC)-Associated Renal Angiomyolipoma
A randomized (2:1), double-blind, placebo-controlled trial (EXIST-2, NCT00790400) of everolimus tablets was conducted in 118 patients with renal angiomyolipoma as a feature of TSC (n = 113) or sporadic lymphangioleiomyomatosis (n = 5). The key eligibility requirements for this trial were at least one angiomyolipoma of ≥ 3 cm in longest diameter on CT/MRI based on local radiology assessment, no immediate indication for surgery, and age ≥ 18 years. Patients received everolimus tablets 10 mg or matching placebo orally once daily until disease progression or unacceptable toxicity. CT or MRI scans for disease assessment were obtained at baseline, 12, 24, and 48 weeks and annually thereafter. Clinical and photographic assessment of skin lesions were conducted at baseline and every 12 weeks thereafter until treatment discontinuation. The major efficacy outcome measure was angiomyolipoma response rate based on independent central radiology review, which was defined as a ≥ 50% reduction in angiomyolipoma volume, absence of new angiomyolipoma lesion ≥ 1 cm, absence of kidney volume increase ≥ 20%, and no angiomyolipoma related bleeding of ≥ Grade 2. Key supportive efficacy outcome measures were time to angiomyolipoma progression and skin lesion response rate. The primary analyses of efficacy outcome measures were limited to the blinded treatment period and conducted 6 months after the last patient was randomized. The comparative angiomyolipoma response rate analysis was stratified by use of enzyme-inducing antiepileptic drugs (EIAEDs) at randomization (yes vs. no).
Of the 118 patients enrolled, 79 were randomized to everolimus tablets and 39 to placebo. The median age was 31 years (18 to 61 years), 34% were male, and 89% were white. At baseline, 17% of patients were receiving EIAEDs. On central radiology review at baseline, 92% of patients had at least 1 angiomyolipoma of ≥ 3 cm in longest diameter, 29% had angiomyolipomas ≥ 8 cm, 78% had bilateral angiomyolipomas, and 97% had skin lesions. The median values for the sum of all target renal angiomyolipoma lesions at baseline were 85 cm3 (9 to 1612 cm3) and 120 cm3 (3 to 4520 cm3) in the everolimus tablets and placebo arms, respectively. Forty-six (39%) patients had prior renal embolization or nephrectomy. The median duration of follow-up was 8.3 months (0.7 to 24.8 months) at the time of the primary analysis.
The renal angiomyolipoma response rate was statistically significantly higher in everolimus tablets-treated patients (Table 24). The median response duration was 5.3+ months (2.3+ to 19.6+ months).
There were 3 patients in the everolimus tablets arm and 8 patients in the placebo arm with documented angiomyolipoma progression by central radiologic review (defined as a ≥ 25% increase from nadir in the sum of angiomyolipoma target lesion volumes to a value greater than baseline, appearance of a new angiomyolipoma ≥ 1 cm in longest diameter, an increase in renal volume ≥ 20% from nadir for either kidney and to a value greater than baseline, or Grade ≥ 2 angiomyolipoma-related bleeding). The time to angiomyolipoma progression was statistically significantly longer in the everolimus tablets arm (HR 0.08 [95% CI: 0.02, 0.37]; p < 0.0001).
Table 24: Angiomyolipoma Response Rate in TSC-Associated Renal Angiomyolipoma in EXIST-2
Everolimus Tablets
N = 79
Placebo
N = 39
p-value
Primary analysis
   Angiomyolipoma response ratePer independent central radiology review. - %
41.8
0
< 0.0001
   95% CI
(30.8, 53.4)
(0.0, 9.0)
Skin lesion response rates were assessed by local investigators for 77 patients in the everolimus tablets arm and 37 patients in the placebo arm who presented with skin lesions at study entry. The skin lesion response rate was statistically significantly higher in the everolimus tablets arm (26% vs. 0, p = 0.0011); all skin lesion responses were partial responses, defined as visual improvement in 50% to 99% of all skin lesions durable for at least 8 weeks (Physician's Global Assessment of Clinical Condition).
