Kerendia Dailymed
Generic: finerenone is used for the treatment of Adrenal Insufficiency Myocardial Infarction Renal Insufficiency


Go PRO for all pill images
1 Indications And Usage
Kerendia is indicated to reduce the risk of sustained eGFR decline, end-stage kidney disease, cardiovascular death, non-fatal myocardial infarction, and hospitalization for heart failure in adult patients with chronic kidney disease (CKD) associated with type 2 diabetes (T2D).
 Kerendia is a non-steroidal mineralocorticoid receptor antagonist (MRA) indicated to reduce the risk of sustained eGFR decline, end stage kidney disease, cardiovascular death, non-fatal myocardial infarction, and hospitalization for heart failure in adult patients with chronic kidney disease (CKD) associated with type 2 diabetes (T2D). (1 )
2 Dosage And Administration
• The recommended starting dosage is 10 mg or 20 mg orally once daily based on estimated glomerular filtration rate (eGFR) and serum potassium thresholds. (2.1 )• Increase dosage after 4 weeks to the target dose of 20 mg once daily, based on eGFR and serum potassium thresholds. (2.3 )• Tablets may be taken with or without food (2.2 )2.1 Prior to Initiation of Kerendia
Measure serum potassium levels and estimated glomerular filtration rate (eGFR) before initiation. Do not initiate treatment if serum potassium is > 5.0 mEq/L [see Warnings and Precautions (5.1)].
2.2 Recommended Starting Dosage
The recommended starting dose of Kerendia is based on eGFR and is presented in Table 1.
Table 1: Recommended Starting Dosage
eGFR (mL/min/1.73m2)
Starting Dose
≥ 60
20 mg once daily
≥ 25 to < 60
10 mg once daily
< 25
Initiation is not Recommended
For patients who are unable to swallow whole tablets, Kerendia may be crushed and mixed with water or soft foods such as applesauce immediately prior to use and administered orally [see Clinical Pharmacology ( 12.3)].
2.3 Monitoring and Dose Adjustment
The target daily dose of Kerendia is 20 mg.
Measure serum potassium 4 weeks after initiating treatment and adjust dose (see Table 2); if serum potassium levels are > 4.8 to 5.0 mEq/L, initiation of Kerendia treatment may be considered with additional serum potassium monitoring within the first 4 weeks based on clinical judgment and serum potassium levels [see Warnings and Precautions (5.1)]. Monitor serum potassium 4 weeks after a dose adjustment and throughout treatment, and adjust the dose as needed (see Table 2) [see Warnings and Precautions ( 5.1) and Drug Interactions (7.1)].
Table 2: Dose Adjustment Based on Current Serum Potassium Concentration and Current Dose
Current Kerendia Dose
10 mg once daily
20 mg once daily
Current Serum Potassium (mEq/L)
≤ 4.8
Increase the dose to 20 mg once daily.*
Maintain 20 mg once daily.
> 4.8 – 5.5
Maintain 10 mg once daily.
Maintain 20 mg once daily.
> 5.5
Withhold Kerendia.
Consider restarting at 10 mg once daily when serum potassium ≤ 5.0 mEq/L.
Withhold Kerendia.
Restart at 10 mg once daily when serum potassium ≤ 5.0 mEq/L.
* If eGFR has decreased by more than 30% compared to previous measurement, maintain 10 mg dose.
2.4 Missed doses
Direct a patient to take a missed dose as soon as possible after it is noticed, but only on the same day. If this is not possible, the patient should skip the dose and continue with the next dose as prescribed.
3 Dosage Forms And Strengths
Kerendia is available as a film-coated, oblong tablet in two strengths.
10 mg tablet: pink, with “FI” on one side, “10” on the other side.
20 mg tablet: yellow, with “FI” on one side, “20” on the other side.
 Tablets: 10 mg and 20 mg (3 )
4 Contraindications
Kerendia is contraindicated in patients:
• Who are receiving concomitant treatment with strong CYP3A4 inhibitors [see Drug Interactions ( 7.1)].• With adrenal insufficiency.
• Concomitant use with strong CYP3A4 inhibitors. (4 ,7.1 )• Patients with adrenal insufficiency. (4 )
5 Warnings And Precautions
• Hyperkalemia. Patients with decreased kidney function and higher baseline potassium levels are at increased risk. Monitor serum potassium levels and adjust dose as needed. (2.1 ,2.2 ,2.3 ,5.1 )5.1 Hyperkalemia
Kerendia can cause hyperkalemia [see Adverse Reactions (6.1)].
