Lorbrena (lorlatinib 100 mg) Dailymed
Generic: lorlatinib
IMPRINT: PFIZER LLN 100
SHAPE: oval
COLOR: purple

Go PRO for all pill images
1indications And Usage
LORBRENA¬ģ is indicated for the treatment of adult patients with metastatic non-small cell lung cancer (NSCLC) whose tumors are anaplastic lymphoma kinase (ALK)-positive as detected by an FDA-approved test.
LORBRENA is a kinase inhibitor indicated for the treatment of adult patients with metastatic non-small cell lung cancer (NSCLC) whose tumors are anaplastic lymphoma kinase (ALK)-positive as detected by an FDA-approved test. (1 ,2.1 )
2dosage And Administration
     
Recommended dosage: 100 mg orally once daily. (2.2 )
Severe Renal Impairment: 75 mg orally once daily. (2.8 ,8.7 ,12.3 )
2.1Patient Selection
Select patients for the treatment of metastatic NSCLC with LORBRENA based on the presence of ALK positivity in tumor specimens [see Indications and Usage (1) and Clinical Studies (14)].
Information on FDA-approved tests for the detection of ALK rearrangements in NSCLC is available at http://www.fda.gov/CompanionDiagnostics.
2.2Recommended Dosage
The recommended dosage of LORBRENA is 100 mg orally once daily, with or without food, until disease progression or unacceptable toxicity [see Clinical Pharmacology (12.3)].
Swallow tablets whole. Do not chew, crush or split tablets. Do not ingest if tablets are broken, cracked, or otherwise not intact.
Take LORBRENA at the same time each day. If a dose is missed, then take the missed dose unless the next dose is due within 4 hours. Do not take 2 doses at the same time to make up for a missed dose.
Do not take an additional dose if vomiting occurs after LORBRENA but continue with the next scheduled dose.
2.3Dosage Modifications for Adverse Reactions
The recommended dose reductions are:
‚ÄĘ First dose reduction: LORBRENA 75 mg orally once daily‚ÄĘ Second dose reduction: LORBRENA 50 mg orally once daily
Permanently discontinue LORBRENA in patients who are unable to tolerate 50 mg orally once daily.
Dosage modifications for adverse reactions of LORBRENA are provided in Table 1.
Table 1 Recommended LORBRENA Dosage Modifications for Adverse Reactions Adverse Reaction Grade based on National Cancer Institute (NCI) Common Terminology Criteria for Adverse Events (CTCAE) version 4.03. Dosage Modifications Abbreviation: AV=atrioventricular; DBP=diastolic blood pressure; SBP=systolic blood pressure.
Central Nervous System Effects [see Warnings and Precautions (5.2)]
Grade 1
Continue at the same dose or withhold the dose until recovery to baseline. Resume LORBRENA at the same dose or at a reduced dose.
Grade 2 OR Grade 3
Withhold dose until Grade 0 or 1. Resume LORBRENA at a reduced dose.
Grade 4
Permanently discontinue LORBRENA.
Hyperlipidemia [see Warnings and Precautions (5.3)]
Grade 4 hypercholesterolemia OR Grade 4 hypertriglyceridemia
Withhold LORBRENA until recovery of hypercholesterolemia and/or hypertriglyceridemia to less than or equal to Grade 2. Resume LORBRENA at the same dose. If severe hypercholesterolemia and/or hypertriglyceridemia recurs, resume LORBRENA at a reduced dose.
Atrioventricular (AV) Block [see Warnings and Precautions (5.4)]
Second-degree AV block
Withhold LORBRENA until PR interval is less than 200 ms. Resume LORBRENA at a reduced dose.
First occurrence of complete AV block
Withhold LORBRENA until
1. pacemaker placed OR2. PR interval less than 200 ms.
If a pacemaker is placed, resume LORBRENA at the same dose. If no pacemaker is placed, resume LORBRENA at a reduced dose.
Recurrent complete AV block
Place pacemaker or permanently discontinue LORBRENA.
Interstitial Lung Disease (ILD)/Pneumonitis [see Warnings and Precautions (5.5)]
Any Grade treatment‚Äďrelated ILD/Pneumonitis
Permanently discontinue LORBRENA.
Hypertension [see Warnings and Precautions (5.6)]
Grade 3 (SBP greater than or equal to 160 mmHg or DBP greater than or equal to 100 mmHg; medical intervention indicated; more than one antihypertensive drug, or more intensive therapy than previously used indicated)
Withhold LORBRENA until hypertension has recovered to Grade 1 or less (SBP less than 140 mmHg and DBP less than 90 mmHg), then resume LORBRENA at the same dose.If Grade 3 hypertension recurs, withhold LORBRENA until recovery to Grade 1 or less, and resume at a reduced dose.If adequate hypertension control cannot be achieved with optimal medical management, permanently discontinue LORBRENA.
Grade 4 (life-threatening consequences, urgent intervention indicated)
Withhold LORBRENA until recovery to Grade 1 or less, and resume at a reduced dose or permanently discontinue LORBRENA.If Grade 4 hypertension recurs, permanently discontinue LORBRENA.
Hyperglycemia [see Warnings and Precautions (5.7)]
Grade 3 (greater than 250 mg/dL) despite optimal anti-hyperglycemic therapy OR Grade 4
Withhold LORBRENA until hyperglycemia is adequately controlled, then resume LORBRENA at the next lower dosage.If adequate hyperglycemic control cannot be achieved with optimal medical management, permanently discontinue LORBRENA.
Other Adverse Reactions
Grade 1 OR Grade 2
Continue LORBRENA at same dose or reduced dose.
Grade 3 OR Grade 4
Withhold LORBRENA until symptoms resolve to less than or equal to Grade 2 or baseline. Resume LORBRENA at reduced dose.
2.4Concomitant Use of Strong CYP3A Inducers
LORBRENA is contraindicated in patients taking strong CYP3A inducers. Discontinue strong CYP3A inducers for 3 plasma half-lives of the strong CYP3A inducer prior to initiating LORBRENA [see Contraindications (4), Warnings and Precautions (5.1), Drug Interactions (7.1), Clinical Pharmacology (12.3)].
2.5Concomitant Use of Moderate CYP3A Inducers
Avoid concomitant use of moderate CYP3A inducers with LORBRENA. If concomitant use with moderate CYP3A inducers is unavoidable, increase the LORBRENA dose to 125 mg once daily [see Drug Interactions (7.1), Clinical Pharmacology (12.3)].
2.6Dosage Modification for Strong CYP3A Inhibitors
Avoid concomitant use of LORBRENA with strong CYP3A inhibitors. If concomitant use with a strong CYP3A inhibitor is unavoidable, reduce the starting dose of LORBRENA from 100 mg orally once daily to 75 mg orally once daily.
In patients who have had a dose reduction to 75 mg orally once daily due to adverse reactions and who initiate a strong CYP3A inhibitor, reduce the LORBRENA dose to 50 mg orally once daily.
If concomitant use of a strong CYP3A inhibitor is discontinued, increase the LORBRENA dose (after 3 plasma half-lives of the strong CYP3A inhibitor) to the dose that was used before starting the strong inhibitor [see Drug Interactions (7.1), Clinical Pharmacology (12.3)].
2.7Dosage Modification for Fluconazole
Avoid concomitant use of LORBRENA with fluconazole [see Clinical Pharmacology (12.3)]. If concomitant use is unavoidable, reduce the starting dose of LORBRENA from 100 mg orally once daily to 75 mg orally once daily [see Drug Interactions (7.1), Clinical Pharmacology (12.3)].
2.8Dosage Modification for Severe Renal Impairment
Reduce the recommended dosage of LORBRENA for patients with severe renal impairment (creatinine clearance [CLcr] 15 to < 30 mL/min, estimated by Cockcroft-Gault) from 100 mg to 75 mg orally once daily [see Use in Specific Populations (8.7) and Clinical Pharmacology (12.3)].
3dosage Forms And Strengths
Tablets:
‚ÄĘ 25 mg: 8 mm round, tan, immediate release, film-coated, debossed with "Pfizer" on one side and "25" and "LLN" on the other side‚ÄĘ 100 mg: 8.5 mm √ó 17 mm oval, lavender, immediate release, film-coated, debossed with "Pfizer" on one side and "LLN 100" on the other side
Tablets: 25 mg or 100 mg. (3 )
4contraindications
LORBRENA is contraindicated in patients taking strong CYP3A inducers, due to the potential for serious hepatotoxicity [see Warnings and Precautions (5.1)].