Patients randomized to placebo were permitted to receive everolimus tablets at the time of angiomyolipoma progression or after the time of the primary analysis. After the primary analysis, patients treated with everolimus tablets underwent additional follow-up CT or MRI scans to assess tumor status until discontinuation of treatment or completion of 4 years of follow-up after the last patient was randomized. A total of 112 patients (79 randomized to everolimus tablets and 33 randomized to placebo) received at least one dose of everolimus tablets. The median duration of everolimus tablets treatment was 3.9 years (0.5 months to 5.3 years) and the median duration of follow-up was 3.9 years (0.9 months to 5.4 years). During the follow-up period after the primary analysis, 32 patients (in addition to the 33 patients identified at the time of the primary analysis) had an angiomyolipoma response based upon independent central radiology review. Among the 65 responders out of 112 patients, the median time to angiomyolipoma response was 2.9 months (2.6 to 33.8 months). Fourteen percent of the 112 patients treated with everolimus tablets had angiomyolipoma progression by the end of the follow-up period. No patient underwent a nephrectomy for angiomyolipoma progression and one patient underwent renal embolization while treated with everolimus tablets.
14.5Tuberous Sclerosis Complex (TSC)-Associated Subependymal Giant Cell Astrocytoma (SEGA)
A randomized (2:1), double-blind, placebo-controlled trial (EXIST-1, NCT00789828) of everolimus was conducted in 117 pediatric and adult patients with SEGA and TSC. Eligible patients had at least one SEGA lesion ≥ 1 cm in longest diameter on MRI based on local radiology assessment and one or more of the following: serial radiological evidence of SEGA growth, a new SEGA lesion ≥ 1 cm in longest diameter, or new or worsening hydrocephalus. Patients randomized to the treatment arm received everolimus at a starting dose of 4.5 mg/m2 daily, with subsequent dose adjustments as needed to achieve and maintain everolimus trough concentrations of 5 to 15 ng/mL as tolerated. Everolimus or matched placebo continued until disease progression or unacceptable toxicity. MRI scans for disease assessment were obtained at baseline, 12, 24, and 48 weeks, and annually thereafter.
The main efficacy outcome measure was SEGA response rate based on independent central radiology review. SEGA response was defined as a ≥ 50% reduction in the sum of SEGA volume relative to baseline, in the absence of unequivocal worsening of non-target SEGA lesions, a new SEGA lesion ≥ 1 cm, and new or worsening hydrocephalus. The primary analysis of SEGA response rate was limited to the blinded treatment period and conducted 6 months after the last patient was randomized. The analysis of SEGA response rate was stratified by use of enzyme-inducing antiepileptic drugs (EIAEDs) at randomization (yes vs. no).
Of the 117 patients enrolled, 78 were randomized to everolimus and 39 to placebo. The median age was 9.5 years (0.8 to 26 years); a total of 20 patients were < 3 years, 54 patients were 3 to < 12 years, 27 patients were 12 to < 18 years, and 16 patients were ≥ 18 years; 57% were male, and 93% were white. At baseline, 18% of patients were receiving EIAEDs. Based on central radiology review at baseline, 98% of patients had at least one SEGA lesion ≥ 1.0 cm in longest diameter, 79% had bilateral SEGAs, 43% had ≥ 2 target SEGA lesions, 26% had growth in or into the inferior surface of the ventricle, 9% had evidence of growth beyond the subependymal tissue adjacent to the ventricle, and 7% had radiographic evidence of hydrocephalus. The median values for the sum of all target SEGA lesions at baseline were 1.63 cm3 (0.18 to 25.15 cm3) and 1.30 cm3 (0.32 to 9.75 cm3) in the everolimus and placebo arms, respectively. Eight (7%) patients had prior SEGA-related surgery. The median duration of follow-up was 8.4 months (4.6 to 17.2 months) at the time of primary analysis.
The SEGA response rate was statistically significantly higher in everolimus-treated patients (Table 25). At the time of the primary analysis, all SEGA responses were ongoing and the median duration of response was 5.3 months (2.1 to 8.4 months).