The risk for developing hyperkalemia increases with decreasing kidney function and is greater in patients with higher baseline potassium levels or other risk factors for hyperkalemia. Measure serum potassium and eGFR in all patients before initiation of treatment with Kerendia and dose accordingly [see Dosage and Administration (2.1)]. Do not initiate Kerendia if serum potassium is > 5.0 mEq/L.
Measure serum potassium periodically during treatment with Kerendia and adjust dose accordingly [see Dosage and Administration (2.3)]. More frequent monitoring may be necessary for patients at risk for hyperkalemia, including those on concomitant medications that impair potassium excretion or increase serum potassium [see Drug Interactions ( 7.1, 7.2)].
6 Adverse Reactions
The following serious adverse reactions are discussed elsewhere in the labeling:
• Hyperkalemia [see Warnings and Precautions ( 5.1)]
 Adverse reactions occurring in ≥ 1% of patients on Kerendia and more frequently than placebo are hyperkalemia, hypotension, and hyponatremia. (6.1 )
To report SUSPECTED ADVERSE REACTIONS, contact Bayer HealthCare Pharmaceuticals Inc. at 1-888-842- or FDA at 1-800-FDA-1088 orwww.fda.gov/medwatch.
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The safety of Kerendia was evaluated in 2 randomized, double-blind, placebo-controlled, multicenter pivotal phase 3 studies, FIDELIO-DKD and FIGARO-DKD, in which a total of 6510 patients were treated with 10 or 20 mg once daily over a mean duration of 2.2 years and 2.9 years, respectively.
Overall, serious adverse events occurred in 32% of patients receiving Kerendia and in 34% of patients receiving placebo in the FIDELIO-DKD study; the findings were similar in the FIGARO-DKD study. Permanent discontinuation due to adverse events also occurred in a similar proportion of patients in the two studies (6-7% of patients receiving Kerendia and 5-6% of patients receiving placebo).
The most frequently reported (≥ 10%) adverse reaction in both studies was hyperkalemia [see Warnings and Precautions ( 5.1)]. Hospitalization due to hyperkalemia for the Kerendia group was 0.9% vs 0.2% in the placebo group across both studies. Hyperkalemia led to permanent discontinuation of treatment in 1.7% of patients receiving Kerendia versus 0.6% of patients receiving placebo across both studies.
Table 3 shows adverse reactions that occurred more commonly on Kerendia than on placebo, and in at least 1% of patients treated with Kerendia.
Table 3: Adverse reactions reported in ≥ 1% of patients on Kerendia and more frequently than placebo (pooled data from FIDELIO-DKD and FIGARO-DKD)
Adverse reactions
Kerendia
N = 6510
n (%)
Placebo
N = 6489
n (%)
Hyperkalemia
912 (14.0)
448 (6.9)
Hypotension
302 (4.6)
194 (3.9)
Hyponatremia
82 (1.3)
47 (0.7)
Laboratory Test
Initiation of Kerendia may cause an initial small decrease in estimated GFR that occurs within the first 4 weeks of starting therapy, and then stabilizes. In a study that included patients with chronic kidney disease associated with type 2 diabetes, this decrease was reversible after treatment discontinuation.
Initiation of Kerendia may also cause a small increase in serum uric acid. This increase appears to attenuate over time.
7 Drug Interactions
• Strong CYP3A4 Inhibitors: Use is contraindicated. (7.1 )• Grapefruit or grapefruit juice: Avoid concomitant use. (7.1 )• Moderate or weak CYP3A4 Inhibitors: Monitor serum potassium during drug initiation or dosage adjustment of either Kerendia or the moderate or weak CYP3A4 inhibitor, and adjust Kerendia dosage as appropriate (7.1 )7.1 Effects of other drugs on Kerendia
Strong CYP3A4 Inhibitors
Kerendia is a CYP3A4 substrate. Concomitant use with a strong CYP3A4 inhibitor increases finerenone exposure [see Clinical Pharmacology (12.3)], which may increase the risk of Kerendia adverse reactions. Concomitant use of Kerendia with strong CYP3A4 inhibitors is contraindicated [see Contraindications (4)]. Avoid concomitant intake of grapefruit or grapefruit juice.
