ZEJULA Dailymed
Generic: niraparib is used for the treatment of Fallopian Tube Neoplasms Ovarian Neoplasms Peritoneal Neoplasms

All Imprints
zejula (niraparib) 300 mg - zejula 300 oval green
zejula (niraparib) 200 mg - zejula 200 oval blue
zejula (niraparib) 100 mg - zejula 100 oval grey

Go PRO for all pill images
1 Indications And Usage
ZEJULA is a poly (ADP-ribose) polymerase (PARP) inhibitor indicated:
• for the maintenance treatment of adult patients with advanced epithelial ovarian, fallopian tube, or primary peritoneal cancer who are in a complete or partial response to first-line platinum-based chemotherapy. (1.1 )• for the maintenance treatment of adult patients with deleterious or suspected deleterious germline BRCA-mutated recurrent epithelial ovarian, fallopian tube, or primary peritoneal cancer who are in a complete or partial response to platinum-based chemotherapy. Select patients for therapy based on an FDA-approved companion diagnostic for ZEJULA (1.2 ,2.1 )1.1 First-Line Maintenance Treatment of Advanced Ovarian Cancer
ZEJULA is indicated for the maintenance treatment of adult patients with advanced epithelial ovarian, fallopian tube, or primary peritoneal cancer who are in a complete or partial response to first-line platinum-based chemotherapy.
1.2 Maintenance Treatment of Recurrent Germline -mutated Ovarian Cancer
ZEJULA is indicated for the maintenance treatment of adult patients with deleterious or suspected deleterious germline BRCA-mutated (gBRCAmut) recurrent epithelial ovarian, fallopian tube, or primary peritoneal cancer who are in a complete or partial response to platinum-based chemotherapy.
Select patients for therapy based on an FDA-approved companion diagnostic for ZEJULA [see Dosage and Administration (2.1)].
2 Dosage And Administration
• First-line maintenance treatment of advanced ovarian cancer:
• For patients weighing <77 kg (<170 lbs) OR with a platelet count <150,000/mcL, the recommended dosage is 200 mg taken orally once daily. (2.2 )• For patients weighing ≥77 kg (≥170 lbs) AND a platelet count ≥150,000/ mcL, the recommended dosage is 300 mg taken orally once daily. (2.2 )• Maintenance treatment of recurrent germline BRCA-mutated ovarian cancer: The recommended dosage is 300 mg taken orally once daily. (2.2 )• Continue treatment until disease progression or unacceptable toxicity. (2.2 )• ZEJULA may be taken with or without food. (2.2 )• For adverse reactions, consider interruption of treatment, dose reduction, or dose discontinuation. (2.3 )• For patients with moderate hepatic impairment, reduce the starting dosage of niraparib to 200 mg once daily. (2.4 )2.1 Patient Selection
Maintenance Treatment of Recurrent Germline BRCA-mutated Ovarian Cancer
Select patients for the maintenance treatment of recurrent ovarian cancer with ZEJULA based on the presence of deleterious or suspected deleterious germline BRCA mutations [see Clinical Studies (14.2)].
Information on FDA-approved tests for the detection of deleterious or suspected deleterious germline BRCA mutations for this indication is available at https://www.fda.gov/companiondiagnostics.
2.2 Recommended Dosage
Continue treatment with ZEJULA until disease progression or unacceptable toxicity.
Instruct patients to take their dose of ZEJULA at approximately the same time each day. Advise patients to swallow each tablet whole and not to chew, crush, or split ZEJULA prior to swallowing. ZEJULA may be taken with or without food. Bedtime administration may be a potential method for managing nausea.
In the case of a missed dose of ZEJULA, instruct patients to take their next dose at its regularly scheduled time. If a patient vomits or misses a dose of ZEJULA, an additional dose should not be taken.
First-Line Maintenance Treatment of Advanced Ovarian Cancer
• For patients weighing <77 kg (<170 lbs) OR with a platelet count of <150,000/mcL, the recommended dosage is 200 mg taken orally once daily.• For patients weighing ≥77 kg (≥170 lbs) AND who have a platelet count ≥150,000/mcL, the recommended dosage is 300 mg taken orally once daily.
For the maintenance treatment of advanced ovarian cancer, patients should start treatment with ZEJULA no later than 12Â weeks after their most recent platinum-containing regimen.