Concomitant use with strong CYP3A inducers. (4 )
5warnings And Precautions
‚ÄĘ Risk of Serious Hepatotoxicity with Concomitant Use of Strong CYP3A Inducers: Discontinue strong CYP3A inducers for 3 plasma half-lives of the strong CYP3A inducer prior to initiating LORBRENA. (2.4 ,5.1 )‚ÄĘ Central Nervous System (CNS) Effects: CNS effects include seizures, psychotic effects and changes in cognitive function, mood (including suicidal ideation), speech, mental status, and sleep. Withhold and resume LORBRENA at same or reduced dose or permanently discontinue LORBRENA based on severity. (2.3 ,5.2 )‚ÄĘ Hyperlipidemia: Initiate or increase the dose of lipid-lowering agents. Withhold and resume LORBRENA at same or reduced dose based on severity. (2.3 ,5.3 )‚ÄĘ Atrioventricular Block: Withhold and resume LORBRENA at same or reduced dose based on severity. (2.3 ,5.4 )‚ÄĘ Interstitial Lung Disease/Pneumonitis: Immediately withhold LORBRENA in patients with suspected ILD/pneumonitis. Permanently discontinue LORBRENA for treatment-related ILD/pneumonitis of any severity. (2.3 ,5.5 )‚ÄĘ Hypertension: Monitor blood pressure after 2 weeks and then at least monthly during treatment. For severe hypertension, withhold LORBRENA, then dose reduce or permanently discontinue. (2.3 ,5.6 )‚ÄĘ Hyperglycemia: Assess fasting serum glucose prior to starting LORBRENA and regularly during treatment. If not adequately controlled with optimal medical management, withhold LORBRENA, then consider dose reduction or permanently discontinue, based on severity. (2.3 ,5.7 )‚ÄĘ Embryo-Fetal Toxicity: Can cause fetal harm. Advise females of reproductive potential of the potential risk to a fetus. Advise males and females of reproductive potential to use an effective non-hormonal method of contraception. (5.8 ,7.2 ,8.1 ,8.3 )5.1Risk of Serious Hepatotoxicity with Concomitant Use of Strong CYP3A Inducers
Severe hepatotoxicity occurred in 10 of 12 healthy subjects receiving a single dose of LORBRENA with multiple daily doses of rifampin, a strong CYP3A inducer. Grade 4 alanine aminotransferase (ALT) or aspartate aminotransferase (AST) elevations occurred in 50% of subjects, Grade 3 ALT or AST elevations occurred in 33% and Grade 2 ALT or AST elevations occurred in 8%. ALT or AST elevations occurred within 3 days and returned to within normal limits after a median of 15 days (7 to 34 days); the median time to recovery was 18 days in subjects with Grade 3 or 4 ALT or AST elevations and 7 days in subjects with Grade 2 ALT or AST elevations [see Drug Interactions (7.1)].
LORBRENA is contraindicated in patients taking strong CYP3A inducers. Discontinue strong CYP3A inducers for 3 plasma half-lives of the strong CYP3A inducer prior to initiating LORBRENA [see Contraindications (4), Drug Interactions (7.1)].
5.2Central Nervous System Effects
A broad spectrum of central nervous system (CNS) effects can occur in patients receiving LORBRENA. These include seizures, psychotic effects and changes in cognitive function, mood (including suicidal ideation), speech, mental status, and sleep. Overall, CNS effects occurred in 52% of the 476 patients who received 100 mg LORBRENA once daily in clinical trials [see Adverse Reactions (6.1)]. Cognitive effects occurred in 28% of the 476 patients; 2.9% of these events were severe (Grade 3 or 4). Mood effects occurred in 21% of patients; 1.7% of these events were severe. Speech effects occurred in 11% of patients; 0.6% of these events were severe. Psychotic effects occurred in 7% of patients; 0.6% of these events were severe. Mental status changes occurred in 1.3% of patients; 1.1% of these events were severe. Seizures occurred in 1.9% of patients, sometimes in conjunction with other neurologic findings. Sleep effects occurred in 12% of patients. The median time to first onset of any CNS effect was 1.4 months (1 day to 3.4 years). Overall, 2.1% of patients required permanent discontinuation of LORBRENA for a CNS effect; 10% required temporary discontinuation and 8% required dose reduction.
Withhold and resume at the same dose or at a reduced dose or permanently discontinue LORBRENA based on severity [see Dosage and Administration (2.3)].
5.3Hyperlipidemia
Increases in serum cholesterol and triglycerides can occur in patients receiving LORBRENA [see Adverse Reactions (6.1)]. Grade 3 or 4 elevations in total cholesterol occurred in 18% and Grade 3 or 4 elevations in triglycerides occurred in 19% of the 476 patients who received 100 mg LORBRENA once daily. The median time to onset was 15 days for both hypercholesterolemia and hypertriglyceridemia. Approximately 4% and 7% of patients required temporary discontinuation and 1% and 3% of patients required dose reduction of LORBRENA for elevations in cholesterol and in triglycerides in Study B7461001 and Study B7461006, respectively. Eighty-three percent of patients required initiation of lipid-lowering medications, with a median time to onset of start of such medications of 17 days.
Initiate or increase the dose of lipid-lowering agents in patients with hyperlipidemia. Monitor serum cholesterol and triglycerides before initiating LORBRENA, 1 and 2 months after initiating LORBRENA, and periodically thereafter. Withhold and resume at the same dose for the first occurrence; resume at the same or a reduced dose of LORBRENA for recurrence based on severity [see Dosage and Administration (2.3)].
5.4Atrioventricular Block
PR interval prolongation and atrioventricular (AV) block can occur in patients receiving LORBRENA [see Adverse Reactions (6.1), Clinical Pharmacology (12.2)].In 476 patients who received 100 mg LORBRENA once daily and who had a baseline electrocardiography (ECG), 1.9% experienced AV block and 0.2% experienced Grade 3 AV block and underwent pacemaker placement.
Monitor ECG prior to initiating LORBRENA and periodically thereafter. Withhold and resume at a reduced dose or at the same dose in patients who undergo pacemaker placement. Permanently discontinue for recurrence in patients without a pacemaker [see Dosage and Administration (2.3)].
5.5Interstitial Lung Disease/Pneumonitis
Severe or life-threatening pulmonary adverse reactions consistent with interstitial lung disease (ILD)/pneumonitis can occur with LORBRENA. ILD/pneumonitis occurred in 1.9% of patients who received 100 mg LORBRENA once daily, including Grade 3 or 4 ILD/pneumonitis in 0.6% of patients. Four patients (0.8%) discontinued LORBRENA for ILD/pneumonitis.
Promptly investigate for ILD/pneumonitis in any patient who presents with worsening of respiratory symptoms indicative of ILD/pneumonitis (e.g., dyspnea, cough, and fever). Immediately withhold LORBRENA in patients with suspected ILD/pneumonitis. Permanently discontinue LORBRENA for treatment-related ILD/pneumonitis of any severity [see Dosage and Administration (2.3)].
5.6Hypertension
Hypertension can occur in patients receiving LORBRENA [see Adverse Reactions (6.1) ]. Hypertension occurred in 13% of patients who received 100 mg LORBRENA once daily, including Grade 3 or 4 in 6% of patients. The median time to onset of hypertension was 6.4 months (1 day to 2.8 years), and 2.3% of patients temporarily discontinued LORBRENA for hypertension.
Control blood pressure prior to initiation of LORBRENA. Monitor blood pressure after 2 weeks and at least monthly thereafter during treatment with LORBRENA. Withhold and resume at a reduced dose or permanently discontinue LORBRENA based on severity [see Dosage and Administration (2.3)].
5.7Hyperglycemia
Hyperglycemia can occur in patients receiving LORBRENA [see Adverse Reactions (6.1)]. Hyperglycemia occurred in 9% of patients who received 100 mg LORBRENA, including Grade 3 or 4 in 3.2% of patients. The median time to onset of hyperglycemia was 4.8 months (1 day to 2.9 years), and 0.8% of patients temporarily discontinued LORBRENA for hyperglycemia.
Assess fasting serum glucose prior to initiation of LORBRENA and monitor periodically thereafter. Withhold and resume at a reduced dose or permanently discontinue LORBRENA based on severity [see Dosage and Administration (2.3)].
5.8Embryo-Fetal Toxicity
Based on findings from animal studies and its mechanism of action, LORBRENA can cause fetal harm when administered to a pregnant woman. Administration of lorlatinib to pregnant rats and rabbits by oral gavage during the period of organogenesis resulted in malformations, increased post-implantation loss, and abortion at maternal exposures that were equal to or less than the human exposure at the recommended dose of 100 mg once daily based on area under the curve (AUC).
Advise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to use an effective non-hormonal method of contraception, since LORBRENA can render hormonal contraceptives ineffective, during treatment with LORBRENA and for at least 6 months after the final dose. Advise males with female partners of reproductive potential to use effective contraception during treatment with LORBRENA and for 3 months after the final dose [see Drug Interactions (7.2), Use in Specific Populations (8.1,8.3), Nonclinical Toxicology (13.1)].
6 Adverse Reactions
The following adverse reactions are described elsewhere in the labeling:
‚ÄĘ Risk of Serious Hepatotoxicity with Concomitant Use of Strong CYP3A Inducers [see Warnings and Precautions (5.1)]‚ÄĘ Central Nervous System Effects [see Warnings and Precautions (5.2)]‚ÄĘ Hyperlipidemia [see Warnings and Precautions (5.3)]‚ÄĘ Atrioventricular Block [see Warnings and Precautions (5.4)]‚ÄĘ Interstitial Lung Disease/Pneumonitis [see Warnings and Precautions (5.5)]‚ÄĘ Hypertension [see Warnings and Precautions (5.6)]‚ÄĘ Hyperglycemia [see Warnings and Precautions (5.7)]
Most common (incidence ‚Č•20%) adverse reactions and Grade 3‚Äď4 laboratory abnormalities are edema, peripheral neuropathy, weight gain, cognitive effects, fatigue, dyspnea, arthralgia, diarrhea, mood effects, hypercholesterolemia, hypertriglyceridemia, and cough. (6.1 )
To report SUSPECTED ADVERSE REACTIONS, contact Pfizer Inc. at 1-800-438-1985 orwww.pfizer.com or FDA at 1-800-FDA-1088 orwww.fda.gov/medwatch .
