Namenda (memantine hydrochloride 10 mg) Dailymed
Generic: memantine hydrochloride is used for the treatment of Alzheimer Disease

IMPRINT: FL 10
SHAPE: oval
COLOR: white
All Imprints
memantine hydrochloride 5 mg - 5 fl fl 5 oval brown
namenda (memantine hydrochloride) tablet namenda (memantine hydrochloride) solution namenda (memantine hydrochloride) kit - fl 10 oval white
memantine hydrochloride 5 mg oral tablet [namenda] - 5 fl oval brown
memantine hydrochloride 5 mg - 5 fl oval brown
memantine hydrochloride 10 mg - 10 fl oval grey

Go PRO for all pill images
1
NAMENDA (memantine hydrochloride) is indicated for the treatment of moderate to severe dementia of the Alzheimer’s type.
NAMENDA is a N-methyl-D-aspartate (NMDA) receptor antagonist indicated for the treatment of moderate to severe dementia of the Alzheimer’s type. (1 )
2
The recommended starting dose of NAMENDA is 5 mg once daily. The dose should be increased in 5 mg increments to 10 mg/day (5 mg twice daily), 15 mg/day (5 mg and 10 mg as separate doses), and 20 mg/day (10 mg twice daily). The minimum recommended interval between dose increases is one week. The dosage shown to be effective in controlled clinical trials is 20 mg/day.
NAMENDA can be taken with or without food. If a patient misses a single dose of NAMENDA, that patient should not double up on the next dose. The next dose should be taken as scheduled.
If a patient fails to take NAMENDA for several days, dosing may need to be resumed at lower doses and retitrated as described above.
Specific Populations
Renal Impairment
A target dose of 5 mg twice daily is recommended in patients with severe renal impairment (creatinine clearance of 5 ‚Äď 29 mL/min based on the Cockcroft-Gault equation).
Hepatic Impairment
NAMENDA should be administered with caution to patients with severe hepatic impairment [see Clinical Pharmacology ( 12.3 )].
- May be taken with or without food. (
2 )- Initial dose is 5 mg once daily. Increase dose in 5 mg increments to a maintenance dose of 10 mg twice daily. A minimum of 1 week of treatment with the previous dose should be observed before increasing the dose. (
2 )- Severe renal impairment: recommended dose is 5 mg twice daily. (
2 ) 
3
NAMENDA 5 mg Tablet: capsule-shaped, film-coated tablets are tan, with the strength ‚Äú5‚ÄĚ debossed on one side and ‚ÄúFL‚ÄĚ on the other side.
NAMENDA 10 mg Tablet: capsule-shaped, film-coated tablets are gray, with the strength ‚Äú10‚ÄĚ debossed on one side and ‚ÄúFL‚ÄĚ on the other side.
Tablets: 5 mg and 10 mg (3 )
4
NAMENDA (memantine hydrochloride) is contraindicated in patients with known hypersensitivity to memantine hydrochloride or to any excipients used in the formulation.
- NAMENDA is contraindicated in patients with known hypersensitivity to memantine hydrochloride or to any excipients used in the formulation. (
4 )
5
- Conditions that raise urine pH may decrease the urinary elimination of memantine, resulting in increased plasma levels of memantine. (
5.1 , 7.1 )
Conditions that raise urine pH may decrease the urinary elimination of memantine resulting in increased plasma levels of memantine [see Drug Interactions ( 7.1 )]. 
6
Most common adverse reactions (‚Č•¬†5% and greater than placebo) are dizziness, headache, confusion and constipation. (6.1 )
To report SUSPECTED ADVERSE REACTIONS, contact Allergan at 1-800-678-1605 or FDA at 1-800-FDA-1088 or www.fda.gov/medwatch.
6.1
NAMENDA was evaluated in eight double-blind placebo-controlled trials involving a total of 1862 dementia (Alzheimer’s disease, vascular dementia) patients (940 patients treated with NAMENDA and 922 patients treated with placebo) for a treatment period up to 28 weeks.
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in clinical practice.
Adverse Events Leading to Discontinuation
In placebo-controlled trials in which dementia patients received doses of NAMENDA up to 20 mg/day, the likelihood of discontinuation because of an adverse reaction was the same in the NAMENDA group (10.1%) as in the placebo group (11.5%). No individual adverse reaction was associated with the discontinuation of treatment in 1% or more of NAMENDA-treated patients and at a rate greater than placebo.
Most Common Adverse Reactions
In double-blind placebo-controlled trials involving dementia patients, the most common adverse reactions (incidence ‚Č• 5% and higher than placebo) in patients treated with NAMENDA were dizziness, headache, confusion and constipation.¬†Table 1 uls all adverse reactions that occurred in at least 2% of patients treated with NAMENDA¬†and at an incidence greater than placebo.
Table 1: Adverse Reactions Reported in Controlled Clinical Trials in at Least 2% of Patients Receiving NAMENDA and at a Higher Frequency than Placebo-Treated Patients Adverse Reaction Placebo (N=922) %       NAMENDA       (N=940) % Body as a Whole       Fatigue 1 2       Pain 1 3 Cardiovascular System       Hypertension 2 4 Central and Peripheral Nervous System       Dizziness 5 7       Headache 3 6 Gastrointestinal System       Constipation 3 5       Vomiting 2 3 Musculoskeletal System       Back pain 2 3 Psychiatric Disorders       Confusion 5 6       Somnolence 2 3       Hallucination 2 3 Respiratory System       Coughing 3 4       Dyspnea 1 2
The overall profile of adverse reactions and the incidence rates for individual adverse reactions in the subpopulation of patients with moderate to severe Alzheimer’s disease were not different from the profile and incidence rates described above for the overall dementia population.