Maintenance Treatment of Recurrent Germline BRCA-mutated Ovarian Cancer
The recommended dosage of ZEJULA is 300 mg taken orally once daily.
For the maintenance treatment of recurrent ovarian cancer, patients should start treatment with ZEJULA no later than 8Â weeks after their most recent platinum-containing regimen.
2.3 Dosage Adjustments for Adverse Reactions
To manage adverse reactions, consider interruption of treatment, dose reduction, or dose discontinuation. The recommended dose modifications for adverse reactions are uled in Tables 1, 2, and 3.
Table 1. Recommended Dose Modifications for Adverse Reactions a If further dose reduction below 100 mg/day is required, discontinue ZEJULA.
Starting Dose Level
200 mg
300 mg
First dose reduction
100 mg/daya
200 mg/day
Second dose reduction
Discontinue ZEJULA.
100 mg/daya
Table 2. Dose Modifications for Non-Hematologic Adverse Reactions CTCAE = Common Terminology Criteria for Adverse Events.
Non-hematologic CTCAE ≥Grade 3 adverse reaction that persists despite medical management
• Withhold ZEJULA for a maximum of 28 days or until resolution of adverse reaction.• Resume ZEJULA at a reduced dose per Table 1.
CTCAE ≥Grade 3 treatment-related adverse reaction lasting more than 28 days while patient is administered ZEJULA 100 mg/day
Discontinue ZEJULA.
Table 3. Dose Modifications for Hematologic Adverse Reactions a If myelodysplastic syndrome or acute myeloid leukemia (MDS/AML) is confirmed, discontinue ZEJULA [see Warnings and Precautions (5.1, 5.2)].
Monitor complete blood counts weekly for the first month, monthly for the next 11 months of treatment, and periodically after this time [see Warnings and Precautions (5.1)].
Platelet count <100,000/mcL
First occurrence:
• Withhold ZEJULA for a maximum of 28 days and monitor blood counts weekly until platelet counts return to ≥100,000/mcL.• Resume ZEJULA at same or reduced dose per Table 1.• If platelet count is <75,000/mcL, resume at a reduced dose.
Second occurrence:
• Withhold ZEJULA for a maximum of 28 days and monitor blood counts weekly until platelet counts return to ≥100,000/mcL.• Resume ZEJULA at a reduced dose per Table 1.• Discontinue ZEJULA if the platelet count has not returned to acceptable levels within 28 days of the dose interruption period or if the patient has already undergone dose reduction to 100 mg once daily.a
Neutrophil <1,000/mcL or hemoglobin <8 g/dL
• Withhold ZEJULA for a maximum of 28 days and monitor blood counts weekly until neutrophil counts return to ≥1,500/mcL or hemoglobin returns to ≥9 g/dL.• Resume ZEJULA at a reduced dose per Table 1.• Discontinue ZEJULA if neutrophils and/or hemoglobin have not returned to acceptable levels within 28 days of the dose interruption period or if the patient has already undergone dose reduction to 100 mg once daily.a
Hematologic adverse reaction requiring transfusion
• For patients with platelet count ≤10,000/mcL, platelet transfusion should be considered. If there are other risk factors such as coadministration of anticoagulation or antiplatelet drugs, consider interrupting these drugs and/or transfusion at a higher platelet count.• Resume ZEJULA at a reduced dose.2.4 Dosage Adjustment for Hepatic Impairment
Moderate Hepatic Impairment
For patients with moderate hepatic impairment, reduce the starting dosage of ZEJULA to 200 mg once daily. Monitor patients for hematologic toxicity and reduce the dose further, if needed [see Dosage and Administration (2.3), Use in Specific Populations (8.7), Clinical Pharmacology (12.3)].
3 Dosage Forms And Strengths
• Tablets: 100-mg gray, oval-shaped, film-coated tablet debossed with “100” on one side and “Zejula” on the other side.• Tablets: 200-mg blue, oval-shaped, film-coated tablet debossed with “200” on one side and “Zejula” on the other side.• Tablets: 300-mg green, oval-shaped, film-coated tablet debossed with “300” on one side and “Zejula” on the other side.
Tablets: 100 mg, 200 mg, 300 mg (3 )
4 Contraindications
None.