Moderate and Weak CYP3A4 Inhibitors
Kerendia is a CYP3A4 substrate. Concomitant use with a moderate or weak CYP3A4 inhibitor increases finerenone exposure [see Clinical Pharmacology (12.3)], which may increase the risk of Kerendia adverse reactions. Monitor serum potassium during drug initiation or dosage adjustment of either Kerendia or the moderate or weak CYP3A4 inhibitor, and adjust Kerendia dosage as appropriate [see Dosing and Administration ( 2.3) and Drug Interaction (7.2)].
Strong and Moderate CYP3A4 Inducers
Kerendia is a CYP3A4 substrate. Concomitant use of Kerendia with a strong or moderate CYP3A4 inducer decreases finerenone exposure [see Clinical Pharmacology (12.3)], which may reduce the efficacy of Kerendia. Avoid concomitant use of Kerendia with strong or moderate CYP3A4 inducers.
7.2 Drugs That Affect Serum Potassium
More frequent serum potassium monitoring is warranted in patients receiving concomitant therapy with drugs or supplements that increase serum potassium. [see Dosage and Administration ( 2.3) and Warnings and Precautions (5.1)].
8 Use In Specific Populations
Lactation: Breastfeeding not recommended (8.2 )
8.1 Pregnancy
Risk Summary
There are no available data on Kerendia use in pregnancy to evaluate for a drug-associated risk of major birth defects, miscarriage, or adverse maternal or fetal outcomes. Animal studies have shown developmental toxicity at exposures about 4 times those expected in humans. (see Data). The clinical significance of these findings is unclear.
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
Data
In the embryo-fetal toxicity study in rats, finerenone resulted in reduced placental weights and signs of fetal toxicity, including reduced fetal weights and retarded ossification at the maternal toxic dose of 10 mg/kg/day corresponding to an AUCunbound of 19 times that in humans. At 30 mg/kg/day, the incidence of visceral and skeletal variations was increased (slight edema, shortened umbilical cord, slightly enlarged fontanelle) and one fetus showed complex malformations including a rare malformation (double aortic arch) at an AUCunbound of about 25 times that in humans. The doses free of any findings (low dose in rats, high dose in rabbits) provide safety margins of 10 to 13 times for the AUCunbound expected in humans.
When rats were exposed during pregnancy and lactation in the pre- and postnatal developmental toxicity study, increased pup mortality and other adverse effects (lower pup weight, delayed pinna unfolding) were observed at about 4 times the AUCunbound expected in humans. In addition, the offspring showed slightly increased locomotor activity, but no other neurobehavioral changes starting at about 4 times the AUCunbound expected in humans. The dose free of findings provides a safety margin of about 2 times for the AUCunbound expected in humans.
8.2 Lactation
Risk Summary
There are no data on the presence of finerenone or its metabolite in human milk, the effects on the breastfed infant or the effects of the drug on milk production. In a pre- and postnatal developmental toxicity study in rats, increased pup mortality and lower pup weight were observed at about 4 times the AUCunbound expected in humans. These findings suggest that finerenone is present in rat milk [see Use in Specific Populations ( 8.1) and Data]. When a drug is present in animal milk, it is likely that the drug will be present in human milk. Because of the potential risk to breastfed infants from exposure to Kerendia, avoid breastfeeding during treatment and for 1 day after treatment.
8.4 Pediatric Use
The safety and efficacy of Kerendia have not been established in patients below 18 years of age.
8.5 Geriatric Use
Of the 6510 patients who received Kerendia in the FIDELIO-DKD and FIGARO-DKD studies, 55% of patients were 65 years and older, and 14% were 75 years and older. No overall differences in safety or efficacy were observed between these patients and younger patients. No dose adjustment is required.
8.6 Hepatic Impairment
Avoid use of Kerendia in patients with severe hepatic impairment (Child Pugh C).
No dosage adjustment is recommended in patients with mild or moderate hepatic impairment (Child Pugh A or B).
Consider additional serum potassium monitoring in patients with moderate hepatic impairment (Child Pugh B) [see Dosing and Administration (2.3) and Clinical Pharmacology (12.3)].
10 Overdosage
In the event of suspected overdose, immediately interrupt or reduce Kerendia treatment. The most likely manifestation of overdose is hyperkalemia. If hyperkalemia develops, standard treatment should be initiated.
Finerenone is unlikely to be efficiently removed by hemodialysis given its fraction bound to plasma proteins of about 90%.