With a median follow-up of 8.4 months, SEGA progression was detected in 15.4% of the 39 patients randomized to receive placebo and none of the 78 patients randomized to receive everolimus. No patient in either treatment arm required surgical intervention.
Table 25: Subependymal Giant Cell Astrocytoma Response Rate in TSC-Associated SEGA in EXIST-1
Everolimus
N = 78
Placebo
N = 39
p-value
Primary analysis
   SEGA response ratePer independent central radiology review. - (%)
35
0
< 0.0001
   95% CI
24, 46
0, 9
Patients randomized to placebo were permitted to receive everolimus at the time of SEGA progression or after the primary analysis, whichever occurred first. After the primary analysis, patients treated with everolimus underwent additional follow-up MRI scans to assess tumor status until discontinuation of treatment or completion of 4 years of follow-up after the last patient was randomized. A total of 111 patients (78 patients randomized to everolimus and 33 patients randomized to placebo) received at least one dose of everolimus. Median duration of everolimus treatment and follow-up was 3.9 years (0.2 to 4.9 years).
By four years after the last patient was enrolled, 58% of the 111 patients treated with everolimus had a ≥ 50% reduction in SEGA volume relative to baseline, including 27 patients identified at the time of the primary analysis and 37 patients with a SEGA response after the primary analysis. The median time to SEGA response was 5.3 months (2.5 to 33.1 months). Twelve percent of the 111 patients treated with everolimus had documented disease progression by the end of the follow-up period and no patient required surgical intervention for SEGA during the study.
Study 2485 (NCT00411619) was an open-label, single-arm trial conducted to evaluate the antitumor activity of everolimus 3 mg/m2/orally once daily in patients with SEGA and TSC. Serial radiological evidence of SEGA growth was required for entry. Tumor assessments were performed every 6 months for 60 months after the last patient was enrolled or disease progression, whichever occurred earlier. The major efficacy outcome measure was the reduction in volume of the largest SEGA lesion with 6 months of treatment, as assessed via independent central radiology review. Progression was defined as an increase in volume of the largest SEGA lesion over baseline that was ≥ 25% over the nadir observed on study.
A total of 28 patients received everolimus for a median duration of 5.7 years (5 months to 6.9 years); 82% of the 28 patients remained on everolimus for at least 5 years. The median age was 11 years (3 to 34 years), 61% male, 86% white.
At the primary analysis, 32% of the 28 patients (95% CI: 16%, 52%) had an objective response at 6 months, defined as at least a 50% decrease in volume of the largest SEGA lesion. At the completion of the study, the median duration of durable response was 12 months (3 months to 6.3 years).
By 60 months after the last patient was enrolled, 11% of the 28 patients had documented disease progression. No patient developed a new SEGA lesion while on everolimus. Nine additional patients were identified as having a ≥ 50% volumetric reduction in their largest SEGA lesion between 1 to 4 years after initiating everolimus, including 3 patients who had surgical resection with subsequent regrowth prior to receiving everolimus.
15 References
1. OSHA Hazardous Drugs. OSHA. http://www.osha.gov/SLTC/hazardousdrugs/index.html.
16 How Supplied/storage And Handling
Everolimus Tablets are available containing 2.5 mg, 5 mg, 7.5 mg or 10 mg of everolimus.
The 2.5 mg tablets are white to slightly yellow, capsule shaped, unscored tablets debossed with M on one side of the tablet and EVR2 on the other side. They are available as follows:
NDC 0378-3096-28bottles of 28 tablets
NDC 0378-3096-85cartons of 28 tablets
The 5 mg tablets are white to slightly yellow, capsule shaped, unscored tablets debossed with M on one side of the tablet and EVR5 on the other side. They are available as follows:
NDC 0378-3097-28bottles of 28 tablets
NDC 0378-3097-85cartons of 28 tablets
The 7.5 mg tablets are white to slightly yellow, capsule shaped, unscored tablets debossed with M on one side of the tablet and EVR7 on the other side. They are available as follows:
NDC 0378-3098-28bottles of 28 tablets
NDC 0378-3098-85cartons of 28 tablets
The 10 mg tablets are white to slightly yellow, capsule shaped, unscored tablets debossed with M on one side of the tablet and EVR10 on the other side. They are available as follows:
NDC 0378-3099-28bottles of 28 tablets
NDC 0378-3099-85cartons of 28 tablets
Store at 20° to 25°C (68° to 77°F). [See USP Controlled Room Temperature.]