None. (4 )
5 Warnings And Precautions
• Myelodysplastic Syndrome/Acute Myeloid Leukemia (MDS/AML): MDS/AML occurred in patients exposed to ZEJULA, and some cases were fatal. Monitor patients for hematological toxicity and discontinue if MDS/AML is confirmed. (5.1 )• Bone Marrow Suppression: Test complete blood counts weekly for the first month, monthly for the next 11 months, and periodically thereafter for clinically significant changes. (5.2 )• Hypertension and Cardiovascular Effects: Monitor blood pressure and heart rate at least weekly for the first 2 months, then monthly for the first year and periodically thereafter during treatment with ZEJULA. Manage with antihypertensive medications and adjustment of the dose of ZEJULA, if necessary. (5.3 )• Posterior Reversible Encephalopathy Syndrome (PRES): PRES has occurred in patients treated with ZEJULA. Discontinue ZEJULA if PRES is confirmed. (5.4 )• Embryo-Fetal Toxicity: ZEJULA can cause fetal harm. Advise females of reproductive potential of the potential risk to a fetus and to use effective contraception. (5.5 ,8.1 ,8.3 )5.1 Myelodysplastic Syndrome/Acute Myeloid Leukemia
Myelodysplastic syndrome/acute myeloid leukemia (MDS/AML), including cases with a fatal outcome, have been reported in patients who received ZEJULA.
In PRIMA, MDS/AML occurred in 6 out of 484 (1.2%) patients treated with ZEJULA and in 3 out of 244 (1.2%) patients treated with placebo [see Adverse Reactions (6.1)]. The duration of therapy with ZEJULA in patients who developed secondary MDS/cancer therapy-related AML varied from 3.7 months to 2.5Â years.
In NOVA, of patients within the gBRCAmut cohort, MDS/AML occurred in 10 out of 136 (7%) patients treated with ZEJULA and in 2 out of 65 (3%) patients treated with placebo [see Adverse Reactions (6.1)]. The duration of therapy with ZEJULA in patients who developed secondary MDS/cancer-therapy related AML varied from 3.6 months to 5.9 years.
All patients who developed secondary MDS/cancer-therapy–related AML had received previous chemotherapy with platinum agents and/or other DNA-damaging agents, including radiotherapy.
For suspected MDS/AML or prolonged hematological toxicities, refer the patient to a hematologist for further evaluation. Discontinue ZEJULA if MDS/AML is confirmed.
5.2 Bone Marrow Suppression
Hematologic adverse reactions, including thrombocytopenia, anemia, neutropenia, and/or pancytopenia have been reported in patients treated with ZEJULA [see Adverse Reactions (6)].
In PRIMA, the overall incidences of ≥Grade 3 thrombocytopenia, anemia, and neutropenia were reported in 39%, 31%, and 21%, respectively, of patients receiving ZEJULA. Discontinuation due to thrombocytopenia, anemia, and neutropenia occurred in 4%, 2%, and 2%, respectively, of patients. In patients who were administered a starting dose of ZEJULA based on baseline weight or platelet count, ≥Grade 3 thrombocytopenia, anemia, and neutropenia were reported in 22%, 23%, and 15%, respectively, of patients receiving ZEJULA. Discontinuation due to thrombocytopenia, anemia, and neutropenia occurred in 3%, 3%, and 2%, respectively, of patients.
In NOVA, ≥Grade 3 thrombocytopenia, anemia, and neutropenia were reported in 29%, 25%, and 20%, respectively, of patients receiving ZEJULA. Discontinuation due to thrombocytopenia, anemia, and neutropenia occurred in 3%, 1%, and 2%, respectively, of patients.
Do not start ZEJULA until patients have recovered from hematological toxicity caused by previous chemotherapy (≤Grade 1). Monitor complete blood counts weekly for the first month, monthly for the next 11 months of treatment, and periodically after this time. If hematological toxicities do not resolve within 28 days following interruption, discontinue ZEJULA and refer the patient to a hematologist for further investigations, including bone marrow analysis and blood sample for cytogenetics [see Dosage and Administration (2.3)].
5.3 Hypertension and Cardiovascular Effects
Hypertension and hypertensive crisis have been reported in patients treated with ZEJULA [see Adverse Reactions (6)].
In PRIMA, Grade 3 to 4 hypertension occurred in 6% of patients treated with ZEJULA compared with 1% of placebo-treated patients with a median time from first dose to first onset of 43 days (range: 1 to 531 days) and with a median duration of 12 days (range: 1 to 61 days). There were no discontinuations due to hypertension.