Seizures
NAMENDA has not been systematically evaluated in patients with a seizure disorder. In clinical trials of NAMENDA, seizures occurred in 0.2% of patients treated with NAMENDA and 0.5% of patients treated with placebo.
POSTMARKETING EXPERIENCE SECTION
The following adverse reactions have been identified during post-approval use of memantine. 
Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure. These reactions include:
Blood and Lymphatic System Disorders - agranulocytosis, leukopenia (including neutropenia), pancytopenia, thrombocytopenia, thrombotic thrombocytopenic purpura.
Cardiac Disorders - cardiac failure congestive.
Gastrointestinal Disorders - pancreatitis.
Hepatobiliary Disorders - hepatitis.
Psychiatric Disorders - suicidal ideation.
Renal and Urinary Disorders - acute renal failure (including increased creatinine and renal insufficiency).
Skin Disorders - Stevens Johnson syndrome.
7
The clearance of memantine was reduced by about 80% under alkaline urine conditions at pH 8. Therefore, alterations of urine pH towards the alkaline condition may lead to an accumulation of the drug with a possible increase in adverse effects. Urine pH is altered by diet, drugs (e.g. carbonic anhydrase inhibitors, sodium bicarbonate) and clinical state of the patient (e.g. renal tubular acidosis or severe infections of the urinary tract). Hence, memantine should be used with caution under these conditions.
7.2
The combined use of NAMENDA with other NMDA antagonists (amantadine, ketamine, and dextromethorphan) has not been systematically evaluated and such use should be approached with caution.
8
PREGNANCY SECTION
Risk Summary
There are no adequate data on the developmental risk associated with the use of NAMENDA in pregnant women.
Adverse developmental effects (decreased body weight, and skeletal ossification) were observed in the offspring of rats administered memantine during pregnancy at doses associated with minimal maternal toxicity. These doses are higher than those used in humans at the maximum recommended daily dose of NAMENDA [see Data].  
In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2-4% and 15-20%, respectively. The background risk of major birth defects and miscarriage for the indicated population is unknown.
Data
Animal Data
Oral administration of memantine (0, 2, 6, or 18 mg/kg/day) to rats during the period of organogenesis resulted in decreased skeletal ossification in fetuses at the highest dose tested. The higher no-effect dose for adverse developmental effects (6 mg/kg) is 3 times the maximum recommended human daily dose (MRHD) of NAMENDA (20 mg) on a body surface area (mg/m2) basis.
Oral administration of memantine to rabbits (0, 3, 10, or 30 mg/kg/day) during the period of organogenesis resulted in no adverse developmental effects. The highest dose tested is approximately 30 times the MRHD of NAMENDA on a mg/m2 basis.
In rats, memantine (0, 2, 6, or 18 mg/kg/day) was administered orally prior to and throughout mating and, in females, through the period of organogenesis or continuing throughout lactation to weaning. Decreased skeletal ossification in fetuses and decreased body weight in pups were observed at the highest dose tested. The higher no-effect dose for adverse developmental effects (6 mg/kg/day) is 3 times the MRHD of NAMENDA on a mg/m2 basis.
Oral administration of memantine (0, 2, 6, or 18 mg/kg/day) to rats from late gestation throughout lactation to weaning, resulted in decreased pup weights at the highest dose tested. The higher no-effect dose (6 mg/kg/day) is approximately 3 times the MRHD of NAMENDA on a mg/m2 basis.
LACTATION SECTION
Risk Summary
There are no data on the presence of memantine in human milk, the effects on the breastfed infant, or the effects of NAMENDA on milk production.
The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for NAMENDA and any potential adverse effects on the breastfed infant from NAMENDA or from the underlying maternal condition.
PEDIATRIC USE SECTION
Safety and effectiveness in pediatric patients have not been established. 
Memantine failed to demonstrate efficacy in two 12-week controlled clinical studies of¬†578 pediatric patients aged 6-12 years with autism spectrum disorders (ASD), including autism, Asperger‚Äôs disorder and Pervasive Development Disorder - Not Otherwise Specified (PDD-NOS). Memantine has not been studied in pediatric patients under 6 years of age or over 12 years of age. Memantine treatment was initiated at 3 mg/day and the dose was escalated to the target dose (weight-based) by week 6. Oral doses of memantine 3, 6, 9, or 15 mg extended-release capsules were administered once daily to patients with weights < 20 kg, 20-39 kg, 40-59 kg and ‚Č• 60 kg, respectively.
In a randomized, 12-week double-blind, placebo-controlled parallel study (Study A) in patients with autism, there was no statistically significant difference in the Social Responsiveness Scale (SRS) total raw score between patients randomized to memantine (n=54) and those randomized to placebo (n=53). In a 12-week responder-enriched randomized withdrawal study (Study B) in 471 patients with ASD, there was no statistically significant difference in the loss of therapeutic response rates between patients randomized to remain on full-dose memantine (n=153) and those randomized to switch to placebo (n=158).