Store in the original container, protect from light and moisture.
Follow special handling and disposal procedures for anti-cancer pharmaceuticals.1
17 Patient Counseling Information
Advise the patient to read the FDA-approved patient labeling (Patient Information).
Non-infectious Pneumonitis: Advise patients of the risk of developing non-infectious pneumonitis and to immediately report any new or worsening respiratory symptoms to their healthcare provider [see Warnings and Precautions (5.1)].
Infections: Advise patients that they are more susceptible to infections and that they should immediately report any signs or symptoms of infections to their healthcare provider [see Warnings and Precautions (5.2)].
Hypersensitivity Reactions: Advise patients of the risk of clinically significant hypersensitivity reactions and to promptly contact their healthcare provider or seek emergency care for signs of hypersensitivity reaction, including rash, itching, hives, difficulty breathing or swallowing, flushing, chest pain, or dizziness [see Contraindications (4), Warnings and Precautions (5.3)].
Angioedema with Concomitant Use of ACE Inhibitors: Advise patients to avoid ACE inhibitors and to promptly contact their healthcare provider or seek emergency care for signs or symptoms of angioedema [see Warnings and Precautions (5.4)].
Stomatitis: Advise patients of the risk of stomatitis and to use alcohol-free mouthwashes during treatment [see Warnings and Precautions (5.5)].
Renal Impairment: Advise patients of the risk of developing kidney failure and the need to monitor their kidney function periodically during treatment [see Warnings and Precautions (5.6)].
Risk of Impaired Wound Healing:Â Advise patients that everolimus may impair wound healing. Advise patients to inform their healthcare provider of any planned surgical procedure [see Warnings and Precautions (5.7)].
Geriatric Patients: Inform patients that in a study conducted in patients with breast cancer, the incidence of deaths and adverse reactions leading to permanent discontinuation was higher in patients ≥ 65 years compared to patients < 65 years [see Warnings and Precautions (5.8), Use in Specific Populations (8.5)].
Metabolic Disorders: Advise patients of the risk of metabolic disorders and the need to monitor glucose and lipids periodically during therapy [see Warnings and Precautions (5.9)].
Myelosuppression: Advise patients of the risk of myelosuppression and the need to monitor CBCs periodically during therapy [see Warnings and Precautions (5.10)].
Risk of Infection or Reduced Immune Response With Vaccination: Advise patients to avoid the use of live vaccines and close contact with those who have received live vaccines [see Warnings and Precautions (5.11)].
Embryo-Fetal Toxicity: Advise females of reproductive potential of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment and for 8 weeks after the last dose. Advise patients to inform their healthcare provider of a known or suspected pregnancy. Advise males with female partners of reproductive potential to use effective contraception during treatment and for 4 weeks after the last dose [see Warnings and Precautions (5.13) , Use in Specific Populations (8.1, 8.3)].
Radiation Sensitization and Radiation Recall:Â Radiation sensitization and recall can occur in patients treated with radiation prior to, during, or subsequent to everolimus tablets treatment. Advise patients to inform their healthcare provider if they have had or are planning to receive radiation therapy [see Warnings and Precautions (5.12)].
Lactation: Advise women not to breastfeed during treatment with everolimus tablets and for 2 weeks after the last dose [see Use in Specific Populations (8.2)].
Infertility: Advise males and females of reproductive potential of the potential risk for impaired fertility [see Use in Specific Populations (8.3)].
Manufactured for: Mylan Pharmaceuticals Inc. Morgantown, WV 26505 U.S.A.