In NOVA, Grade 3 to 4 hypertension occurred in 9% of patients treated with ZEJULA compared with 2% of placebo-treated patients with a median time from first dose to first onset of 77 days (range: 4 to 504 days) and with a median duration of 15 days (range: 1 to 86 days). Discontinuation due to hypertension occurred in <1% of patients.
Monitor blood pressure and heart rate at least weekly for the first 2 months, then monthly for the first year and periodically thereafter during treatment with ZEJULA. Closely monitor patients with cardiovascular disorders, especially coronary insufficiency, cardiac arrhythmias, and hypertension. Medically manage hypertension with antihypertensive medications and adjustment of the dose of ZEJULA, if necessary [see Dosage and Administration (2.3), Nonclinical Toxicology (13.2)].
5.4 Posterior Reversible Encephalopathy Syndrome
Posterior reversible encephalopathy syndrome (PRES) occurred in 0.1% of 2,165 patients treated with ZEJULA in clinical trials and has also been described in postmarketing reports [see Adverse Reactions (6.2)]. Signs and symptoms of PRES include seizure, headache, altered mental status, visual disturbance, or cortical blindness, with or without associated hypertension. A diagnosis of PRES requires confirmation by brain imaging, preferably magnetic resonance imaging.
Monitor all patients treated with ZEJULA for signs and symptoms of PRES. If PRES is suspected, promptly discontinue ZEJULA and administer appropriate treatment. The safety of reinitiating ZEJULA in patients previously experiencing PRES is not known.
5.5 Embryo-Fetal Toxicity
Based on its mechanism of action, ZEJULA can cause fetal harm when administered to a pregnant woman [see Clinical Pharmacology (12.1)]. ZEJULA has the potential to cause teratogenicity and/or embryo-fetal death since niraparib is genotoxic and targets actively dividing cells in animals and patients (e.g., bone marrow) [see Warnings and Precautions (5.2), Nonclinical Toxicology (13.1)]. Due to the potential risk to a fetus based on its mechanism of action, animal developmental and reproductive toxicology studies were not conducted with niraparib.
Apprise pregnant women of the potential risk to a fetus. Advise females of reproductive potential to use effective contraception during treatment and for 6 months after the last dose of ZEJULA [see Use in Specific Populations (8.1, 8.3)].
6 Adverse Reactions
The following clinically significant adverse reactions are described elsewhere in the labeling:
• MDS/AML [see Warnings and Precautions (5.1)]• Bone marrow suppression [see Warnings and Precautions (5.2)]• Hypertension and cardiovascular effects [see Warnings and Precautions (5.3)]• Posterior reversible encephalopathy syndrome [see Warnings and Precautions (5.4)]
Most common adverse reactions (incidence ≥10%) in patients who received ZEJULA were nausea, thrombocytopenia, anemia, fatigue, constipation, musculoskeletal pain, abdominal pain, vomiting, neutropenia, decreased appetite, leukopenia, insomnia, headache, dyspnea, rash, diarrhea, hypertension, cough, dizziness, acute kidney injury, urinary tract infection, and hypomagnesemia. (6.1 )
To report SUSPECTED ADVERSE REACTIONS, contact GlaxoSmithKline at 1-888-825-5249 or FDA at 1-800-FDA-1088 orwww.fda.gov/medwatch.
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared with rates in the clinical trials of another drug and may not reflect the rates observed in practice.
In a pooled safety population of patients (n = 1,314) with advanced ovarian, fallopian tube, or primary peritoneal cancer treated with ZEJULA monotherapy including PRIMA (n = 484), NOVA (n = 367), and another clinical trial (n = 463), the most common adverse reactions >10% were nausea (65%), thrombocytopenia (60%), anemia (56%), fatigue (55%), constipation (39%), musculoskeletal pain (36%), abdominal pain (35%), vomiting (33%), neutropenia (31%), decreased appetite (24%), leukopenia (24%), insomnia (23%), headache (23%), dyspnea (22%), rash (21%), diarrhea (18%), hypertension (17%), cough (16%), dizziness (14%), acute kidney injury (13%), urinary tract infection (12%), and hypomagnesemia (11%).
First-Line Maintenance Treatment of Advanced Ovarian Cancer
The safety of ZEJULA for the treatment of patients with advanced ovarian cancer following first-line treatment with platinum-based chemotherapy was studied in the PRIMA trial, a placebo-controlled, double-blind study in which 728 patients received niraparib or placebo. Among patients who received ZEJULA, the median duration of treatment was 11.1Â months (range: 0.03 to 29 months).