11 Description
Kerendia contains finerenone, a nonsteroidal mineralocorticoid receptor antagonist. Finerenone’s chemical name is (4S)-4-(4-cyano-2- methoxyphenyl)-5-ethoxy-2,8-dimethyl-1,4-dihydro-1,6-naphthyridine-3-carboxamide. The molecular formula is C21H22N4O3 and the molecular weight is 378.43 g/mol. The structural formula is:
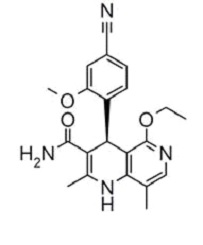
Finerenone is a white to yellow crystalline powder. It is practically insoluble in water; and sparingly soluble in 0.1 M HCl, ethanol, and acetone.
Each Kerendia tablet contains 10 mg or 20 mg of finerenone. The inactive ingredients of Kerendia are lactose monohydrate, cellulose microcrystalline, croscarmellose sodium, hypromellose, magnesium stearate, and sodium lauryl sulfate. The film coating contains hypromellose, titanium dioxide and talc, in addition to ferric oxide red (10 mg strength tablets) or ferric oxide yellow (20 mg strength tablets).
12 Clinical Pharmacology
12.1 Mechanism of Action
Finerenone is a nonsteroidal, selective antagonist of the mineralocorticoid receptor (MR), which is activated by aldosterone and cortisol and regulates gene transcription. Finerenone blocks MR mediated sodium reabsorption and MR overactivation in both epithelial (e.g., kidney) and nonepithelial (e.g., heart, and blood vessels) tissues. MR overactivation is thought to contribute to fibrosis and inflammation. Finerenone has a high potency and selectivity for the MR and has no relevant affinity for androgen, progesterone, estrogen, and glucocorticoid receptors.
12.2 Pharmacodynamics
In FIDELIO-DKD and FIGARO-DKD, randomized, double-blind, placebo-controlled, multicenter studies in adult patients with chronic kidney disease associated with type 2 diabetes, the placebo-corrected relative reduction in urinary albumin-to-creatinine ratio (UACR) in patients randomized to finerenone was 31% (95% CI 29-34%) and 32% (95% CI 30-35%) respectively at month 4 and remained stable for the duration of the trial.
In ARTS DN, a randomized, double-blind, placebo-controlled, multicenter phase IIb dose finding study in adults with CKD and T2D, the placebo-corrected relative reduction in UACR at Day 90 was 25% and 38% in patients treated with finerenone 10 mg and 20 mg once daily, respectively.
In patients treated with Kerendia, the mean systolic blood pressure decreased by 3 mmHg and the mean diastolic blood pressure decreased by 1-2 mmHg at month 1, remaining stable thereafter.
At a dose 4 times the maximum approved recommended dose, finerenone does not prolong the QT interval to any clinically relevant extent.
12.3 Pharmacokinetics
Finerenone exposure increased proportionally over a dose range of 1.25 to 80 mg (0.06 to 4 times the maximum approved recommended dosage). Steady state of finerenone was achieved after 2 days of dosing. The estimated steady-state geometric mean Cmax,md was 160 µg/L and steady-state geometric mean AUCτ,md was 686 µg.h/L following administration of finerenone 20 mg to patients.
Finerenone is completely absorbed after oral administration but undergoes metabolism resulting in absolute bioavailability of 44%. Finerenone Cmax was achieved between 0.5 and 1.25 hours after dosing.
There was no clinically significant effect on finerenone AUC following administration with high fat, high calorie food.
The volume of distribution at steady-state (Vss) of finerenone is 52.6 L. Plasma protein binding of finerenone is 92%, primarily to serum albumin, in vitro.
The terminal half-life of finerenone is about 2 to 3 hours, and the systemic blood clearance is about 25 L/h.
Finerenone is primarily metabolized by CYP3A4 (90%) and to a lesser extent by CYP2C8 (10%) to inactive metabolites.
About 80% of the administered dose is excreted in urine (<1% as unchanged) and approximately 20% in feces (< 0.2% as unchanged).
There are no clinically significant effects of age (18 to 79 years), sex, race/ethnicity (White, Asian, Black, and Hispanic), or weight (58 to 121 kg) on the pharmacokinetics of finerenone.