6.1Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
The pooled safety population described in the Warnings and Precautions section reflects exposure to LORBRENA in 476 patients who received 100 mg LORBRENA once daily in Study B7461001 (N=327) and Study B7461006 (N=149). Among 476 patients who received LORBRENA, 75% were exposed for 6 months or longer and 61% were exposed for greater than 1 year. In this pooled safety population, the most frequent adverse reactions in ‚Č• 20% of 476 patients who received LORBRENA were edema (56%), peripheral neuropathy (44%), weight gain (31%), cognitive effects (28%), fatigue (27%), dyspnea (27%), arthralgia (24%), diarrhea (23%), mood effects (21%), and cough (21%). The most frequent Grade 3‚Äď4 laboratory abnormalities in ‚Č• 20% of 476 patients who received LORBRENA were hypercholesterolemia (21%) and hypertriglyceridemia (21%).
Previously Untreated ALK-Positive Metastatic NSCLC (CROWN Study)
The safety of LORBRENA was evaluated in 149 patients with ALK-positive NSCLC in a randomized, open-label, active-controlled trial for the treatment of patients with ALK-positive, locally advanced or metastatic, NSCLC who had not received previous systemic treatment for advanced disease [see Clinical Studies (14)]. The median duration of exposure to LORBRENA was 16.7 months (4 days to 34.3 months) and 76% received LORBRENA for at least 12 months.
Serious adverse reactions occurred in 34% of patients treated with LORBRENA; the most frequently reported serious adverse reactions were pneumonia (4.7%), dyspnea (2.7%), respiratory failure (2.7%), cognitive effects (2.0%), and pyrexia (2.0%). Fatal adverse reactions occurred in 3.4% of patients treated with LORBRENA and included pneumonia (0.7%), respiratory failure (0.7%), cardiac failure acute (0.7%), pulmonary embolism (0.7%), and sudden death (0.7%).
Permanent discontinuation of LORBRENA due to adverse reactions occurred in 6.7% of patients. The most frequent adverse reaction that led to permanent discontinuation of LORBRENA was cognitive effects (1.3%). Adverse reactions leading to dose interruptions occurred in 49% of patients treated with LORBRENA. The most frequent adverse reactions that led to dose interruptions of LORBRENA were hypertriglyceridemia (7%), edema (5%), pneumonia (4.7%) cognitive effects (4.0%), mood effects (4.0%), and hypercholesterolemia (3.4%). Adverse reactions leading to dose reductions occurred in 21% of patients treated with LORBRENA. The most frequent adverse reactions that led to dose reductions were edema (5%), hypertriglyceridemia (4.0%), and peripheral neuropathy (3.4%).
Tables 2 and 3 summarize most frequent adverse reactions and laboratory abnormalities, respectively, in patients treated with LORBRENA in Study B7461006.
Table 2 Adverse Reactions (‚Č•10% for all NCI CTCAE Grades or ‚Č•2% for Grades 3‚Äď4) in Patients Treated with LORBRENA in Study B7461006 Adverse reactions were graded using NCI CTCAE version 4.03. Adverse Reaction LORBRENA N=149 Crizotinib N=142 All Grades (%) Grade 3 or 4 (%) All Grades (%) Grade 3 or 4 (%) Abbreviations: NCI CTCAE=National Cancer Institute Common Terminology Criteria for Adverse Events; SOC=System organ class.
Psychiatric
  Mood effectsMood effects (including affective disorder, affect lability, agitation, anger, anxiety, bipolar I disorder, depressed mood, depression, depressive symptom, euphoric mood, intentional self-injury, irritability, mood altered, mood swings, stress).
16
2
5
0
Nervous system
  Peripheral neuropathyPeripheral neuropathy (including dysesthesia, gait disturbance, hypoesthesia, motor dysfunction, muscular weakness, neuralgia, neuropathy peripheral, paresthesia, peripheral motor neuropathy, peripheral sensory neuropathy).
34
2
15
0.7
  Cognitive effectsCognitive effects (including events from SOC Nervous system disorders: amnesia, cognitive disorder, disturbance in attention, memory impairment, mental impairment; and also including events from SOC Psychiatric disorders: confusional state, delirium, disorientation).
21
2
6
0
  Headache
17
0
18
0.7
  Dizziness
11
0
14
0
  Sleep effectsSleep effects (including insomnia, nightmare, sleep disorder, somnambulism).
11
1.3
10
0
Respiratory
  Dyspnea
20
2.7
16
2.1
  Cough
16
0
18
0
  Respiratory failure
2.7
2
0
0
Vascular disorders
  Hypertension
18
10
2.1
0
Ocular
  Vision disorderVision disorder (including diplopia, photophobia, photopsia, vision blurred, visual acuity reduced, visual impairment, vitreous floaters).
18
0
39
0.7
Gastrointestinal
  Diarrhea
21
1.3
52
0.7
  Nausea
15
0.7
52
2.1
  Constipation
17
0
30
0.7
  Vomiting
13
0.7
39
1.4
Musculoskeletal and connective tissue
  Arthralgia
19
0.7
11
0
  MyalgiaMyalgia (including musculoskeletal pain, myalgia).
15
0.7
7
0
  Back pain
15
0.7
11
0
  Pain in extremity
17
0
8
0
General
  EdemaEdema (including edema, edema peripheral, eyelid edema, face edema, generalized edema, localized edema, periorbital edema, peripheral swelling, swelling).
56
4
40
1.4
  Weight gain
38
17
13
2.1
  FatigueFatigue (including asthenia, fatigue).
19
1.3
32
2.8
  Pyrexia
17
1.3
13
1.4
  Chest pain
11
1.3
14
0.7
Infections
  Upper respiratory tract infectionUpper respiratory tract infection (including upper respiratory infection).
11
0.7
7.7
1.4
  Pneumonia
7.4
2
8.5
3.5
  Bronchitis
6.7
2
2.1
0
Skin
  RashRash (including dermatitis acneiform, maculopapular rash, rash).
11
0
8.5
0
Additional clinically significant adverse reactions occurring at an incidence between 1% and 10% were speech effects (6.7%) and psychotic effects (3.4%).
Table 3 Laboratory Abnormalities Worsening from Baseline in ‚Č•20% of Patients in Study B7461006 Laboratory Abnormality LORBRENA N=149 Crizotinib N=142 All Grades (%) Grade 3 or 4 (%) All Grades (%) Grade 3 or 4 (%)
Abbreviations: ALT=alanine aminotransferase; AST=aspartate aminotransferase; CPK=creatine phosphokinase; GGT=gamma glutamyl transferase; NCI CTCAE=National Cancer Institute Common Terminology Criteria for Adverse Events; PTT=partial thromboplastin time.
N=number of patients who had at least one on-study assessment for the parameter of interest.
Chemistry
  HypertriglyceridemiaN=149 (LORBRENA). N=141 (crizotinib).
95
22
27
0
  Hypercholesterolemia
91
19
12
0
  Increased creatinine
81
0.7
99
2.1
  Increased GGT
52
6
41
6
  Increased AST
48
2
75
3.5
  Hyperglycemia
48
7
27
2.1
  Increased ALT
44
2.7
75
4.3
  Increased CPK
39
2
64
5
  Hypoalbuminemia
36
0.7
61
6
  Increased lipase
28
7
34
5
  Increased alkaline phosphatase
23
0
50
0.7
  Hyperkalemia
21
1.3
27
2.1
  Increased amylaseN=148 (LORBRENA).
20
1.4
32
1.4
Hematology
  Anemia
48
2
38
2.8
  Activated PTTN=138 (LORBRENA). N=135 (crizotinib).
25
0
14
0
  Lymphopenia
23
2.7
43
6
  Thrombocytopenia
23
0
7
0.7
Previously Treated ALK-Positive Metastatic NSCLC
The data described below reflect exposure to LORBRENA in 295 patients with ALK-positive or ROS1-positive metastatic NSCLC who received LORBRENA 100 mg orally once daily in Study B7461001, a multi-cohort, non-comparative trial [see Clinical Studies (14)]. The median duration of exposure to LORBRENA was 12.5 months (1 day to 35 months) and 52% received LORBRENA for ‚Č•12 months. Patient characteristics were: median age of 53 years (19 to 85 years), age ‚Č•65 years (18%), female (58%), White (49%), Asian (37%), and ECOG performance status 0 or 1 (96%).
The most frequent (‚Č•20%) adverse reactions were edema, peripheral neuropathy, cognitive effects, dyspnea, fatigue, weight gain, arthralgia, mood effects, and diarrhea. Of the worsening laboratory values occurring in ‚Č•20% of patients, the most frequent were hypercholesterolemia, hypertriglyceridemia, anemia, hyperglycemia, increased AST, hypoalbuminemia, increased ALT, increased lipase, and increased alkaline phosphatase.