All Patients Receiving ZEJULA in PRIMA: Serious adverse reactions occurred in 32% of patients receiving ZEJULA. Serious adverse reactions in >2% of patients were thrombocytopenia (16%), anemia (6%), and small intestinal obstruction (2.9%). Fatal adverse reactions occurred in 0.4% of patients, including intestinal perforation and pleural effusion (1 patient each). MDS/AML occurred in 1.2% of patients receiving ZEJULA.
Permanent discontinuation due to adverse reactions occurred in 12% of patients who received ZEJULA. Adverse reactions resulting in permanent discontinuation in >1% of patients who received ZEJULA included thrombocytopenia (3.7%), anemia (1.9%), and nausea and neutropenia (1.2% each).
Adverse reactions led to dose reduction or interruption in 80% of patients, most frequently from thrombocytopenia (56%), anemia (33%), and neutropenia (20%).
Table 4 and Table 5 summarize the common adverse reactions and abnormal laboratory findings, respectively, observed in all patients treated with ZEJULA in the PRIMA study.
Table 4. Adverse Reactions Reported in ≥10% of All Patients Receiving ZEJULA in PRIMAa AST/ALT = Aspartate transaminase/alanine aminotransferase. a All adverse reactions in the table consist of grouped preferred terms except for nausea, vomiting, decreased appetite, headache, and insomnia, which are single preferred terms. b Common Terminology Criteria for Adverse Events version 4.02. c Includes neutropenia, neutropenic infection, neutropenic sepsis, and febrile neutropenia. d Includes leukopenia, lymphocyte count decreased, lymphopenia, and white blood cell count decreased. e Includes blood creatinine increased, blood urea increased, acute kidney injury, renal failure, and blood creatine increased.
Adverse Reaction
Grades 1-4b
Grades 3-4b
ZEJULA
(n = 484)
%
Placebo
(n = 244)
%
ZEJULA
(n = 484)
%
Placebo
(n = 244)
%
Blood and lymphatic system disorders
     Thrombocytopenia
66
5
39
0.4
     Anemia
64
18
31
2
     Neutropeniac
42
8
21
1
     Leukopeniad
28
9
5
0.4
Gastrointestinal disorders
     Nausea
57
28
1
1
     Constipation
40
20
1
0.4
     Vomiting
22
12
1
1
General disorders and administration site conditions
     Fatigue
51
41
3
1
Musculoskeletal and connective tissue disorders
     Musculoskeletal pain
39
38
1
0
Nervous system disorders
     Headache
26
15
0.4
0
     Dizziness
19
13
0
0.4
Psychiatric disorders
     Insomnia
25
15
1
0.4
Respiratory, thoracic, and mediastinal disorders
     Dyspnea
22
13
0.4
1
     Cough
18
15
0
0.4
Metabolism and nutrition disorders
     Decreased appetite
19
8
1
0
Vascular disorders
     Hypertension
18
7
6
1
Investigations
     AST/ALT elevation
14
7
3
0.8
Renal and urinary disorders
     Acute kidney injurye
12
5
0.2
0
Table 5. Abnormal Laboratory Findings in ≥25% of All Patients Receiving ZEJULA in PRIMA
Abnormal Laboratory Finding
Grades 1-4
Grades 3-4
ZEJULA
(n = 484)
%
Placebo
(n = 244)
%
ZEJULA
(n = 484)
%
Placebo
(n = 244)
%
Decreased hemoglobin
87
66
29
1
Decreased platelets
74
13
37
0
Decreased leukocytes
71
36
9
0
Increased glucose
66
57
3
3
Decreased neutrophils
66
25
23
1
Decreased lymphocytes
51
29
7
3
Increased alkaline phosphatase
46
21
1
0
Increased creatinine
40
23
0
0
Decreased magnesium
36
34
1
0
Increased aspartate aminotransferase
35
17
1
0.4
Increased alanine aminotransferase
29
17
2
1
Patients Receiving ZEJULA with Dose Based on Baseline Weight or Platelet Count in PRIMA: Among patients who received ZEJULA with the dose based on weight and platelet count, the median duration of treatment was 11 months (range: 1 day to 16 months).
Serious adverse reactions occurred in 27% of patients receiving ZEJULA. Serious adverse reactions in >2% of patients were anemia (8%), and thrombocytopenia (7%). No fatal adverse reactions occurred.