The overall risk profile of memantine in pediatric patients was generally consistent with the known risk profile in adults [see Adverse Reactions ( 6.1 )].
In Study A, the adverse reactions in the memantine group (n=56) that were reported in at least 5% of patients and at least twice the frequency of the placebo group (N=58) are uled in Table 2:
Table¬†2: Study A Commonly Reported Adverse¬†Reactions with a Frequency ‚Č• 5% and Twice That of Placebo¬†¬† A d vers e R eact i on M e m a n t in e N=56 P l a c e b o N=58 Cough 8.9% 3.4% Influenza 7.1% 3.4% Rhinorrhea 5.4% 0% Agitation 5.4% 1.7% D i sco n t inu at i o n s ¬† d u e ¬† to ¬† A d verse ¬† R e a ct i o n s a Aggression 3.6% 1.7% Irritability 1.8% 3.4% a Reported adverse reactions leading to discontinuation in more than one patient ineither treatment group.
The adverse reactions that were reported in at least 5% of patients in the 12-48 week open-label study to identify responders to enroll in Study B are uled in Table 3:
Table 3: 12-48 Week Open Label Lead-In study to Study B Commonly Reported Adverse Reactions with a Frequency ‚Č• 5% A d vers e R eact i on M e m a n t in e N=903 Headache 8.0% Nasopharyngitis 6.3% Pyrexia 5.8% Irritability 5.4% D i sco n t inu a t i o n s d u e to ¬† A dverse ¬† R e a ction s a Irritability 1.2% Aggression 1.0% a ¬† A t l ea st 1 % ¬† in c i d e n c e o f ¬† a d ve rse r eac tion s l ea din g ¬† t o ¬† p r e m a t u re d is c onti n u a t i on.
In the randomized withdrawal study (Study B), the adverse reaction in patients randomized to placebo (n=160) and reported in at least 5% of patients and at twice the frequency of the full-dose memantine treatment group (n=157) was irritability (5.0% vs 2.5%).
Juvenile Animal Study
In a study in which memantine (0, 15, 30 or 45 mg/kg/day) was orally administered to rats during the juvenile period of development (postnatal days [PND] 14 through 70), delays in sexual maturation were noted in males and females at all but the lowest dose tested, and body weight was reduced at the high dose. In rats orally administered memantine as a single dose (PND 14) or three daily doses (PND 14-16), neuronal lesions were observed in several areas of the brain at all but the lowest dose tested. Adverse neurobehavioral effects (decreased auditory startle habituation) were observed at the high dose. The no-effect dose for developmental toxicity was the lowest dose tested (15 mg/kg/day).
In a second juvenile animal study, memantine (0, 1, 3, 8, 15, 30, and 45 mg/kg/day) was orally administered to male and female rats beginning on PND 7 and continuing for various periods during postnatal development. Because of early memantine-related mortality, the 30 and 45 mg/kg/day groups were terminated without further evaluation. Apoptosis or neuronal degeneration in the brain was observed on PNDs 8-17 at a dose of 15 mg/kg/day. The no-effect dose for apoptosis and neuronal degeneration was 8 mg/kg/day. In animals in which memantine (0, 1, 3, 8, or 15 mg/kg/day) was orally administered on PNDs 7-70, adverse neurobehavioral effects (increased locomotor motor activity, increased auditory startle response and decreased habituation, and deficit in learning and memory) were observed at all but the lowest dose tested. Effects on auditory startle persisted after drug discontinuation. The no-effect dose for developmental toxicity was the lowest dose tested (1 mg/kg/day).
GERIATRIC USE SECTION
The majority of people with Alzheimer‚Äôs disease are 65 years of age and older. In the clinical studies of NAMENDA the mean age of patients was approximately 76 years; over 90% of patients were 65 years and older, 60% were 75 years and older, and 12% were at or above 85 years of age. The efficacy and safety data presented in the clinical trial sections were obtained from these patients. There were no clinically meaningful differences in most adverse events reported by patient groups ‚Č•¬†65 years old and <¬†65 years old.
RENAL IMPAIRMENT SUBSECTION
No dosage adjustment is needed in patients with mild or moderate renal impairment. A dosage reduction is recommended in patients with severe renal impairment [ see Dosage and Administration ( 2 ) and Clinical Pharmacology ( 12.3 ) ]. 
HEPATIC IMPAIRMENT SUBSECTION
No dosage adjustment is needed in patients with mild or moderate hepatic impairment. NAMENDA should be administered with caution to patients with severe hepatic impairment [ see Dosage and Administration ( 2 ) and Clinical Pharmacology ( 12.3 ) ].
10
Signs and symptoms most often accompanying memantine overdosage in clinical trials and from worldwide marketing experience, alone or in combination with other drugs and/or alcohol, include agitation, asthenia, bradycardia, confusion, coma, dizziness, ECG changes, increased blood pressure, lethargy, loss of consciousness, psychosis, restlessness, slowed movement, somnolence, stupor, unsteady gait, visual hallucinations, vertigo, vomiting, and weakness. The largest known ingestion of memantine worldwide was 2 grams in a patient who took memantine in conjunction with unspecified antidiabetic medications. This patient experienced coma, diplopia, and agitation, but subsequently recovered. Fatal outcome has been very rarely reported with memantine, and the relationship to memantine was unclear.