Manufactured by: Auro PR Inc. RD 156 Caguas West Industrial Park, Lot 24Caguas, PR 00725 U.S.A.
Patient Information
Everolimus Tablets
(e″ ver oh′ li mus)
Read this Patient Information leaflet that comes with everolimus tablets before you start taking them and each time you get a refill. There may be new information. This information does not take the place of talking to your healthcare provider about your medical condition or treatment.
What is the most important information I should know about everolimus tablets?
Â
Everolimus tablets can cause serious side effects, including:
1. You may develop lung or breathing problems. In some people lung or breathing problems may be severe and can lead to death. Tell your healthcare provider right away if you have any of these symptoms:
• New or worsening cough• Shortness of breath• Chest pain• Difficulty breathing or wheezing
2. You may be more likely to develop an infection, such as pneumonia, or a bacterial, fungal or viral infection. Viral infections may include active hepatitis B in people who have had hepatitis B in the past (reactivation). In some people (including adults and children) these infections may be severe and can lead to death. You may need to be treated as soon as possible.
Â
   Tell your healthcare provider right away if you have a temperature of 100.5°F or above, chills, or do not feel well.
Â
   Symptoms of hepatitis B or infection may include the following:
• Fever• Chills• Skin rash• Joint pain and swelling• Tiredness
• Loss of appetite• Nausea• Pale stools or dark urine• Yellowing of the skin• Pain in the upper right side of the stomach
3. Severe allergic reactions. Call your healthcare provider or get medical help right away if you get signs and symptoms of a severe allergic reaction, including: rash, itching, hives, flushing, trouble breathing or swallowing, chest pain or dizziness.
Â
4. Possible increased risk for a type of allergic reaction called angioedema, in people who take an Angiotensin-Converting Enzyme (ACE) inhibitor medicine during treatment with everolimus tablets. Talk with your healthcare provider before taking everolimus tablets if you are not sure if you take an ACE inhibitor medicine. Get medical help right away if you have trouble breathing or develop swelling of your tongue, mouth, or throat during treatment with everolimus tablets.
Â
5. Mouth ulcers and sores. Mouth ulcers and sores are common during treatment with everolimus tablets but can also be severe. When you start treatment with everolimus tablets, your healthcare provider may tell you to also start a prescription mouthwash to reduce the likelihood of getting mouth ulcers or sores and to reduce their severity. Follow your healthcare provider’s instructions on how to use this prescription mouthwash. If you develop pain, discomfort, or open sores in your mouth, tell your healthcare provider. Your healthcare provider may tell you to restart this mouthwash or to use a special mouthwash or mouth gel that does not contain alcohol, peroxide, iodine, or thyme.
Â
6. You may develop kidney failure. In some people this may be severe and can lead to death. Your healthcare provider should do tests to check your kidney function before and during your treatment with everolimus tablets.
Â
If you have any of the serious side effects uled above, you may need to stop taking everolimus tablets for a while or use a lower dose. Follow your healthcare provider’s instructions.
What are everolimus tablets?
Everolimus tablets are a prescription medicine used to treat:
• advanced hormone receptor-positive, HER2-negative breast cancer, along with the medicine exemestane, in postmenopausal women who have already received certain other medicines for their cancer.• adults with a type of cancer known as neuroendocrine tumor (NET) of the stomach and intestine (gastrointestinal), or lung that has progressed and cannot be treated with surgery.
Everolimus tablets are not for use in people with carcinoid tumors that actively produce hormones.
• people with the following types of tumors that are seen with a genetic condition called tuberous sclerosis complex (TSC):
o adults with a kidney tumor called angiomyolipoma when their kidney tumor does not require surgery right away.o adults and children 1 year of age and older with a brain tumor called subependymal giant cell astrocytoma (SEGA) when the tumor cannot be removed completely by surgery.
It is not known if everolimus tablets are safe and effective in children to treat:
• hormone receptor-positive, HER-2 negative breast cancer• a type of cancer called neuroendocrine tumors (NET)• kidney cancer (renal cell carcinoma)• a kidney tumor called angiomyolipoma, that can happen in children with a genetic condition called tuberous sclerosis complex (TSC).