Renal Impairment
There were no clinically relevant differences in finerenone AUC or Cmax values in patients with eGFR 15 to < 90 mL/min/1.73m2 compared to eGFR ≥ 90 mL/min/1.73 m2. For dosing recommendations based on eGFR and serum potassium levels see Dosage and Administration ( 2).
Hepatic Impairment
There was no clinically significant effect on finerenone exposure in cirrhotic patients with mild hepatic impairment (Child Pugh A).
Finerenone mean AUC was increased by 38% and Cmax was unchanged in cirrhotic patients with moderate hepatic impairment (Child Pugh B) compared to healthy control subjects.
The effect of severe hepatic impairment (Child Pugh C) on finerenone exposure was not studied.
Clinical Studies and Model-Informed Approaches
Strong CYP3A Inhibitors: Concomitant use of itraconazole (strong CYP3A4 inhibitor) increased finerenone AUC by >400%.
Moderate CYP3A Inhibitors: Concomitant use of erythromycin (moderate CYP3A4 inhibitor) increased finerenone mean AUC and Cmax by 248% and 88%, respectively.
Weak CYP3A Inhibitors: Concomitant use of amiodarone (weak CYP3A4 inhibitor) increased finerenone AUC by 21%.
Strong or Moderate CYP3A Inducers: Concomitant use of efavirenz (moderate CYP3A4 inducer) and rifampicin (strong CYP3A4 inducer) decreased finerenone AUC by 80% and 90%, respectively.
Other Drugs: There was no clinically significant difference in finerenone pharmacokinetics when used concomitantly with gemfibrozil (strong CYP2C8 inhibitor), omeprazole (proton pump inhibitor), or an aluminium hydroxide and magnesium hydroxide antacid. There were no clinically significant pharmacokinetic differences for either finerenone or concomitant digoxin (P-gp substrate) or warfarin (CYP2C9 substrate). There were no clinically significant differences in the pharmacokinetics of either midazolam (CYP3A4 substrate) or repaglinide (CYP2C8 substrate) when used concomitantly with finerenone. Multiple doses of 40 mg finerenone once-daily had no clinically relevant effect on AUC or Cmax of the BCRP and OATP substrate rosuvastatin.
13 Nonclinical Toxicology
Click here to enter Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Finerenone was non-genotoxic in an in vitro bacterial reverse mutation (Ames) assay, the in vitro chromosomal aberration assay in cultured Chinese hamster V79 cells, or the in vivo micronucleus assay in mice.
In 2-year carcinogenicity studies, finerenone did not show a statistically significant increase in tumor response in Wistar rats or in CD1 mice. In male mice, Leydig cell adenoma was numerically increased at a dose representing 26 times the AUCunbound in humans and is not considered clinically relevant. Finerenone did not impair fertility in male rats but impaired fertility in female rats at 20 times AUC to the maximum human exposure.
14 Clinical Studies
FIDELIO-DKD (NCT: 02540993) and FIGARO-DKD (NCT: 02545049) studies were randomized, double-blind, placebo-controlled, multicenter studies in adult patients with chronic kidney disease (CKD) associated with type 2 diabetes (T2D). In FIDELIO-DKD, patients needed to either have an UACR of 30 to < 300 mg/g, eGFR 25 to < 60 mL/min/1.73 m2 and diabetic retinopathy, or an UACR of ≥ 300 mg/g and an eGFR of 25 to < 75 mL/min/1.73 m2 to qualify for enrollment. In FIGARO-DKD, patients needed to have an UACR of 30 mg/g to < 300 mg/g and an eGFR of 25 to 90 mL/min/1.73m2, or an UACR ≥ 300 mg/g and an eGFR ≥ 60 mL/min/1.73m2.
Both trials excluded patients with known significant non-diabetic kidney disease. All patients were to have a serum potassium ≤4.8 mEq/L at screening and be receiving standard of care background therapy, including a maximum tolerated labeled dose of an angiotensin-converting enzyme inhibitor (ACEi) or angiotensin receptor blocker (ARB). Patients with a clinical diagnosis of chronic heart failure with reduced ejection fraction and persistent symptoms (New York Heart Association class II to IV) were excluded. The starting dose of Kerendia was based on screening eGFR (10 mg once daily in patients with an eGFR of 25 to <60 mL/min/1.73 m2 and 20 mg once daily in patients with an eGFR ≥60 mL/min/1.73 m2). The dose of Kerendia could be titrated during the study, with a target dose of 20 mg daily.