Because strategies for the management of overdose are continually evolving, it is advisable to contact a poison control center to determine the latest recommendations for the management of an overdose of any drug. As in any cases of overdose, general supportive measures should be utilized, and treatment should be symptomatic. Elimination of memantine can be enhanced by acidification of urine.
11
NAMENDA (memantine hydrochloride) is an orally active NMDA receptor antagonist. The chemical name for memantine hydrochloride is 1-amino-3,5-dimethyladamantane hydrochloride with the following structural formula:

The molecular formula is C12H21N‚ÄĘHCl and the molecular weight is 215.76. Memantine hydrochloride¬†occurs as a fine white to off-white powder and is soluble in water.
NAMENDA tablets are available for oral administration as capsule-shaped, film-coated tablets containing 5 mg and 10 mg of memantine hydrochloride. NAMENDA tablets also contain the following inactive ingredients: microcrystalline cellulose/colloidal silicon dioxide, talc, croscarmellose sodium, and magnesium stearate. In addition, the following inactive ingredients are also present as components of the film coat: hypromellose, titanium dioxide, polyethylene glycol 400, FD&C yellow #6 and FD&C blue #2 (5 mg tablets), and hypromellose, titanium dioxide, macrogol/polyethylene glycol 400 and iron oxide black (10 mg tablets).
12
12.1
Persistent activation of central nervous system N-methyl-D-aspartate (NMDA) receptors by the excitatory amino acid glutamate has been hypothesized to contribute to the symptomatology of Alzheimer’s disease. Memantine is postulated to exert its therapeutic effect through its action as a low to moderate affinity uncompetitive (open-channel) NMDA receptor antagonist which binds preferentially to the NMDA receptor-operated cation channels. There is no evidence that memantine prevents or slows neurodegeneration in patients with Alzheimer’s disease.
12.2
Memantine showed low to negligible affinity for GABA, benzodiazepine, dopamine, adrenergic, histamine and glycine receptors and for voltage-dependent Ca2+, Na+, or K+ channels. Memantine also showed antagonistic effects at the 5HT3 receptor with a potency similar to that for the NMDA receptor and blocked nicotinic acetylcholine receptors with one-sixth to one-tenth the potency.
In vitro studies have shown that memantine does not affect the reversible inhibition of acetylcholinesterase by donepezil, galantamine, or tacrine.
12.3
Absorption
Following oral administration memantine is highly absorbed with peak concentrations reached in about 3-7 hours. Memantine has linear pharmacokinetics over the therapeutic dose range. Food has no effect on the absorption of memantine.
Distribution
The mean volume of distribution of memantine is 9-11 L/kg and the plasma protein binding is low (45%).
Metabolism  
Memantine undergoes partial hepatic metabolism. The hepatic microsomal CYP450 enzyme system does not play a significant role in the metabolism of memantine.
Elimination
Memantine is excreted predominantly (about 48%) unchanged in urine and has a terminal elimination half-life of about 60-80 hours.
The remainder is converted primarily to three polar metabolites which possess minimal NMDA receptor antagonistic activity: the N-glucuronide conjugate, 6-hydroxy memantine, and 1-nitroso-deaminated memantine. A total of 74% of the administered dose is excreted as the sum of the parent drug and the N-glucuronide conjugate. Renal clearance involves active tubular secretion moderated by pH dependent tubular reabsorption.
Speci fic   Populations
Gender 
Following multiple dose administration of NAMENDA 20 mg daily, females had about 45% higher exposure than males, but there was no difference in exposure when body weight was taken into account.
Elderly
The pharmacokinetics of NAMENDA in young and elderly subjects are similar.
Renal Impairment
Memantine pharmacokinetics were evaluated following single oral administration of 20 mg memantine hydrochloride in 8 subjects with mild renal impairment (creatinine clearance, CLcr, >¬†50 ‚Äď 80 mL/min), 8 subjects with moderate renal impairment (CLcr 30 ‚Äď 49 mL/min), 7 subjects with severe renal impairment (CLcr 5 ‚Äď 29 mL/min) and 8 healthy subjects (CLcr > 80 mL/min) matched as closely as possible by age, weight and gender to the subjects with renal impairment. Mean AUC0- ‚ąě increased by 4%, 60%, and 115% in subjects with mild, moderate, and severe renal impairment, respectively, compared to healthy subjects. The terminal elimination half-life increased by 18%, 41%, and 95% in subjects with mild, moderate, and severe renal impairment, respectively, compared to healthy subjects.
No dosage adjustment is recommended for patients with mild and moderate renal impairment. Dosage should be reduced in patients with severe renal impairment [ see Dosage and Administration ( 2 ) ].
Hepatic Impairment
Memantine pharmacokinetics were evaluated following the administration of single oral doses of 20 mg in 8 subjects with moderate hepatic impairment (Child-Pugh Class B, score 7-9) and 8 subjects who were age-, gender-, and weight-matched to the hepatically-impaired subjects. There was no change in memantine exposure (based on Cmax and AUC) in subjects with moderate hepatic impairment as compared with healthy subjects. However, terminal elimination half-life increased by about 16% in subjects with moderate hepatic impairment as compared with healthy subjects. No dose adjustment is recommended for patients with mild and moderate hepatic impairment. Memantine should be administered with caution to patients with severe hepatic impairment as the pharmacokinetics of memantine have not been evaluated in that population.