Do not take everolimus tablets if you have had a severe allergic reaction to everolimus.
Â
Talk to your healthcare provider before taking this medicine if you are allergic to:
• a medicine that contains sirolimus• a medicine that contains temsirolimus
Ask your healthcare provider if you do not know.
Before taking everolimus tablets, tell your healthcare provider about all of your medical conditions, including if you:
• Have or have had kidney problems• Have or have had liver problems• Have diabetes or high blood sugar• Have high blood cholesterol levels• Have any infections• Previously had hepatitis B• Are scheduled to receive any vaccinations. You should not receive a “live vaccine” or be around people who have recently received a “live vaccine” during your treatment with everolimus tablets. If you are not sure about the type of immunization or vaccine, ask your healthcare provider. For children with TSC and SEGA, work with your healthcare provider to complete the recommended childhood series of vaccines before your child starts treatment with everolimus tablets.• Are pregnant, can become pregnant, or have a partner who can become pregnant. Everolimus tablets can cause harm to your unborn baby. Females who are able to become pregnant:o Your healthcare provider will give you a pregnancy test before you start treatment with everolimus tablets.o You should use effective birth control during treatment and for 8 weeks after your last dose of everolimus tablets. Â Males with a female partner, you should use effective birth control during treatment and for 4 weeks after your last dose of everolimus tablets. Â Talk to your healthcare provider about birth control methods that may be right for you during this time. If you become pregnant or think you are pregnant, tell your healthcare provider right away.• Are breastfeeding or plan to breastfeed. It is not known if everolimus passes into your breast milk. Do not breastfeed during treatment and for 2 weeks after your last dose of everolimus tablets.• Are planning to have surgery or if you have had a recent surgery. You should stop taking everolimus tablets at least 1 week before planned surgery. See “What are the possible side effects of everolimus tablets?”• Have received radiation therapy or are planning to receive radiation therapy in the future. See “What are the possible side effects of everolimus tablets?”
Tell your healthcare provider about all of the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements.
Â
Everolimus tablets may affect the way other medicines work, and other medicines can affect how everolimus tablets work. Taking everolimus tablets with other medicines can cause serious side effects.
Â
Know the medicines you take. Keep a ul of them and show it to your healthcare provider and pharmacist when you get a new medicine. Especially tell your healthcare provider if you take:
• St. John’s Wort (Hypericum perforatum)• Medicine for:
o Fungal infectionso Bacterial infectionso Tuberculosiso Seizureso HIV-AIDSo Heart conditions or high blood pressure• Medicines that weaken your immune system (your body’s ability to fight infections and other problems)
Ask your healthcare provider or pharmacist if you are not sure if your medicine is one of those taken for the conditions uled above. If you are taking any medicines for the conditions uled above, your healthcare provider might need to prescribe a different medicine or your dose of everolimus tablets may need to be changed. You should also tell your healthcare provider before you start taking any new medicine.
How should I take everolimus tablets?
• Your healthcare provider will prescribe the dose of everolimus tablets that is right for you.• Take everolimus tablets exactly as your healthcare provider tells you to.• Your healthcare provider may change your dose of everolimus tablets or tell you to temporarily interrupt dosing, if needed.• Take only everolimus tablets or AFINITOR DISPERZ. Do not mix everolimus tablets and AFINITOR DISPERZ together.• Use scissors to open the buler pack.• Take everolimus tablets 1 time each day at about the same time.• Take everolimus tablets the same way each time, either with food or without food.• If you take too many everolimus tablets, contact your healthcare provider or go to the nearest hospital emergency room right away. Take the pack of everolimus tablets with you.• If you miss a dose of everolimus tablets, you may take it if it is less than 6 hours after the time you normally take it. If it is more than 6 hours after you normally take your everolimus tablets, skip the dose for that day. The next day, take everolimus tablets at your usual time. Do not take 2 doses to make up for a missed dose. If you are not sure about what to do, call your healthcare provider.• You should have blood tests before you start everolimus tablets and as needed during your treatment. These will include tests to check your blood cell count, kidney and liver function, cholesterol, and blood sugar levels.• If you take everolimus tablets to treat SEGA, you will also need to have blood tests regularly to measure how much medicine is in your blood. This will help your healthcare provider decide how many everolimus tablets you need to take.