Permanent discontinuation due to adverse reactions occurred in 14% of patients who received ZEJULA. Adverse reactions resulting in permanent discontinuation in >2% of patients who received ZEJULA included thrombocytopenia and anemia (3% each) and nausea (2.4%).
Adverse reactions led to dose reduction or interruption in 72% of patients, most frequently from thrombocytopenia (40%), anemia (23%), and neutropenia (15%).
Table 6 and Table 7 summarize adverse reactions and abnormal laboratory findings in the group of patients who received ZEJULA.
Table 6. Adverse Reactions Reported in ≥10% of Patients Receiving ZEJULA Based on Baseline Weight or Platelet Count in PRIMAa a All adverse reactions in the table consist of grouped preferred terms except for nausea, vomiting, decreased appetite, headache, and insomnia, which are single preferred terms. b Common Terminology Criteria for Adverse Events version 4.02. c Includes neutropenia, neutropenic infection, neutropenic sepsis, and febrile neutropenia. d Includes leukopenia, lymphocyte count decreased, lymphopenia, and white blood cell count decreased. e Includes blood creatinine increased, blood urea increased, acute kidney injury, renal failure, and blood creatine increased.
Adverse Reaction
Grades 1-4b
Grades 3-4b
ZEJULA
(n = 169)
%
Placebo
(n = 86)
%
ZEJULA
(n = 169)
%
Placebo
(n = 86)
%
Blood and lymphatic system disorders
     Thrombocytopenia
54
5
21
1
     Anemia
50
28
23
1
     Neutropeniac
36
8
15
1
     Leukopeniad
28
11
5
0
Gastrointestinal disorders
Nausea
53
21
1
0
     Constipation
31
15
1
1
     Vomiting
17
9
0
1
General disorders and administration site conditions
     Fatigue
48
36
3
0
Nervous system disorders
     Headache
22
17
1
0
     Dizziness
14
13
0
0
Psychiatric disorders
     Insomnia
21
14
0
0
Metabolism and nutrition disorders
     Decreased appetite
19
5
1
0
Respiratory, thoracic, and mediastinal disorders
     Dyspnea
18
10
0
1
Vascular disorders
     Hypertension
17
9
5
2
Renal and urinary disorders
     Acute kidney injurye
12
5
1
0
Table 7. Abnormal Laboratory Findings in ≥25% of All Patients Receiving ZEJULA Based on Baseline Weight or Platelet Count in PRIMA
Abnormal Laboratory Finding
Grades 1-4
Grades 3-4
ZEJULA
(n = 169)
%
Placebo
(n = 86)
%
ZEJULA
(n = 169)
%
Placebo
(n = 86)
%
Decreased hemoglobin
81
70
21
0
Decreased leukocytes
70
36
6
0
Decreased platelets
63
15
18
0
Increased glucose
63
56
2
1
Decreased neutrophils
60
27
15
0
Decreased lymphocytes
52
30
5
4
Decreased magnesium
44
30
0
0
Increased alkaline phosphatase
43
17
1
0
Increased creatinine
41
22
0
0
Increased aspartate aminotransferase
31
19
1
0
Increased alanine aminotransferase
28
15
2
2
Maintenance Treatment of Recurrent Germline BRCA-mutated Ovarian Cancer
The safety of monotherapy with ZEJULA 300 mg once daily has been studied in 136 patients with platinum-sensitive recurrent gBRCAmut ovarian, fallopian tube, and primary peritoneal cancer in the NOVA trial. The percentages of patients who experienced adverse reactions in NOVA that led to dose reduction and dose interruption were 79% and 68%, respectively, most frequently from thrombocytopenia (41% and 35%, respectively) and anemia (23% and 20%, respectively). The permanent discontinuation rate due to adverse reactions in NOVA was 13%. The median exposure to ZEJULA in these patients was 367 days.
Table 8 and Table 9 summarize the common adverse reactions and abnormal laboratory findings, respectively, observed in patients treated with ZEJULA in the gBRCAmut cohort in NOVA.
Table 8. Adverse Reactions Reported in ≥10% of Patients Receiving ZEJULA in NOVA gBRCAmut Cohort a Common Terminology Criteria for Adverse Events version 4.02. b Includes platelet count decreased. c Includes hemoglobin decreased. d Includes neutrophil count decreased. e Includes asthenia, malaise, lethargy.