Drug-Drug Interactions
Use with Cholinesterase Inhibitors
Coadministration of memantine with the AChE inhibitor donepezil hydrochloride did not affect the pharmacokinetics of either compound. Furthermore, memantine did not affect AChE inhibition by donepezil. In a 24-week controlled clinical study in patients with moderate to severe Alzheimer’s disease, the adverse event profile observed with a combination of NAMENDA and donepezil was similar to that of donepezil alone.
Effect of NAMENDA on the Metabolism of Other Drugs
I n vitro studies conducted with marker substrates of CYP450 enzymes (CYP1A2, -2A6, -2C9, -2D6, -2E1, -3A4) showed minimal inhibition of these enzymes by memantine. In addition, in vitro studies indicate that at concentrations exceeding those associated with efficacy, memantine does not induce the cytochrome P450 isozymes CYP1A2, -2C9, -2E1 and -3A4/5. No pharmacokinetic interactions with drugs metabolized by these enzymes are expected.
Pharmacokinetic studies evaluated the potential of memantine for interaction with warfarin, and bupropion. Memantine did not affect the pharmacokinetics of the CYP2B6 substrate bupropion or its metabolite hydroxy-bupropion. Furthermore, memantine did not affect the pharmacokinetics or pharmacodynamics of warfarin as assessed by the prothrombin INR.
Effect of Other Drugs on N AMENDA
Memantine is predominantly renally eliminated, and drugs that are substrates and/or inhibitors of the CYP450 system are not expected to alter the metabolism of memantine.
Drugs Eliminated via Renal Mechanisms
Because memantine is eliminated in part by tubular secretion, coadministration of drugs that use the same renal cationic system, including hydrochlorothiazide (HCTZ), triamterene (TA), metformin, cimetidine, ranitidine, quinidine, and nicotine, could potentially result in altered plasma levels of both agents. However, coadministration of NAMENDA and HCTZ/TA did not affect the bioavailability of either memantine or TA, and the bioavailability of HCTZ decreased by 20%. In addition, coadministration of memantine with the antihyperglycemic drug Glucovance¬ģ (glyburide and metformin hydrochloride) did not affect the pharmacokinetics of memantine, metformin and glyburide. Furthermore, memantine did not modify the serum glucose lowering effect of Glucovance¬ģ, indicating the absence of a pharmacodynamic interaction.
Drugs Highly Bound to Plasma Proteins
Because the plasma protein binding of memantine is low (45%), an interaction with drugs that are highly bound to plasma proteins, such as warfarin and digoxin, is unlikely.
13
CARCINOGENESIS & MUTAGENESIS & IMPAIRMENT OF FERTILITY SECTION
There was no evidence of carcinogenicity in a 113-week oral study in mice at doses up to 40 mg/kg/day (10 times the maximum recommended human dose [MRHD] on a mg/m2 basis). There was also no evidence of carcinogenicity in rats orally dosed at up to 40 mg/kg/day for 71 weeks followed by 20 mg/kg/day (20 and 10 times the MRHD on a mg/m2 basis, respectively) through 128 weeks.
Memantine produced no evidence of genotoxic potential when evaluated in the in vitro S. typhimurium or E. coli reverse mutation assay, an in vitro chromosomal aberration test in human lymphocytes, an in vivo cytogenetics assay for chromosome damage in rats, and the in vivo mouse micronucleus assay. The results were equivocal in an in vitro gene mutation assay using Chinese hamster V79 cells.
No impairment of fertility or reproductive performance was seen in rats administered up to 18 mg/kg/day (9 times the MRHD on a mg/m2 basis) orally from 14 days prior to mating through gestation and lactation in females, or for 60 days prior to mating in males.
13.2
Memantine induced neuronal lesions (vacuolation and necrosis) in the multipolar and pyramidal cells in cortical layers III and IV of the posterior cingulate and retrosplenial neocortices in rats, similar to those which are known to occur in rodents administered other NMDA receptor antagonists. Lesions were seen after a single dose of memantine. In a study in which rats were given daily oral doses of memantine for 14 days, the no-effect dose for neuronal necrosis was 6 times the maximum recommended human dose of 20 mg/day on a mg/m2 basis.
In acute and repeat-dose neurotoxicity studies in female rats, oral administration of memantine and donepezil in combination resulted in increased incidence, severity, and distribution of neurodegeneration compared with memantine alone. The no-effect levels of the combination were associated with clinically relevant plasma memantine and donepezil exposures.
The relevance of these findings to humans is unknown.
14
The effectiveness of NAMENDA as a treatment for patients with moderate to severe Alzheimer’s disease was demonstrated in 2 randomized, double-blind, placebo-controlled clinical studies (Studies 1 and 2) conducted in the United States that assessed both cognitive function and day to day function. The mean age of patients participating in these two trials was 76 years with a range of 50-93 years. Approximately 66% of patients were female and 91% of patients were Caucasian. A third study (Study 3), carried out in Latvia, enrolled patients with severe dementia, but did not assess cognitive function as a planned endpoint. Study Outcome Measures: In each U.S. study, the effectiveness of NAMENDA was determined using both an instrument designed to evaluate overall function through caregiver-related assessment, and an instrument that measures cognition. Both studies showed that patients on NAMENDA experienced significant improvement on both measures compared to placebo.