Everolimus tablets:
• Swallow everolimus tablets whole with a glass of water. Do not take any tablet that is broken or crushed.
What should I avoid while taking everolimus tablets?
You should not drink grapefruit juice or eat grapefruit during your treatment with everolimus tablets. It may make the amount of everolimus in your blood increase to a harmful level.
What are the possible side effects of everolimus tablets?
Everolimus tablets can cause serious side effects, including:
• See “What is the most important information I should know about everolimus tablets?” for more information.• Risk of wound healing problems. Wounds may not heal properly during everolimus tablets treatment. Tell your healthcare provider if you plan to have any surgery before starting or during treatment with everolimus tablets.
o You should stop taking everolimus tablets at least 1 week before planned surgery.o Your healthcare provider should tell you when you may start taking everolimus tablets again after surgery.• Increased blood sugar and fat (cholesterol and triglyceride) levels in the blood. Your healthcare provider should do blood tests to check your fasting blood sugar, cholesterol, and triglyceride levels in the blood before you start and during treatment with everolimus tablets.• Decreased blood cell counts. Everolimus tablets can cause you to have decreased red blood cells, white blood cells, and platelets. Your healthcare provider should do blood tests to check your blood cell counts before you start and during treatment with everolimus tablets.• Worsening side effects from radiation treatment, that can sometimes be severe. Tell your healthcare provider if you have had or are planning to receive radiation therapy.
The most common side effects of everolimus tablets in people with advanced hormone receptor-positive, HER2-negative breast cancer and advanced neuroendocrine tumors of the stomach and intestine (gastrointestinal) or lung include:
• Infections• Rash• Feeling weak or tired• Diarrhea• Swelling of arms, hands, feet, ankles, face or other parts of the body
• Stomach-area (abdominal) pain• Nausea• Fever• Cough• Headache• Decreased appetite
The most common side effects of everolimus tablets in people who have SEGA or renal angiomyolipoma include respiratory tract infections.
Â
Other side effects that may occur with everolimus tablets:
• Absence of menstrual periods (menstruation). You may miss 1 or more menstrual periods. Tell your healthcare provider if this happens.• Everolimus tablets may affect fertility in females and may affect your ability to become pregnant. Talk to your healthcare provider if this is a concern for you.• Everolimus tablets may affect fertility in males and may affect your ability to father a child. Talk to your healthcare provider if this is a concern for you.
Tell your healthcare provider if you have any side effect that bothers you or does not go away.
Â
These are not all the possible side effects of everolimus tablets. For more information, ask your healthcare provider or pharmacist.
Â
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store everolimus tablets?
• Store everolimus tablets at room temperature, between 20° to 25°C (68° to 77°F).• Keep everolimus tablets in the pack they come in.• Open the bottle or buler pack just before taking everolimus tablets.• Keep everolimus tablets dry and away from light.• Do not use everolimus tablets that are out of date or no longer needed.
Keep everolimus tablets and all medicines out of the reach of children.
General information about the safe and effective use of everolimus tablets
Medicines are sometimes prescribed for purposes other than those uled in a Patient Information leaflet. Do not use everolimus tablets for a condition for which they were not prescribed. Do not give everolimus tablets to other people, even if they have the same problem you have. They may harm them. This leaflet summarizes the most important information about everolimus tablets. If you would like more information, talk with your healthcare provider. You can ask your healthcare provider or pharmacist for information written for healthcare professionals.
What are the ingredients in everolimus tablets?
Active ingredient: everolimus.
Inactive ingredients: anhydrous lactose, butylated hydroxytoluene, colloidal silicon dioxide, croscarmellose sodium, hypromellose and magnesium stearate.