Serious adverse reactions occurred in 32% of the 295 patients; the most frequently reported serious adverse reactions were pneumonia (3.4%), dyspnea (2.7%), pyrexia (2%), mental status changes (1.4%), and respiratory failure (1.4%). Fatal adverse reactions occurred in 2.7% of patients and included pneumonia (0.7%), myocardial infarction (0.7%), acute pulmonary edema (0.3%), embolism (0.3%), peripheral artery occlusion (0.3%), and respiratory distress (0.3%). Permanent discontinuation of LORBRENA for adverse reactions occurred in 8% of patients.
The most frequent adverse reactions that led to permanent discontinuation were respiratory failure (1.4%), dyspnea (0.7%), myocardial infarction (0.7%), cognitive effects (0.7%) and mood effects (0.7%). Approximately 48% of patients required dose interruption. The most frequent adverse reactions that led to dose interruptions were edema (7%), hypertriglyceridemia (6%), peripheral neuropathy (5%), cognitive effects (4.4%), increased lipase (3.7%), hypercholesterolemia (3.4%), mood effects (3.1%), dyspnea (2.7%), pneumonia (2.7%), and hypertension (2.0%). Approximately 24% of patients required at least 1 dose reduction for adverse reactions. The most frequent adverse reactions that led to dose reductions were edema (6%), peripheral neuropathy (4.7%), cognitive effects (4.1%), and mood effects (3.1%).
Tables 4 and 5 summarize most frequent adverse reactions and laboratory abnormalities, respectively, in patients treated with LORBRENA in Study B7461001.
Table 4 Adverse Reactions Occurring in ‚Č•10% of Patients in Study B7461001 Adverse reactions were graded using NCI CTCAE version 4.03. Adverse Reaction LORBRENA (N=295) All Grades (%) Grade 3 or 4 (%) Abbreviations: NCI CTCAE=National Cancer Institute Common Terminology Criteria for Adverse Events; SOC=System organ class.
Psychiatric
  Mood effectsMood effects (including affective disorder, affect lability, aggression, agitation, anxiety, depressed mood, depression, euphoric mood, irritability, mania, mood altered, mood swings, personality change, stress, suicidal ideation).
23
1.7
Nervous system
  Peripheral neuropathyPeripheral neuropathy (including burning sensation, carpal tunnel syndrome, dysesthesia, formication, gait disturbance, hypoesthesia, muscular weakness, neuralgia, neuropathy peripheral, neurotoxicity, paresthesia, peripheral sensory neuropathy, sensory disturbance).
47
2.7
  Cognitive effectsCognitive effects (including events from SOC Nervous system disorders: amnesia, cognitive disorder, dementia, disturbance in attention, memory impairment, mental impairment; and also including events from SOC Psychiatric disorders: attention deficit/hyperactivity disorder, confusional state, delirium, disorientation, reading disorder).
27
2
  Headache
18
0.7
  Dizziness
16
0.7
  Speech effectsSpeech effects (including aphasia, dysarthria, slow speech, speech disorder)
12
0.3
  Sleep effectsSleep effects (including abnormal dreams, insomnia, nightmare, sleep disorder, sleep talking, somnambulism)
10
0
Respiratory
  Dyspnea
27
5
  Cough
18
0
Ocular
  Vision disorderVision disorder (including blindness, diplopia, photophobia, photopsia, vision blurred, visual acuity reduced, visual impairment, vitreous floaters).
15
0.3
Gastrointestinal
  Diarrhea
22
0.7
  Nausea
18
0.7
  Constipation
15
0
  Vomiting
12
1
Musculoskeletal and connective tissue
  Arthralgia
23
0.7
  MyalgiaMyalgia (including musculoskeletal pain, myalgia).
17
0
  Back pain
13
0.7
  Pain in extremity
13
0.3
General
  EdemaEdema (including edema, edema peripheral, eyelid edema, face edema, generalized edema, localized edema, periorbital edema, peripheral swelling, swelling).
57
3.1
  FatigueFatigue (including asthenia, fatigue).
26
0.3
  Weight gain
24
4.4
  Pyrexia
12
0.7
Infections
  Upper respiratory tract infectionUpper respiratory infection (including fungal upper respiratory infection, upper respiratory infection, viral upper respiratory infection).
12
0
Skin
  RashRash (including dermatitis acneiform, maculopapular rash, pruritic rash, rash).
14
0.3
Additional clinically significant adverse reactions occurring at an incidence between 1% and 10% were psychotic effects (7%).
Table 5 Worsening Laboratory Values Occurring in ‚Č•20% of Patients in Study B7461001 Grades using NCI CTCAE version 4.03. Laboratory Abnormality LORBRENA All Grades (%) Grade 3 or 4 (%) Abbreviations: ALT=alanine aminotransferase; AST=aspartate aminotransferase; NCI CTCAE=National Cancer Institute Common Terminology Criteria for Adverse Events. N=number of patients who had at least one on-study assessment for the parameter of interest.
Chemistry
  HypercholesterolemiaN=292.
96
18
  Hypertriglyceridemia
90
18
  HyperglycemiaN=293.
52
5
  Increased AST
37
2.1
  HypoalbuminemiaN=291.
33
1
  Increased ALT
28
2.1
  Increased lipaseN=290.
24
10
  Increased alkaline phosphatase
24
1
  Increased amylaseN=284.
22
3.9
  Hypophosphatemia
21
4.8
  Hyperkalemia
21
1
  Hypomagnesemia
21
0
Hematology
  Anemia
52
4.8
  Thrombocytopenia
23
0.3
  Lymphopenia
22
3.4
7 Drug Interactions
‚ÄĘ Strong CYP3A Inducers: Contraindicated. (2.4 ,7.1 )‚ÄĘ Moderate CYP3A Inducers: Avoid concomitant use. If coadministration cannot be avoided, increase the LORBRENA dose. (2.5 ,7.1 )‚ÄĘ Strong CYP3A Inhibitors: Avoid concomitant use; reduce LORBRENA dose if concomitant use cannot be avoided. (2.6 ,7.1 )‚ÄĘ Fluconazole: Avoid concomitant use; reduce LORBRENA dose if concomitant use cannot be avoided. (2.7 ,7.1 )‚ÄĘ Certain CYP3A Substrates: Avoid concomitant use with CYP3A substrates for which minimal concentration changes may lead to serious therapeutic failures. (7.2 )‚ÄĘ Certain P-gp Substrates: Avoid concomitant use with P-gp substrates for which minimal concentration changes may lead to serious therapeutic failures. (7.2 )7.1Effect of Other Drugs on LORBRENA
Strong CYP3A Inducers
Concomitant use of LORBRENA with a strong CYP3A inducer decreased lorlatinib plasma concentrations [see Clinical Pharmacology (12.3)], which may decrease the efficacy of LORBRENA.
Severe hepatotoxicity occurred in healthy subjects receiving LORBRENA with rifampin, a strong CYP3A inducer. In 12 healthy subjects receiving a single 100 mg dose of LORBRENA with multiple daily doses of rifampin, Grade 3 or 4 increases in ALT or AST occurred in 83% of subjects and Grade 2 increases in ALT or AST occurred in 8%. A possible mechanism for hepatotoxicity is through activation of the pregnane X receptor (PXR) by LORBRENA and rifampin, which are both PXR agonists.
LORBRENA is contraindicated in patients taking strong CYP3A inducers [see Contraindication (4)]. Discontinue strong CYP3A inducers for 3 plasma half-lives of the strong CYP3A inducer prior to initiating LORBRENA [see Dosage and Administration (2.3)].
Moderate CYP3A Inducers
Concomitant use of LORBRENA with a moderate CYP3A inducer decreased lorlatinib plasma concentrations, which may decrease the efficacy of LORBRENA [see Clinical Pharmacology (12.3)]. Avoid concomitant use of moderate CYP3A inducers with LORBRENA. If concomitant use is unavoidable, increase the LORBRENA dose [see Dosage and Administration (2.4)].
Strong CYP3A Inhibitors
Concomitant use with a strong CYP3A inhibitor increased lorlatinib plasma concentrations [see Clinical Pharmacology (12.3)], which may increase the incidence and severity of adverse reactions of LORBRENA. Avoid concomitant use of LORBRENA with a strong CYP3A inhibitor. If concomitant use cannot be avoided, reduce the LORBRENA dosage [see Dosage and Administration (2.5)].
Fluconazole
Concomitant use of LORBRENA with fluconazole may increase lorlatinib plasma concentrations [see Clinical Pharmacology (12.3)], which may increase the incidence and severity of adverse reactions of LORBRENA. Avoid concomitant use of LORBRENA with fluconazole. If concomitant use cannot be avoided, reduce the LORBRENA dosage [see Dosage and Administration (2.7)].
7.2Effect of LORBRENA on Other Drugs
Certain CYP3A Substrates
LORBRENA is a moderate CYP3A inducer. Concomitant use of LORBRENA decreases the concentration of CYP3A substrates [see Clinical Pharmacology (12.3)], which may reduce the efficacy of these substrates. Avoid concomitant use of LORBRENA with certain CYP3A substrates, for which minimal concentration changes may lead to serious therapeutic failures. If concomitant use is unavoidable, increase the CYP3A substrate dosage in accordance with approved product labeling.