Day-to-day function was assessed in both studies using the modified Alzheimer’s disease Cooperative Study - Activities of Daily Living inventory (ADCS-ADL). The ADCS-ADL consists of a comprehensive battery of ADL questions used to measure the functional capabilities of patients. Each ADL li is rated from the highest level of independent performance to complete loss. The investigator performs the inventory by interviewing a caregiver familiar with the behavior of the patient. A subset of 19 lis, including ratings of the patient’s ability to eat, dress, bathe, telephone, travel, shop, and perform other household chores has been validated for the assessment of patients with moderate to severe dementia. This is the modified ADCS-ADL, which has a scoring range of 0 to 54, with the lower scores indicating greater functional impairment.
The ability of NAMENDA to improve cognitive performance was assessed in both studies with the Severe Impairment Battery (SIB), a multi-li instrument that has been validated for the evaluation of cognitive function in patients with moderate to severe dementia. The SIB examines selected aspects of cognitive performance, including elements of attention, orientation, language, memory, visuospatial ability, construction, praxis, and social interaction. The SIB scoring range is from 0 to 100, with lower scores indicating greater cognitive impairment.
Study 1 (Twenty-Eight-Week Study)
In a study of 28 weeks duration, 252 patients with moderate to severe probable Alzheimer‚Äôs disease (diagnosed by DSM-IV and NINCDS-ADRDA criteria, with Mini-Mental State Examination scores ‚Č•¬†3 and ‚ȧ¬†14 and Global Deterioration Scale Stages 5-6) were randomized to NAMENDA or placebo. For patients randomized to NAMENDA, treatment was initiated at 5 mg once daily and increased weekly by 5 mg/day in divided doses to a dose of 20 mg/day (10 mg twice a day).
Effects on the ADCS-ADL
Figure 1 shows the time course for the change from baseline in the ADCS-ADL score for patients in the two treatment groups completing the 28 weeks of the study. At 28 weeks of treatment, the mean difference in the ADCS-ADL change scores for the NAMENDA-treated patients compared to the patients on placebo was 3.4 units. Using an analysis based on all patients and carrying their last study observation forward (LOCF analysis), NAMENDA treatment was statistically significantly superior to placebo.

Figure 1: Time course of the change from baseline in ADCS-ADL score for patients completing 28 weeks of treatment
Figure 2 shows the cumulative percentages of patients from each of the treatment groups who had attained at least the change in the ADCS-ADL shown on the X axis. The curves show that both patients assigned to NAMENDA and placebo have a wide range of responses and generally show deterioration (a negative change in ADCS-ADL compared to baseline), but that the NAMENDA group is more likely to show a smaller decline or an improvement. (In a cumulative distribution display, a curve for an effective treatment would be shifted to the left of the curve for placebo, while an ineffective or deleterious treatment would be superimposed upon or shifted to the right of the curve for placebo).

Figure 2: Cumulative percentage of patients completing 28 weeks of
double-blind treatment with specified changes from baseline in ADCS-ADL scores
Ef fects on the SIB
Figure 3 shows the time course for the change from baseline in SIB score for the two treatment groups over the 28 weeks of the study. At 28 weeks of treatment, the mean difference in the SIB change scores for the NAMENDA-treated patients compared to the patients on placebo was 5.7 units. Using an LOCF analysis, NAMENDA treatment was statistically significantly superior to placebo.
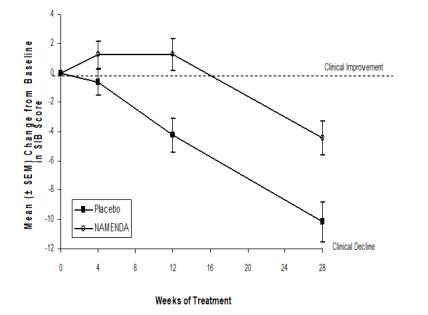
Figure 3: Time course of the change from baseline in
SIB score for patients completing 28 weeks of treatment
Figure 4 shows the cumulative percentages of patients from each treatment group who had attained at least the measure of change in SIB score shown on the X axis. The curves show that both patients assigned to NAMENDA and placebo have a wide range of responses and generally show deterioration, but that the NAMENDA group is more likely to show a smaller decline or an improvement.

Figure 4: Cumulative percentage of patients completing 28 weeks of
double-blind treatment with specified changes from baseline in SIB scores
Study 2 (Twenty-Four-Week Study)
In a study of 24 weeks duration, 404 patients with moderate to severe probable Alzheimer‚Äôs disease (diagnosed by NINCDS-ADRDA criteria, with Mini-Mental State Examination scores ‚Č•¬†5 and ‚ȧ¬†14) who had been treated with donepezil for at least 6 months and who had been on a stable dose of donepezil for the last 3 months were randomized to NAMENDA or placebo while still receiving donepezil. For patients randomized to NAMENDA, treatment was initiated at 5 mg once daily and increased weekly by 5 mg/day in divided doses to a dose of 20 mg/day (10 mg twice a day).
Effects on the ADCS-ADL
Figure 5 shows the time course for the change from baseline in the ADCS-ADL score for the two treatment groups over the 24 weeks of the study. At 24 weeks of treatment, the mean difference in the ADCS-ADL change scores for the NAMENDA/donepezil treated patients (combination therapy) compared to the patients on placebo/donepezil (monotherapy) was 1.6 units. Using an LOCF analysis, NAMENDA/donepezil treatment was statistically significantly superior to placebo/donepezil.