Â
Manufactured for: Mylan Pharmaceuticals Inc., Morgantown, WV 26505 U.S.A.
Â
The brands uled are trademarks of their respective owners.
Â
For more information, call Mylan at 1-877-446-3679 (1-877-4-INFO-RX).
This Patient Information has been approved by the U.S. Food and Drug Administration.
Manufactured for: Mylan Pharmaceuticals Inc. Morgantown, WV 26505 U.S.A.
Manufactured by: Auro PR Inc. RD 156 Caguas West Industrial Park, Lot 24Caguas, PR 00725 U.S.A.
Revised: 10/2023EVERT:RX7
Principal Display Panel 2.5 Mg
NDC 0378-3096-85
EverolimusTablets2.5 mg
Each tablet contains everolimus, 2.5 mg
Rx only     28 Tablets (carton contains 4 individual buler cards of 7 tablets)
For oral use.
Usual Dosage:Â See accompanying prescribing information.
Store at 20° to 25°C (68° to 77°F).[See USP ControlledRoom Temperature.]
Store in the original container; protect from light and moisture.
Keep this and allmedication out of thereach of children.
Manufactured for: Mylan Pharmaceuticals Inc. Morgantown, WV 26505 U.S.A.
Made in Puerto Rico
Mylan.com
M:3096:28C:R3
Principal Display Panel 5 Mg
NDC 0378-3097-85
EverolimusTablets5 mg
Each tablet contains everolimus, 5 mg
Rx only     28 Tablets (carton contains 4 individual buler cards of 7 tablets)
For oral use.
Usual Dosage:Â See accompanying prescribing information.
Store at 20° to 25°C (68° to 77°F).[See USP ControlledRoom Temperature.]
Store in the original container; protect from light and moisture.
Keep this and allmedication out of thereach of children.
Manufactured for: Mylan Pharmaceuticals Inc. Morgantown, WV 26505 U.S.A.
Made in Puerto Rico
Mylan.com
M:3097:28C:R3
Principal Display Panel 7.5 Mg
NDC 0378-3098-85
EverolimusTablets7.5 mg
Each tablet contains everolimus, 7.5 mg
Rx only     28 Tablets (carton contains 4 individual buler cards of 7 tablets)
For oral use.
Usual Dosage:Â See accompanying prescribing information.
Store at 20° to 25°C (68° to 77°F).[See USP ControlledRoom Temperature.]
Store in the original container; protect from light and moisture.
Keep this and allmedication out of thereach of children.
Manufactured for: Mylan Pharmaceuticals Inc. Morgantown, WV 26505 U.S.A.
Made in Puerto Rico
Mylan.com
M:3098:28C:R3
Principal Display Panel 10 Mg
NDC 0378-3099-85
EverolimusTablets10 mg
Each tablet contains everolimus, 10 mg
Rx only 28 Tablets (carton contains 4 individual buler cards of 7 tablets)
For oral use.
Usual Dosage: See accompanying prescribing information.
Store at 20° to 25°C (68° to 77°F).[See USP ControlledRoom Temperature.]
Store in the original container; protect from light and moisture.
Keep this and allmedication out of thereach of children.
Manufactured for: Mylan Pharmaceuticals Inc. Morgantown, WV 26505 U.S.A.
Made in Puerto Rico
Mylan.com
M:3099:28C:R3
DISCLAIMER:
"This tool does not provide medical advice, and is for informational and educational purposes only, and is not a substitute for professional medical advice, treatment or diagnosis. Call your doctor to receive medical advice. If you think you may have a medical emergency, please dial 911."
"Do not rely on openFDA to make decisions regarding medical care. While we make every effort to ensure that data is accurate, you should assume all results are unvalidated. We may limit or otherwise restrict your access to the API in line with our Terms of Service."
"This product uses publicly available data from the U.S. National Library of Medicine (NLM), National Institutes of Health, Department of Health and Human Services; NLM is not responsible for the product and does not endorse or recommend this or any other product."
PillSync may earn a commission via links on our site