The primary objective of the FIDELIO-DKD study was to determine whether Kerendia reduced the incidence of a sustained decline in eGFR of ≥40%, kidney failure (defined as chronic dialysis, kidney transplantation, or a sustained decrease in eGFR to < 15 mL/min/1.73m2), or renal death. The secondary outcome was a composite of time to first occurrence of CV death, non-fatal MI, non-fatal stroke or hospitalization for heart failure. The primary objective of the FIGARO-DKD study was to determine whether Kerendia reduced the time to first occurrence of CV death, non-fatal MI, non-fatal stroke or hospitalization for heart failure. The secondary outcome was a composite of time to kidney failure, a sustained decline in eGFR of 40% or more compared to baseline over at least 4 weeks, or renal death.
In FIDELIO-DKD, a total of 5674 patients were randomized to receive Kerendia (N=2833) or placebo (N=2841) and were followed for a median of 2.6 years. The mean age of the study population was 66 years, and 70% of patients were male. This global trial population was 63% White, 25% Asian, and 5% Black (24% Black in the US). At baseline, the mean eGFR was 44 mL/min/1.73m2, with 55% of patients having an eGFR <45 mL/min/1.73m2. Median urine albumin-to-creatinine ratio (UACR) was 852 mg/g, mean glycated hemoglobin A1c (HbA1c) was 7.7%. and the mean blood pressure was 138/76 mmHg. Approximately 46% of patients had a history of atherosclerotic cardiovascular disease and 8% had a history of heart failure.
At baseline, 99.8% of patients were treated with an ACEi or ARB. Approximately 97% were on an antidiabetic agent (insulin [64.1%], biguanides [44%], glucagon-like peptide-1 [GLP-1] receptor agonists [7%], sodium-glucose cotransporter 2 [SGLT2] inhibitors [5%]), 74% were on a statin, and 57% were on an antiplatelet agent.
In FIGARO-DKD, a total of 7352 patients were randomized to receive Kerendia (N=3683) or placebo (N=3666) and were followed for 3.4 years. As compared to FIDELIO-DKD, baseline eGFR was higher in FIGARO-DKD (mean eGFR 68, with 62% of patients having an eGFR ≥ 60 mL/min/1.73 m2) and median UACR was lower (308 mg/g). Otherwise, baseline patient characteristics and background therapies were similar in the two trials.
In FIDELIO-DKD, Kerendia reduced the incidence of the primary composite endpoint of a sustained decline in eGFR of ≥40%, kidney failure, or renal death (HR 0.82, 95% CI 0.73-0.93, p=0.001) as shown in Table 4 and Figure 1. The treatment effect reflected a reduction in a sustained decline in eGFR of ≥40% and progression to kidney failure. There were few renal deaths during the trial.
Kerendia also reduced the incidence of the secondary composite endpoint of cardiovascular (CV) death, non-fatal myocardial infarction (MI), non-fatal stroke or hospitalization for heart failure (HR 0.86, 95% CI 0.75-0.99, p=0.034) as shown in Table 4 and Figure 3. The treatment effect reflected a reduction in CV death, non-fatal MI, and hospitalization for heart failure.
The treatment effect on the primary and secondary composite endpoints was generally consistent across subgroups.
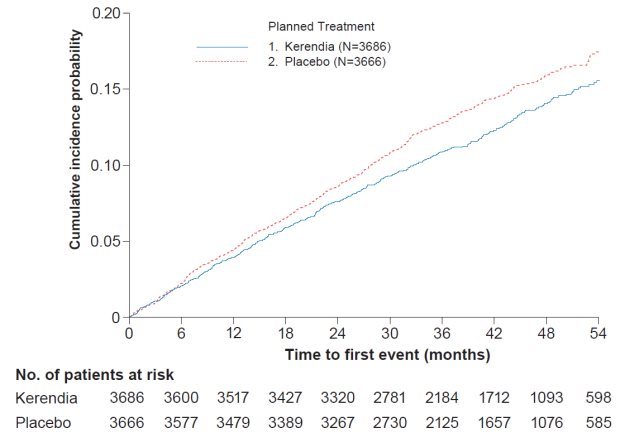
In FIGARO-DKD, Kerendia reduced the incidence of the primary composite endpoint of CV death, non-fatal MI, non-fatal stroke or hospitalization for heart failure (HR 0.87, 95% CI 0.76-0.98, p = 0.026) as shown in Table 4 and Figure 4. The treatment effect was mainly driven by an effect on hospitalization for heart failure, though CV death also contributed to the treatment effect. The treatment effect on the primary composite endpoint was generally consistent across subgroups, including patients with and without pre-existing cardiovascular disease. The findings for the renal composite endpoint are shown in Table 4 and Figure 2.