Certain P-glycoprotein (P-gp) Substrates
LORBRENA is a moderate P-gp inducer. Concomitant use of LORBRENA decreases the concentration of P-gp substrates [see Clinical Pharmacology (12.3)], which may reduce the efficacy of these substrates. Avoid concomitant use of LORBRENA with certain P-gp substrates for which minimal concentration changes may lead to serious therapeutic failures. If concomitant use is unavoidable, increase the P-gp substrate dosage in accordance with approved product labeling.
8use In Specific Populations
Lactation: Advise not to breastfeed. (8.2 )
8.1Pregnancy
Risk Summary
Based on findings from animal studies and its mechanism of action [see Clinical Pharmacology (12.1)], LORBRENA can cause embryo-fetal harm when administered to a pregnant woman. There are no available data on LORBRENA use in pregnant women. Administration of lorlatinib to pregnant rats and rabbits by oral gavage during the period of organogenesis resulted in malformations, increased post-implantation loss, and abortion at maternal exposures that were equal to or less than the human exposure at the recommended dose of 100 mg once daily based on AUC (see Data). Advise a pregnant woman of the potential risk to a fetus.
In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies are 2 to 4% and 15 to 20%, respectively.
Data
Animal Data
Preliminary embryo-fetal development studies investigating the administration of lorlatinib during the period of organogenesis were conducted in rats and rabbits. In rabbits, lorlatinib administration resulted in abortion and total loss of pregnancy at doses of 15 mg/kg (approximately 3 times the human exposure at the recommended dose of 100 mg) or greater. At a dose of 4 mg/kg (approximately 0.6 times the human exposure at the recommended dose of 100 mg) toxicities included increased post-implantation loss and malformations including rotated limbs, malformed kidneys, domed head, high arched palate, and dilation of the cerebral ventricles. In rats, administration of lorlatinib resulted in total loss of pregnancy at doses of 4 mg/kg (approximately 5 times the human exposure at the recommended dose of 100 mg) or greater. At a dose of 1 mg/kg (approximately equal to the human exposure at the recommended dose of 100 mg) there was increased post-implantation loss, decreased fetal body weight, and malformations including gastroschisis, rotated limbs, supernumerary digits, and vessel abnormalities.
8.2Lactation
Risk Summary
There are no data on the presence of lorlatinib or its metabolites in either human or animal milk or its effects on the breastfed infant or on milk production. Because of the potential for serious adverse reactions in breastfed infants, instruct women not to breastfeed during treatment with LORBRENA and for 7 days after the final dose.
8.3Females and Males of Reproductive Potential
Pregnancy Testing
Verify pregnancy status in females of reproductive potential prior to initiating LORBRENA [see Use in Specific Populations (8.1)].
Contraception
LORBRENA can cause embryo-fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)].
Females
Advise female patients of reproductive potential to use effective non-hormonal contraception during treatment with LORBRENA and for at least 6 months after the final dose. Advise females of reproductive potential to use a non-hormonal method of contraception, because LORBRENA can render hormonal contraceptives ineffective [see Drug Interactions (7.2)].
Males
Based on genotoxicity findings, advise males with female partners of reproductive potential to use effective contraception during treatment with LORBRENA and for at least 3 months after the final dose [see Nonclinical Toxicology (13.1)].
Infertility
Males
Based on findings from animal studies, LORBRENA may transiently impair male fertility [see Nonclinical Toxicology (13.1)].
8.4Pediatric Use
The safety and effectiveness of LORBRENA in pediatric patients have not been established.
8.5Geriatric Use
Of the patients in Study B7461001 (N=295) and Study B7461006 (N=149) who received 100 mg LORBRENA orally once daily, 18% and 40% of patients, respectively, were aged 65 years or older. No clinically important differences in safety or efficacy were observed between patients aged 65 years or older and younger patients.
8.6Hepatic Impairment
No dose adjustment is recommended for patients with mild hepatic impairment (total bilirubin ‚ȧ upper limit of normal [ULN] with AST > ULN or total bilirubin >1 to 1.5 √ó ULN with any AST). The recommended dose of LORBRENA has not been established for patients with moderate (total bilirubin ‚Č• 1.5 to 3.0 √ó ULN with any AST) or severe (total bilirubin > 3.0 √ó ULN with any AST) hepatic impairment [see Clinical Pharmacology (12.3)].
8.7Renal Impairment
Reduce the dose when administering LORBRENA to patients with severe (CLcr 15 to <30 mL/min, estimated by Cockcroft Gault) renal impairment [see Dosage and Administration (2.8) and Clinical Pharmacology (12.3)].
No dose adjustment is recommended for patients with mild or moderate (CLcr 30 to 89 mL/min, estimated by Cockcroft-Gault) renal impairment [see Clinical Pharmacology (12.3)].
11description
LORBRENA (lorlatinib) is a kinase inhibitor for oral administration. The molecular formula is C21H19FN6O2 (anhydrous form) and the molecular weight is 406.41 Daltons. The chemical name is (10R)-7-amino-12-fluoro-2,10,16-trimethyl-15-oxo-10,15,16,17-tetrahydro-2H-4,8-methenopyrazolo[4,3-h][2,5,11] benzoxadiazacyclotetradecine-3-carbonitrile. The chemical structure is shown below:
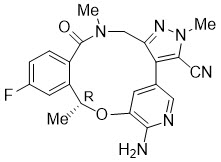
Lorlatinib is a white to off-white powder with a pKa of 4.92. The solubility of lorlatinib in aqueous media decreases over the range pH 2.55 to pH 8.02 from 32.38 mg/mL to 0.17 mg/mL. The log of the distribution coefficient (octanol/water) at pH 9 is 2.45.
LORBRENA is supplied as tablets containing 25 mg or 100 mg of lorlatinib with the following inactive ingredients: microcrystalline cellulose, dibasic calcium phosphate anhydrous, sodium starch glycolate, and magnesium stearate. The film-coating contains hydroxypropyl methylcellulose (HPMC) 2910/hypromellose, lactose monohydrate, macrogol/polyethylene glycol (PEG) 3350, triacetin, titanium dioxide, ferrosoferric oxide/black iron oxide, and iron oxide red.
12clinical Pharmacology
12.1Mechanism of Action
Lorlatinib is a kinase inhibitor with in vitro activity against ALK and ROS1 as well as TYK1, FER, FPS, TRKA, TRKB, TRKC, FAK, FAK2, and ACK. Lorlatinib demonstrated in vitro activity against multiple mutant forms of the ALK enzyme, including some mutations detected in tumors at the time of disease progression on crizotinib and other ALK inhibitors.
In mice subcutaneously implanted with tumors harboring EML4 fusions with either ALK variant 1 or ALK mutations, including the G1202R and I1171T mutations detected in tumors at the time of disease progression on ALK inhibitors, administration of lorlatinib resulted in antitumor activity. Lorlatinib also demonstrated anti-tumor activity and prolonged survival in mice implanted intracranially with EML4-ALK-driven tumor cell lines. The overall antitumor activity of lorlatinib in in vivo models was dose-dependent and correlated with inhibition of ALK phosphorylation.
12.2Pharmacodynamics
Exposure-response relationships for Grade 3 or 4 hypercholesterolemia and for any Grade 3 or 4 adverse reaction were observed at steady-state exposures achieved at the recommended dosage, with higher probability of the occurrence of adverse reactions with increasing lorlatinib exposure.
Cardiac Electrophysiology
In 295 patients who received LORBRENA at the recommended dosage of 100 mg once daily and had an ECG measurement in Study B7461001, the maximum mean change from baseline for PR interval was 16.4 ms (2-sided 90% upper confidence interval [CI] 19.4 ms). Among the 284 patients with PR interval <200 ms at baseline, 14% had PR interval prolongation ‚Č•200 ms after starting LORBRENA. The prolongation of PR interval occurred in a concentration-dependent manner. Atrioventricular block occurred in 1% of patients.
In 275 patients who received LORBRENA at the recommended dosage in the activity-estimating portion of Study B7461001, no large mean increases from baseline in the QTcF interval (i.e., >20 ms) were detected.
12.3Pharmacokinetics
Steady-state lorlatinib maximum plasma concentration (Cmax) increases proportionally and AUC increased slightly less than proportionally over the dose range of 10 mg to 200 mg orally once daily (0.1 to 2 times the recommended dosage). At the recommended dosage, the mean (coefficient of variation [CV] %) Cmax was 577 ng/mL (42%) and the AUC0‚Äď24h was 5650 ng‚ąôh/mL (39%) in patients with cancer. Lorlatinib oral clearance increased at steady-state compared to single dose, indicating autoinduction.
Absorption
The median lorlatinib Tmax was 1.2 hours (0.5 to 4 hours) following a single oral 100 mg dose and 2 hours (0.5 to 23 hours) following 100 mg orally once daily at steady-state.
The mean absolute bioavailability is 81% (90% CI 75.7%, 86.2%) after oral administration compared to intravenous administration.
Effect of Food
There was no clinically significant effect on lorlatinib pharmacokinetics following administration of LORBRENA with a high fat, high calorie meal (approximately 1000 calories with 150 calories from protein, 250 calories from carbohydrate, and 500 to 600 calories from fat).