Figure 5: Time course of the change from baseline in
ADCS-ADL score for patients completing 24 weeks of treatment
Figure 6 shows the cumulative percentages of patients from each of the treatment groups who had attained at least the measure of improvement in the ADCS-ADL shown on the X axis. The curves show that both patients assigned to NAMENDA/donepezil and placebo/donepezil have a wide range of responses and generally show deterioration, but that the NAMENDA/donepezil group is more likely to show a smaller decline or an improvement.

Figure 6: Cumulative percentage of patients completing 24 weeks of
double-blind treatment with specified changes from baseline in ADCS-ADL scores
Effects on the SIB
Figure 7 shows the time course for the change from baseline in SIB score for the two treatment groups over the 24 weeks of the study. At 24 weeks of treatment, the mean difference in the SIB change scores for the NAMENDA/donepezil-treated patients compared to the patients on placebo/donepezil was 3.3 units. Using an LOCF analysis, NAMENDA/donepezil treatment was statistically significantly superior to placebo/donepezil.

Figure 7: Time course of the change from baseline in
SIB score for patients completing 24 weeks of treatment
Figure 8 shows the cumulative percentages of patients from each treatment group who had attained at least the measure of improvement in SIB score shown on the X axis. The curves show that both patients assigned to NAMENDA/donepezil and placebo/donepezil have a wide range of responses, but that the NAMENDA/donepezil group is more likely to show an improvement or a smaller decline.

Figure 8: Cumulative percentage of patients completing 24 weeks of
double-blind treatment with specified changes from baseline in SIB scores
Study 3 (Twelve-Week Study)
In a double-blind study of 12 weeks duration, conducted in nursing homes in Latvia, 166 patients with dementia according to DSM-III-R, a Mini-Mental State Examination score of < 10, and Global Deterioration Scale staging of 5 to 7 were randomized to either NAMENDA or placebo. For patients randomized to NAMENDA, treatment was initiated at 5 mg once daily and increased to 10 mg once daily after 1 week. The primary efficacy measures were the care dependency subscale of the Behavioral Rating Scale for Geriatric Patients (BGP), a measure of day-to-day function, and a Clinical Global Impression of Change (CGI-C), a measure of overall clinical effect. No valid measure of cognitive function was used in this study. A statistically significant treatment difference at 12 weeks that favored NAMENDA over placebo was seen on both primary efficacy measures. Because the patients entered were a mixture of Alzheimer’s disease and vascular dementia, an attempt was made to distinguish the two groups and all patients were later designated as having either vascular dementia or Alzheimer’s disease, based on their scores on the Hachinski Ischemic Scale at study entry. Only about 50% of the patients had computerized tomography of the brain. For the subset designated as having Alzheimer’s disease, a statistically significant treatment effect favoring NAMENDA over placebo at 12 weeks was seen on both the BGP and CGI-C.
16
5 mg Tablet s:
Tan, capsule-shaped, film-coated tablets¬†with ‚Äú5‚ÄĚ debossed on one side and ‚ÄúFL‚ÄĚ on the other side.
Bottle of 60       NDC #0456-3205-60
10 x 10 Unit Dose       NDC #0456-3205-63
10 mg Tablet s:
Gray, capsule-shaped, film-coated tablets¬†with ‚Äú10‚ÄĚ debossed on one side and ‚ÄúFL‚ÄĚ on the other side.
Bottle of 60       NDC #0456-3210-60
10 x 10 Unit Dose       NDC #0456-3210-63
Titration Pa c k:             NDC #0456-3200-14
Buler package containing 49 tablets (28 x 5 mg and 21 x 10 mg tablets).
Store NAMENDA¬†Tablets at 25¬įC (77¬įF); excursions permitted to 15-30¬įC (59-86¬įF) [see USP Controlled Room Temperature].
17
Advise the patient to read the FDA-approved patient labeling (Patient Information). 
To assure safe and effective use of NAMENDA, the following information and instructions provided in the patient information section should be discussed with patients and caregivers.
Patients/caregivers should be instructed to follow the dose titration schedule provided by their physician or healthcare professional for NAMENDA. They should be warned not to use any tablets of NAMENDA that are damaged or show signs of tampering.
If a patient misses a single dose of NAMENDA, that patient should not double up on the next dose. The next dose should be taken as scheduled. If a patient fails to take NAMENDA for several days, dosing should not be resumed without consulting that patient’s healthcare professional.
Made in Ireland
Distributed by:
Allergan USA, Inc.
Madison, NJ 07940
Licensed from Merz Pharmaceuticals GmbH
NAMENDA¬ģ is a registered trademark of Merz Pharma GmbH & Co KGaA
All other trademarks are the property of their respective owners.
Allergan¬ģ and its design are trademarks of Allergan, Inc.

© 2019 Allergan. All rights reserved.
v1.1USPI3205
Spl Patient Package Insert Section
Patient Information
NAMENDA ¬ģ ¬† [ Nuh-MEN-dah ]
(mema n tine hydrochloride)
Tablets
Read this Patient Information that comes with NAMENDA before you start taking it and each time you get a refill. There may be new information. This information does not take the place of talking to your doctor about your medical condition or your treatment. 