Table 4: Analysis of the Primary and Secondary Time-to-Event Endpoints (and their Individual Components) in Phase 3 Studies FIDELIO-DKD and FIGARO-DKD
FIDELIO-DKD
FIGARO-DKD
Kerendia
N=2833
Placebo
N=2841
Treatment Effect
Kerendia / Placebo
Kerendia
N=3686
Placebo
N=3666
Treatment Effect
Kerendia / Placebo
Time-to-event Endpoints:
Event Rate (100 pt-yr)
Event Rate (100 pt-yr)
Hazard Ratio
(95% CI)
p-value
Event Rate (100 pt-yr)
Event Rate (100 pt-yr)
Hazard Ratio
(95% CI)
p-value
Composite of kidney failure, sustained eGFR decline ≥40% or renal death
7.6
9.1
0.82[0.73; 0.93]
0.001
3.2
3.6
0.87[0.76; 1.01]
-
Kidney failure
3.0
3.4
0.87[0.72; 1.05]
-
0.4
0.5
0.72[0.49;1.05]
-
Sustained eGFR decline ≥40%
7.2
8.7
0.81[0.72; 0.92]
-
3.0
3.5
0.87[0.75;>1.00]
-
Renal death
-
-
-
-
-
-
-
-
Composite of CV death, non-fatal MI, non-fatal stroke or hospitalization for heart failure
5.1Â
5.9
0.86[0.75; 0.99]
0.034
3.9
4.5
0.87[0.76; 0.98]
0.026
CV death
1.7
2.0
0.86[0.68;1.08]
-
1.6
1.7
0.90[0.74; 1.09]
-
Non-fatal MI
0.9
1.2
0.80[0.58;1.09]
-
0.9
0.9
0.99[0.76; 1.31]
-
Non-fatal stroke
1.2
1.2
1.03[0.76;1.38]
-
0.9
0.9
0.97[0.74; 1.26]
-
Hospitalization for heart failure
1.9Â
2.2
0.86[0.68;1.08]
-
1.0
1.4
0.71[0.56; 0.90]
-
p-value: two-sided p-value from stratified logrank test
CI = confidence interval, CV = cardiovascular, eGFR = estimated glomerular filtration rate, MI = myocardial infarction, N = number of subjects, n = number of subjects with event, pt-yr = patient year.
NOTE: Time to first event was analyzed in a Cox proportional hazards model. For patients with multiple events, only the first event contributed to the composite endpoint. Sums of the numbers of first events for the single components do not add up to the numbers of events in the composite endpoint.
Figure 1: Time to first occurrence of kidney failure, sustained decline in eGFR ≥ 40% from baseline, or renal death in the FIDELIO-DKD study
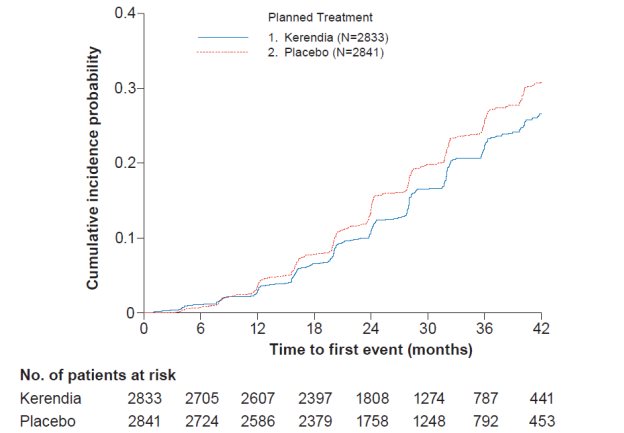
Figure 2: Time to first occurrence of kidney failure, sustained decline in eGFR ≥ 40% from baseline, or renal death in the FIGARO-DKD study
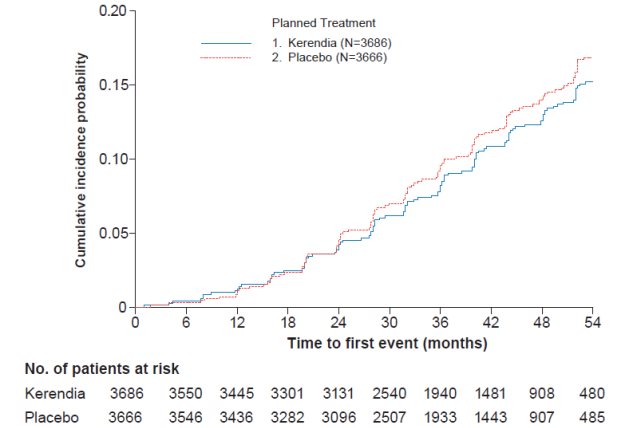
Figure 3: Time to first occurrence of CV death, non-fatal myocardial infarction, non-fatal stroke or hospitalization for heart failure in the FIDELIO-DKD study
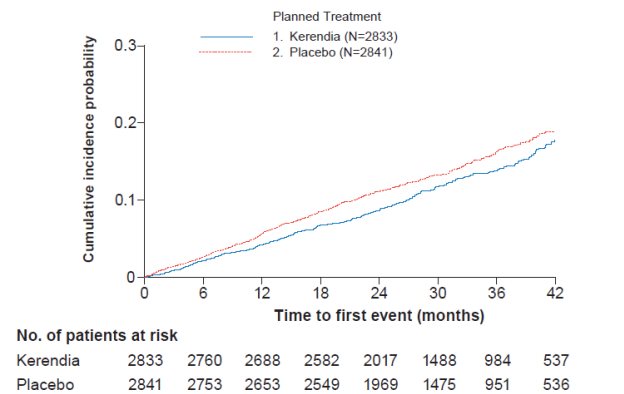
Figure 4: Time to first occurrence of CV death, non-fatal myocardial infarction, non-fatal stroke or hospitalization for heart failure in the FIGARO-DKD study
16 How Supplied/storage And Handling
16.1 How Supplied