Distribution
Lorlatinib was 66% bound to plasma proteins at a concentration of 2.4 ¬ĶM. The blood-to-plasma ratio was 0.99, in vitro. The mean (CV%) steady-state volume of distribution (Vss) was 305 L (28%) following a single intravenous dose.
Elimination
The mean plasma half-life (t¬Ĺ) of lorlatinib was 24 hours (40%) after a single oral 100 mg dose of LORBRENA. The mean oral clearance (CL/F) was 11 L/h (35%) following a single oral 100 mg dose and increased to 18 L/h (39%) at steady-state, suggesting autoinduction.
Metabolism
Lorlatinib is metabolized primarily by CYP3A4 and UGT1A4, with minor contribution from CYP2C8, CYP2C19, CYP3A5, and UGT1A3, in vitro.
In plasma, a benzoic acid metabolite (M8) of lorlatinib resulting from the oxidative cleavage of the amide and aromatic ether bonds of lorlatinib accounted for 21% of the circulating radioactivity. The oxidative cleavage metabolite, M8, is pharmacologically inactive.
Excretion
Following a single oral 100 mg dose of radiolabeled lorlatinib, 48% of the radioactivity was recovered in urine (<1% as unchanged) and 41% in feces (about 9% as unchanged).
Specific Populations
No clinically significant differences in lorlatinib pharmacokinetics were observed based on age (19 to 85 years), sex, race/ethnicity, body weight, mild to moderate renal impairment (CLcr 30 to 89 mL/min, estimated by Cockcroft-Gault), mild hepatic impairment (total bilirubin ‚ȧ ULN and AST > ULN or total bilirubin > 1 to 1.5 √ó ULN and any AST), or metabolizer phenotypes for CYP3A5 and CYP2C19. The effect of moderate to severe hepatic impairment (total bilirubin ‚Č• 1.5 √ó ULN with any AST) on lorlatinib pharmacokinetics is unknown [see Use in Specific Populations (8.6, 8.7)].
Patients with Severe Renal Impairment
Following administration of a single oral 100 mg dose of LORBRENA, lorlatinib AUCinf increased by 42% in subjects with severe renal impairment (CLcr 15 to <30 mL/min, estimated by Cockcroft-Gault) compared to subjects with normal renal function (CLcr ‚Č• 90 mL/min, estimated by Cockcroft-Gault). The pharmacokinetics of lorlatinib have not been studied in patients with end-stage renal disease requiring hemodialysis.
Drug Interaction Studies
Clinical Studies and Model-Informed Approaches
In Vitro Studies
13nonclinical Toxicology
13.1Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies have not been conducted with lorlatinib. Lorlatinib was aneugenic in an in vitro assay in human lymphoblastoid TK6 cells and positive for micronuclei formation in vivo in the bone marrow of rats. Lorlatinib was not mutagenic in an in vitro bacterial reverse mutation (Ames) assay.
Dedicated fertility studies were not conducted with lorlatinib. Findings in male reproductive organs occurred in repeat-dose toxicity studies and included lower testicular, epididymal, and prostate weights; testicular tubular degeneration/atrophy; prostatic atrophy; and/or epididymal inflammation at 15 mg/kg/day and 7 mg/kg/day in rats and dogs, respectively (approximately 8 and 2 times, respectively, the human exposure at the recommended dose of 100 mg based on AUC). The effects on male reproductive organs were reversible.
13.2Animal Toxicology and/or Pharmacology
Distended abdomen, skin rash, and increased cholesterol and triglycerides occurred in animals. These findings were accompanied by hyperplasia and dilation of the bile ducts in the liver and acinar atrophy of the pancreas in rats at 15 mg/kg/day and in dogs at 2 mg/kg/day (approximately 8 and 0.5 times, respectively, the human exposure at the recommended dose of 100 mg based on AUC). All effects were reversible within the recovery period.
14clinical Studies
Previously Untreated ALK-Positive Metastatic NSCLC (CROWN Study)
The efficacy of LORBRENA for the treatment of patients with ALK-positive NSCLC who had not received prior systemic therapy for metastatic disease was established in an open-label, randomized, active-controlled, multicenter study (Study B7461006; NCT03052608). Patients were required to have an ECOG performance status of 0‚Äď2 and ALK-positive NSCLC as identified by the VENTANA ALK (D5F3) CDx assay. Neurologically stable patients with treated or untreated asymptomatic CNS metastases, including leptomeningeal metastases, were eligible. Patients were required to have finished radiation therapy, at least 2 weeks (for stereotactic or partial radiation) or 4 weeks (for whole brain irradiation) prior to randomization. Patients with severe acute or chronic psychiatric conditions, including recent (within the past year) or active suicidal ideation or behavior, were excluded.
Patients were randomized 1:1 to receive LORBRENA 100 mg orally once daily or crizotinib 250 mg orally twice daily. Randomization was stratified by ethnic origin (Asian vs. non-Asian) and the presence or absence of CNS metastases at baseline. Treatment on both arms was continued until disease progression or unacceptable toxicity. The major efficacy outcome measure was progression-free survival (PFS) as determined by Blinded Independent Central Review (BICR) according to Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1 (v1.1). Additional efficacy outcome measures were overall survival (OS) and tumor assessment related data by BICR, including overall response rate (ORR), and duration of response (DOR). In patients with measurable CNS metastases at baseline, additional outcome measures were intracranial overall response rate (IC-ORR) and intracranial duration of response (IC-DOR) by BICR.
A total of 296 patients were randomized to LORBRENA (n=149) or crizotinib (n=147). The demographic characteristics of the overall study population were: median age 59 years (range: 26 to 90 years), age ‚Č•65 years (35%), 59% female, 49% White, 44% Asian, and 0.3% Black. The ECOG performance status at baseline was 0 or 1 in 96% of patients. The majority of patients had adenocarcinoma (95%) and never smoked (59%). CNS metastases were present in 26% (n=78) of patients: of these, 30 patients had measurable CNS lesions.
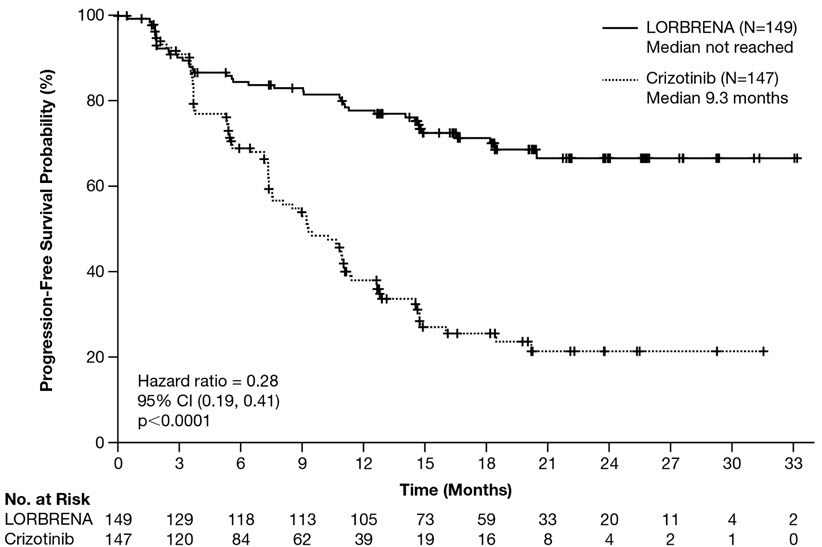
Efficacy results from Study B7461006 as assessed by BICR are summarized in Table 6 and Figure 1. Results demonstrated a significant improvement in PFS for the LORBRENA arm over the crizotinib arm. At the data cutoff point OS data was not mature.
Table 6 Efficacy Results in Study B7461006 (CROWN) Efficacy Parameter LORBRENA N=149 Crizotinib N=147 Abbreviations: CI=confidence interval; N=number of patients; NE=not estimable; PFS=progression free survival.
Progression-free survival
  Number of events, n (%)
41 (28%)
86 (59%)
    Progressive disease, n (%)
32 (22%)
82 (56%)
    Death, n (%)
9 (6%)
4 (3%)
  Median, months (95% CI)Based on the Brookmeyer and Crowley method.
NE (NE, NE)
9.3 (7.6, 11.1)
  Hazard ratio (95% CI)Hazard ratio based on Cox proportional hazards model.
0.28 (0.19, 0.41)
  p-valuep-value based on 1-sided stratified log-rank test.
<0.0001
Overall response rate
  Overall response rate (95% CI)Using exact method based on binomial distribution.
76% (68, 83)
58% (49, 66)
  Complete response
3%
0%
  Partial response
73%
58%
Duration of response
  Number of responders, n
113
85
  Median, months (Range)
NE (0.9, 31.3)
11 (1.1, 27.5)
¬†¬†Response duration ‚Č•6 months, n (%)
101 (89%)
53 (62%)
¬†¬†Response duration ‚Č•12 months, n (%)
79 (70%)
23 (27%)
¬†¬†Response duration ‚Č•18 months, n (%)
34 (30%)
9 (11%)
Figure 1: Kaplan-Meier Plot of Progression-Free Survival by BICR in Study B7461006 (CROWN)
The results of prespecified exploratory analyses of intracranial response rate in 30 patients with measurable CNS lesions at baseline as assessed by BICR are summarized in Table 7.