What is NAMENDA?
NAMENDA¬ģ is a prescription medicine used for the treatment of moderate to severe dementia in people with Alzheimer‚Äôs disease. NAMENDA belongs to a class of medicines called N-methyl-D-aspartate¬†(NMDA) inhibitors.
It is not known if NAMENDA is safe and effective in children.
Who should not take NAMENDA?
Do not take NAMENDA if you are allergic to memantine or any of the ingredients in NAMENDA tablets. See the end of this leaflet for a complete ul of ingredients in NAMENDA tablets.
What should I tell my doctor before taking NAMENDA?
Before you take NAMENDA, tell your doctor if you:
- have or have had seizures
- have or have had problems passing urine
- have or have had bladder or kidney problems
- have liver problems
- have any other medical conditions
- are pregnant or plan to become pregnant. It is not known if NAMENDA will harm your unborn baby.
- are breastfeeding or plan to breastfeed. It is not known if memantine passes into your breast milk. Talk to your doctor about the best way to feed your baby if you take NAMENDA.
Tell your doctor about all the medicines you take, including prescription and non-prescription medicines, vitamins, and herbal supplements.
Taking NAMENDA with certain other medicines may affect each other. Taking NAMENDA with other medicines can cause serious side effects.
Especially tell your doctor if you take:
- other NMDA antagonists such as amantadine, ketamine, and dextromethorphan
- medicines that make your urine alkaline such as carbonic anhydrase inhibitors and sodium bicarbonate
Ask your doctor or pharmacist for a ul of these medicines, if you are not sure.
Know the medicines you take. Keep a ul of them to show your doctor and pharmacist when you get a new medicine.
How should I take NAMENDA?
- Your doctor will tell you how much NAMENDA to take and when to take it.
- Your doctor may change your dose if needed.
- NAMENDA can be taken with food or without food.
- Do not use any tablets of NAMENDA that are damaged or show signs of tampering.
- If you forget to take one dose of NAMENDA, do not double up on the next dose. You should take only the next dose as scheduled.
- If you have forgotten to take NAMENDA for several days, you should not take the next dose until you talk to your doctor.
- If you take too much NAMENDA, call your doctor or poison control center at 1-800-222-1222, or go to the nearest hospital emergency room right away.
What are the possible side effects of NAMENDA?
NAMENDA may cause side effects, including:
The most common side effects of NAMENDA include:
- dizziness
- headache
- confusion
- constipation
These are not all the possible side effects of NAMENDA. For more information, ask your doctor or pharmacist.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store NAMENDA?
- Store NAMENDA at room temperature between 68¬įF to 77¬įF (20¬įC to 25¬įC).
What are the ingredients in NAMENDA?
NAMENDA tablets:
Active ingredient: memantine hydrochloride   
Inactive ingredients: microcrystalline cellulose/colloidal silicon dioxide, talc, croscarmellose sodium, and magnesium stearate
Inactive ingredients of tablet film coating: hypromellose, titanium dioxide, polyethylene glycol 400, FD&C yellow #6 and FD&C blue #2 (5 mg tablets), and hypromellose, titanium dioxide, macrogol/polyethylene glycol 400 and iron oxide black (10 mg tablets)
Keep NAMENDA and all medicines out of the reach of children.
General information about the safe and effective use of NAMENDA.
Medicines are sometimes prescribed for purposes other than those uled in a Patient Information leaflet. Do not take NAMENDA for a condition for which it was not prescribed. Do not give NAMENDA to other people, even if they have the same condition. It may harm them.
This Patient Information leaflet summarizes the most important information about NAMENDA. If you would like more information, talk with your doctor. You can ask your doctor or pharmacist for information about NAMENDA that was written for healthcare professionals.
For more information about NAMENDA, go to www.namenda.com, or call Allergan at 1-800-678-1605.
This Patient Information has been approved by the U.S. Food and Drug Administration.
Made in Ireland
Distributed by:
Allergan USA, Inc.
Madison, NJ 07940
Licensed from Merz Pharmaceuticals GmbH
NAMENDA¬ģ is a registered trademark of Merz Pharma GmbH & Co KGaA
Allergan¬ģ and its design are trademarks of Allergan, Inc.
© 2019 Allergan. All rights reserved.
Content updated: November 2018
v1.1PPI3205
Principal Display Panel
Rx Only NDC 0456-3205-60
Namenda
memantine HCl tablets
5 mg
60 Tablets 
Principal Display Panel
Rx Only NDC 0456-3210-60Namendamemantine HCl tablets10 mg60 Tablets 

Principal Display Panel
Rx Only NDC 0456-3200-14Namendamemantine HCl tabletsTITRATION PAK
 
DISCLAIMER:
"This tool does not provide medical advice, and is for informational and educational purposes only, and is not a substitute for professional medical advice, treatment or diagnosis. Call your doctor to receive medical advice. If you think you may have a medical emergency, please dial 911."
"Do not rely on openFDA to make decisions regarding medical care. While we make every effort to ensure that data is accurate, you should assume all results are unvalidated. We may limit or otherwise restrict your access to the API in line with our Terms of Service."
"This product uses publicly available data from the U.S. National Library of Medicine (NLM), National Institutes of Health, Department of Health and Human Services; NLM is not responsible for the product and does not endorse or recommend this or any other product."
PillSync may earn a commission via links on our site