Kerendia is available as a film-coated tablet in two strengths. The 10 mg is a pink oblong tablet with “FI” on one side of tablet and “10” on the other side of tablet. The 20 mg tablet is a yellow oblong tablet with “FI” on one side of tablet and “20” on the other side of tablet. Kerendia 10 mg and 20 mg are available in bottles of 30 tablets and bottles of 90 tablets.
Bottle Count Strength NDC Code
30 10 mg NDC 50419-540-01
90 10 mg NDC 50419-540-02
30 20 mg NDC 50419-541-01
90 20 mg NDC 50419-541-02
16.2 Storage and Handling
Store at 20°C to 25°C (68°F to 77°F); excursions are permitted from 15°C to 30°C (59°F to 86°F) [see USP Controlled Room Temperature].
17 Patient Counseling Information
Advise patients of the need for periodic monitoring of serum potassium levels. Advise patients receiving Kerendia to consult with their physician before using potassium supplements or salt substitutes containing potassium [see Warnings and Precautions ( 5.1)].
Advise patients to avoid strong or moderate CYP3A4 inducers and to find alternative medicinal products with no or weak potential to induce CYP3A4 [see Drug Interactions ( 7.1)]
Avoid concomitant intake of grapefruit or grapefruit juice as it is expected to increase the plasma concentration of finerenone [see Drug Interactions ( 7.1)].
Advise women that breastfeeding is not recommended at the time of treatment with KERENDIA and for 1 day after treatment [see Use in Specific Populations ( 8.2)].
Package/label Principal Display Panel
NDC 50419-541-01
Rx only
Kerendia
(finerenone) tablets
20 mg
Oral use
30 tablets

Package/label Display Panel
NDC 50419-540-01
Rx only
Kerendia
(finerenone) tablets
10 mg
Oral use
30 tablets

DISCLAIMER:
"This tool does not provide medical advice, and is for informational and educational purposes only, and is not a substitute for professional medical advice, treatment or diagnosis. Call your doctor to receive medical advice. If you think you may have a medical emergency, please dial 911."
"Do not rely on openFDA to make decisions regarding medical care. While we make every effort to ensure that data is accurate, you should assume all results are unvalidated. We may limit or otherwise restrict your access to the API in line with our Terms of Service."
"This product uses publicly available data from the U.S. National Library of Medicine (NLM), National Institutes of Health, Department of Health and Human Services; NLM is not responsible for the product and does not endorse or recommend this or any other product."
PillSync may earn a commission via links on our site