Table 7 Intracranial Response Rate in Patients with Measurable Intracranial Lesions in CROWN Intracranial Tumor Response Assessment LORBRENA N=17 Crizotinib N=13 Abbreviations: CI=confidence interval; N/n=number of patients.
Intracranial response rate (95% CI)Using exact method based on binomial distribution.
82% (57, 96)
23% (5, 54)
  Complete response
71%
8%
Duration of response
  Number of responders, n
14
3
¬†¬†Response duration ‚Č•12 months, n (%)
11 (79%)
0
ALK-Positive Metastatic NSCLC Previously Treated with an ALK Kinase Inhibitor
The efficacy of LORBRENA was demonstrated in a subgroup of patients with ALK-positive metastatic NSCLC previously treated with one or more ALK kinase inhibitors who were enrolled in a non-randomized, dose-ranging and activity-estimating, multi-cohort, multicenter study (Study B7461001; NCT01970865). Patients included in this subgroup were required to have metastatic disease with at least 1 measurable target lesion according to RECIST v1.1, ECOG performance status of 0 to 2, and documented ALK rearrangement in tumor tissue as determined by fluorescence in situ hybridization (FISH) assay or by Immunohistochemistry (IHC), and received LORBRENA 100 mg orally once daily. Patients with asymptomatic CNS metastases, including patients with stable or decreasing steroid use within 2 weeks prior to study entry, were eligible. Patients with severe, acute, or chronic psychiatric conditions including suicidal ideation or behavior were excluded. In addition, for patients with ALK-positive metastatic NSCLC, the extent and type of prior treatment was specified for each individual cohort (see Table 8). The major efficacy outcome measures were ORR and intracranial ORR, according to RECIST v1.1, as assessed by Independent Central Review (ICR) committee. Data were pooled across all subgroups uled in Table 8. Additional efficacy outcome measures included DOR, and intracranial DOR.
A total of 215 patients were enrolled across the subgroups in Table 8. The distribution of patients by type and extent of prior therapy is provided in Table 8. The demographic characteristics across all 215 patients were: 59% female, 51% White, 34% Asian, and the median age was 53 years (29 to 85 years) with 18% of patients ‚Č•65 years. The ECOG performance status at baseline was 0 or 1 in 96% of patients. All patients had metastatic disease and 95% had adenocarcinoma. Brain metastases as identified by ICR were present in 69% of patients; of these, 60% had received prior radiation to the brain and 60% (n=89) had measurable disease per ICR.
Table 8 Extent of Prior Therapy in the Subgroup of Patients with Previously Treated ALK-Positive Metastatic NSCLC in Study B7461001 Extent of prior therapy Number of patients Abbreviations: ALK=anaplastic lymphoma kinase; NSCLC=non-small cell lung cancer.
Prior crizotinib and no prior chemotherapyChemotherapy administered in the metastatic setting.
29
Prior crizotinib and 1‚Äď2 lines of prior chemotherapy
35
Prior ALK inhibitor (not crizotinib) with or without prior chemotherapy
28
Two prior ALK inhibitors with or without prior chemotherapy
75
Three prior ALK inhibitors with or without prior chemotherapy
48
Total
215
Efficacy results for Study B7461001 are summarized in Tables 9 and 10.
Table 9 Efficacy Results in Study B7461001 Efficacy Parameter Overall N=215 Abbreviations: CI=confidence interval; N=number of patients.
Overall response ratePer Independent Central Review. (95% CI)Using exact method based on binomial distribution.
48% (42, 55)
  Complete response
4%
  Partial response
44%
Duration of response
  Median, monthsEstimated using the Kaplan-Meier method. (95% CI)
12.5 (8.4, 23.7)
An assessment of intracranial ORR and the duration of response for CNS metastases in the subgroup of 89 patients in Study B7461001 with baseline measurable lesions in the CNS according to RECIST v1.1 are summarized in Table 10. Of these, 56 (63%) patients received prior brain radiation, including 42 patients (47%) who completed brain radiation treatment at least 6 months before starting treatment with LORBRENA.
Table 10 Intracranial Response Rate in Patients with Measurable Intracranial Lesions in Study B7461001 Efficacy Parameter Intracranial N=89 Abbreviations: CI=confidence interval; N=number of patients; NR=not reached.
Intracranial response ratePer Independent Central Review. (95% CI)Using exact method based on binomial distribution.
60% (49, 70)
  Complete response
21%
  Partial response
38%
Duration of response
  Median, monthsEstimated using the Kaplan-Meier method. (95% CI)
19.5 (12.4, NR)
In exploratory analyses conducted in subgroups defined by prior therapy, the response rates to LORBRENA were:
‚ÄĘ ORR = 39% (95% CI: 30, 48) in 119 patients who received crizotinib and at least one other ALK inhibitor, with or without prior chemotherapy‚ÄĘ ORR = 31% (95% CI: 9, 61) in 13 patients who received alectinib as their only ALK inhibitor, with or without prior chemotherapy‚ÄĘ ORR = 46% (95% CI: 19, 75) in 13 patients who received ceritinib as their only ALK inhibitor, with or without prior chemotherapy
16how Supplied/storage And Handling
Table 11 describes the available strengths and package configurations for LORBRENA:
Table 11 LORBRENA Tablets Package Configuration Strength (mg) NDC Description
30 count bottle with a child-resistant closure
25
0069-0227-01
8 mm round, tan, immediate release film-coated, debossed with "Pfizer" on one side and "25" and "LLN" on the other side
30 count bottle with a child-resistant closure
100
0069-0231-01
8.5 mm √ó 17 mm oval, lavender, immediate release, film-coated, debossed with "Pfizer" on one side and "LLN 100" on the other side
STORAGE AND HANDLING SECTION
Store at 20¬įC to 25¬įC (68¬įF to 77¬įF); excursions permitted between 15¬įC to 30¬įC (59¬įF to 86¬įF) [see USP Controlled Room Temperature].
17patient Counseling Information
Advise the patient to read the FDA-approved patient labeling (Patient Information).
Risk of Serious Hepatotoxicity with Concomitant Use of Strong CYP3A Inducers
Inform patients of the potential risk of hepatotoxicity with the concomitant use of strong CYP3A inducers.
Advise patients to inform their healthcare providers of all medications they are taking, including prescription medicines, over-the-counter drugs, vitamins, and herbal products (e.g., St. John's wort) [see Warnings and Precautions (5.1)].
Central Nervous System (CNS) Effects
Advise patients to notify their healthcare provider if they experience new or worsening CNS symptoms [see Warnings and Precautions (5.2)].
Hyperlipidemia
Inform patients that serum cholesterol and triglycerides will be monitored during treatment. Advise patients that initiation or an increase in the dose of lipid-lowering agents may be required [see Warnings and Precautions (5.3)].
Atrioventricular (AV) Block
Inform patients of the risks of AV block. Advise patients to contact their healthcare provider immediately to report new or worsening cardiac symptoms [see Warnings and Precautions (5.4)].
Interstitial Lung Disease (ILD)/Pneumonitis
Inform patients of the risks of severe ILD/pneumonitis. Advise patients to contact their healthcare provider immediately to report new or worsening respiratory symptoms [see Warnings and Precautions (5.5)].
Hypertension
Advise patients of the risks of hypertension and to promptly report signs or symptoms of hypertension to their healthcare provider. Advise patients with hypertension that antihypertension medications may need to be initiated or adjusted during treatment with LORBRENA [see Warnings and Precautions (5.6)] .
Hyperglycemia
Inform patients of the risks of new or worsening hyperglycemia and the need to periodically monitor glucose levels. Advise patients with newly occurring hyperglycemia during treatment with LORBRENA that antihyperglycemic medications may need to be initiated. Inform patients with diabetes mellitus or glucose intolerance that antihyperglycemic medications may need to be adjusted during treatment with LORBRENA [see Warnings and Precautions (5.7)].
Embryo-Fetal Toxicity
Advise females of reproductive potential of the potential risk to a fetus. Advise females to inform their healthcare provider of a known or suspected pregnancy [see Warnings and Precautions (5.8), Use in Specific Populations (8.1)].
Advise females of reproductive potential to use effective non-hormonal contraception during treatment with LORBRENA and for at least 6 months after the final dose [see Use in Specific Populations (8.3)].
Advise male patients with female partners of reproductive potential to use effective contraception during treatment with LORBRENA and for at least 3 months after the final dose [see Use in Specific Populations (8.3), Nonclinical Toxicology (13.1)].
Lactation
Advise women not to breastfeed during treatment with LORBRENA and for 7 days after the final dose [see Use in Specific Populations (8.2)].
Infertility
Advise males of reproductive potential that LORBRENA may transiently impair fertility [see Use in Specific Populations (8.3), Nonclinical Toxicology (13.1)].
This product's labeling may have been updated. For the most recent prescribing information, please visit www.Pfizer.com. For medical information about LORBRENA, please visit www.pfizermedinfo.com or call 1‚ÄĎ800-438-1985.

LAB-1162-6.0
Spl Patient Package Insert Section
PATIENT INFORMATION LORBRENA (lor-BREN-ah) (lorlatinib) tablets
This Patient Information has been approved by the U.S. Food and Drug Administration.
Revised: April 2023
What is the most important information I should know about LORBRENA?
LORBRENA may cause serious side effects, including:
‚ÄĘ Liver problems due to interactions with other medicines. It is important to know what medicines should not be taken with LORBRENA.‚ÄĘ Central nervous system (CNS) effects. LORBRENA may cause CNS effects, including:
o problems with thinking, such as forgetfulness or confusiono changes in mood, such as depression and thoughts about suicide or dyingo psychotic effects, such as seeing or hearing things that are not real (hallucinations)o seizureso changes in speecho changes in sleep
Tell your healthcare provider if you experience new or worsening symptoms of these CNS effects during treatment with LORBRENA.
‚ÄĘ Increases in the cholesterol and triglycerides (lipid) levels in your blood. Most people will have an increase in the lipid levels in their blood during treatment with LORBRENA.
o If you have increases in the lipid levels in your blood during treatment with LORBRENA, your healthcare provider may need to start you on a medicine to lower the levels. If you are already taking a medicine to lower the lipid levels in your blood, your healthcare provider may need to increase your dose of that medicine.o Your healthcare provider should do blood tests to check the lipid levels in your blood before starting treatment, 1 to 2 months after starting treatment, and during treatment with LORBRENA.‚ÄĘ Heart problems. LORBRENA may cause very slow or abnormal heartbeats. Your healthcare provider should check your heart rhythm (electrocardiogram or EKG) before starting and during treatment with LORBRENA. Tell your healthcare provider right away if you feel dizzy or faint or have abnormal heartbeats. In some people, these problems are severe and your healthcare provider may need to have you stop taking LORBRENA or have a pacemaker placed.‚ÄĘ Lung problems. LORBRENA may cause severe or life-threatening swelling (inflammation) of the lungs during treatment that can lead to death. Symptoms may be similar to those from lung cancer. Tell your healthcare provider right away if you have any new or worsening symptoms of lung problems, including trouble breathing, shortness of breath, cough, or fever.‚ÄĘ High blood pressure (hypertension). Your healthcare provider should check your blood pressure before starting treatment, 2 weeks after starting treatment, and then at least every month during treatment with LORBRENA. Your healthcare provider may need to start or change your blood pressure medicine if you have high blood pressure during treatment with LORBRENA. Tell your healthcare provider right away if you get signs or symptoms of high blood pressure, including: headaches, dizziness, blurred vision, chest pain or shortness of breath.‚ÄĘ High blood sugar (hyperglycemia). LORBRENA may increase your blood sugar levels. Your healthcare provider should do blood tests to check your blood sugar levels before starting and during treatment with LORBRENA. Your healthcare provider may need to start or change your blood sugar medicine to control your blood sugar levels. Tell your healthcare provider right away if you get new or worsening signs and symptoms of high blood sugar, including:
o feeling very thirstyo needing to urinate more than usualo feeling very hungry
o feeling sick to your stomacho feeling weak or tiredo feeling confused
If you have serious side effects during treatment with LORBRENA, your healthcare provider may change your dose, stop your treatment for a period of time, or completely stop treatment with LORBRENA. See "What are possible side effects of LORBRENA?" for more information about side effects.
What is LORBRENA?
LORBRENA is a prescription medicine used to treat adults with non-small cell lung cancer (NSCLC):
‚ÄĘ that is caused by an abnormal anaplastic lymphoma kinase (ALK) gene, and‚ÄĘ that has spread to other parts of your body.
Your healthcare provider will perform a test to make sure that LORBRENA is right for you.It is not known if LORBRENA is safe and effective in children.
Do not take LORBRENA if you take certain other medicines called strong CYP3A inducers. Ask your healthcare provider for a ul of these medicines if you are not sure.
Before taking LORBRENA, tell your healthcare provider about all of your medical conditions, including if you:
‚ÄĘ have kidney problems‚ÄĘ have had episodes of depression or seizures‚ÄĘ have high levels of cholesterol or triglycerides in your blood‚ÄĘ have problems with your heart beat‚ÄĘ have lung or breathing problems‚ÄĘ have high blood pressure‚ÄĘ have diabetes or high blood sugar‚ÄĘ are pregnant or plan to become pregnant. LORBRENA can harm your unborn baby.
o Your healthcare provider will do a pregnancy test before you start treatment with LORBRENA.o Tell your healthcare provider right away if you become pregnant or think you may be pregnant during treatment with LORBRENA.
‚Ė™ Females who are able to become pregnant should use effective non-hormonal birth control during treatment with LORBRENA and for at least 6 months after the final dose of LORBRENA. Birth control pills (oral contraceptives) and other hormonal forms of birth control may not be effective if used during treatment with LORBRENA. Talk to your healthcare provider about birth control choices that are right for you during this time.‚Ė™ Males who have female partners who are able to become pregnant should use effective birth control during treatment with LORBRENA and for at least 3 months after the final dose of LORBRENA.‚ÄĘ are breastfeeding or plan to breastfeed. It is not known if LORBRENA passes into your breast milk. Do not breastfeed during treatment with LORBRENA and for 7 days after the final dose. Talk to your healthcare provider about the best way to feed your baby during this time.
Tell your healthcare provider about all the medicines you take, including prescription medicines, over-the-counter medicines, vitamins, and herbal supplements. LORBRENA may affect the way other medicines work and other medicines may affect the way LORBRENA works causing side effects. Know the medicines you take. Keep a ul of them to show to your healthcare provider and pharmacist when you get a new medicine.
How should I take LORBRENA?
‚ÄĘ Take LORBRENA exactly as your healthcare provider tells you to take it. Do not change your dose or stop taking LORBRENA unless your healthcare provider tells you to.‚ÄĘ Swallow LORBRENA tablets whole. Do not chew, crush, or split LORBRENA tablets. Do not take LORBRENA tablets if they are broken, cracked, or not intact.‚ÄĘ Take LORBRENA 1 time a day, at the same time each day.‚ÄĘ You may take LORBRENA with or without food.‚ÄĘ If you miss a dose, take it as soon as you remember. However, if it is close to the time of your next dose (within 4 hours), just take your next dose at your regular time. Do not take 2 doses of LORBRENA at the same time to make up for the missed dose.‚ÄĘ If you vomit after taking a dose of LORBRENA, do not take an extra dose. Take your next dose at your regular time.
What are the possible side effects of LORBRENA?
The most common side effects of LORBRENA include:
‚ÄĘ swelling in your arms, legs, hands and feet (edema)‚ÄĘ numbness and tingling feeling in your joints or arms and legs (peripheral neuropathy)‚ÄĘ weight gain‚ÄĘ problems with thinking, such as forgetfulness or confusion‚ÄĘ tiredness (fatigue)‚ÄĘ difficulty breathing‚ÄĘ pain in your joints‚ÄĘ diarrhea‚ÄĘ changes in mood, such as depression and irritability‚ÄĘ high cholesterol and triglyceride levels in the blood‚ÄĘ cough
LORBRENA may cause decreased fertility in males. In males, this could affect your ability to father a child. Talk to your healthcare provider if you have concerns about fertility.
These are not all of the possible side effects of LORBRENA. For more information, ask your healthcare provider or pharmacist.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store LORBRENA?
‚ÄĘ Store LORBRENA at room temperature between 68¬įF to 77¬įF (20¬įC to 25¬įC).
Keep LORBRENA and all medicines out of the reach of children.
General information about the safe and effective use of LORBRENA.
Medicines are sometimes prescribed for purposes other than those uled in a Patient Information leaflet. Do not use LORBRENA for a condition for which it was not prescribed. Do not give LORBRENA to other people, even if they have the same symptoms that you have. It may harm them. You can ask your healthcare provider or pharmacist for more information about LORBRENA that is written for health professionals.
What are the ingredients in LORBRENA?
Active ingredient: lorlatinib
Inactive ingredients: microcrystalline cellulose, dibasic calcium phosphate anhydrous, sodium starch glycolate, and magnesium stearate.
Film-coating contains: hydroxypropyl methylcellulose (HPMC) 2910/hypromellose, lactose monohydrate, macrogol/polyethylene glycol (PEG) 3350, triacetin, titanium dioxide, ferrosoferric oxide/black iron oxide, and iron oxide red.
For more information, go to www.pfizer.com.

LAB-1163-5.0
Principal Display Panel - 25 Mg Tablet Bottle Label
PfizerNDC 0069-0227-01
Lorbrena¬ģ (lorlatinib) tablets
25 mg
30 TabletsRx only
Principal Display Panel - 100 Mg Tablet Bottle Label
PfizerNDC 0069-0231-01
Lorbrena¬ģ (lorlatinib) tablets
100 mg
30 TabletsRx only

DISCLAIMER:
"This tool does not provide medical advice, and is for informational and educational purposes only, and is not a substitute for professional medical advice, treatment or diagnosis. Call your doctor to receive medical advice. If you think you may have a medical emergency, please dial 911."
"Do not rely on openFDA to make decisions regarding medical care. While we make every effort to ensure that data is accurate, you should assume all results are unvalidated. We may limit or otherwise restrict your access to the API in line with our Terms of Service."
"This product uses publicly available data from the U.S. National Library of Medicine (NLM), National Institutes of Health, Department of Health and Human Services; NLM is not responsible for the product and does not endorse or recommend this or any other product."
PillSync may earn a commission via links on our site