Adverse Reaction
Grades 1-4a
Grades 3-4a
ZEJULA
(n = 136)
%
Placebo
(n = 65)
%
ZEJULA
(n = 136)
%
Placebo
(n = 65)
%
Gastrointestinal disorders
     Nausea
77
34
5
3
     Vomiting
40
15
4
0
     Constipation
38
18
0.7
2
     Dyspepsia
17
12
0
0
     Dry mouth
13
3
0.7
0
Blood and lymphatic system disorders
     Thrombocytopeniab
71
5
38
2
     Anemiac
52
8
33
0
     Neutropeniad
31
9
21
3
General disorders and administration site conditions
     Fatiguee
61
35
8
2
Nervous system disorders
     Headache
35
8
0.7
0
     Dizziness
18
9
0
0
     Dysgeusia
13
2
0
0
Metabolism and nutrition disorders
     Decreased appetite
22
14
0
0
Vascular disorders
     Hypertension
21
8
8
5
Psychiatric disorders
     Insomnia
18
6
0.7
0
     Anxiety
10
11
0.7
0
Respiratory, thoracic, and mediastinal disorders
     Dyspnea
17
5
2
0
     Cough
16
2
0
0
     Nasopharyngitis
13
5
0
0
Musculoskeletal and connective tissue disorders
     Back pain
16
11
0.7
0
Infections and infestations
     Urinary tract infection
11
9
0
2
Skin and subcutaneous tissue disorders
     Rash
10
2
0
0
The following adverse reactions have been identified in ≥1 to <10% of the 136 patients receiving ZEJULA in the gBRCAmut cohort of the NOVA trial and not included in the table: palpitations (9%), mucositis/stomatitis (9%), MDS/AML (7%), tachycardia (7%), and bronchitis (4%).
Table 9. Abnormal Laboratory Findings in ≥25% of Patients Receiving ZEJULA in NOVA gBRCAmut Cohort
Abnormal Laboratory Finding
Grades 1-4
Grades 3-4
ZEJULA
(n = 136)
%
Placebo
(n = 65)
%
ZEJULA
(n = 136)
%
Placebo
(n = 65)
%
Decrease in hemoglobin
85
62
32
0
Decrease in platelet count
81
25
38
2
Decrease in white blood cell count
71
37
9
2
Decrease in absolute neutrophil count
56
34
23
3
Increase in aspartate aminotransferase
35
25
0.7
0
Increase in alanine aminotransferase
25
15
0.7
2
6.2 Postmarketing Experience
The following adverse reactions have been identified during postapproval use of ZEJULA. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Blood and Lymphatic System Disorders
Pancytopenia.
Immune System Disorders
Hypersensitivity (including anaphylaxis).
Nervous System Disorders
Posterior reversible encephalopathy syndrome (PRES).
Psychiatric Disorders
Confusional state/disorientation, hallucination, cognitive impairment (e.g., memory impairment, concentration impairment).
Respiratory, Thoracic, and Mediastinal Disorders
Non-infectious pneumonitis.
Skin and Subcutaneous Tissue Disorders
Photosensitivity.
Vascular Disorders
Hypertensive crisis.
8 Use In Specific Populations
Lactation: Advise not to breastfeed. (8.2 )
8.1 Pregnancy
Risk Summary
Based on its mechanism of action, ZEJULA can cause fetal harm when administered to pregnant women [see Clinical Pharmacology (12.1)]. There are no data regarding the use of ZEJULA in pregnant women to inform the drug-associated risk. ZEJULA has the potential to cause teratogenicity and/or embryo-fetal death since niraparib is genotoxic and targets actively dividing cells in animals and patients (e.g., bone marrow) [see Warnings and Precautions (5.2), Nonclinical Toxicology (13.1)]. Due to the potential risk to a fetus based on its mechanism of action, animal developmental and reproductive toxicology studies were not conducted with niraparib. Apprise pregnant women of the potential risk to a fetus.
The background risk of major birth defects and miscarriage for the indicated population is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2% to 4% and 15% to 20%, respectively.
8.2 Lactation
Risk Summary
No data are available regarding the presence of niraparib or its metabolites in human milk, or on its effects on the breastfed child or milk production. Because of the potential for serious adverse reactions in a breastfed child, advise a lactating woman not to breastfeed during treatment with ZEJULA and for 1 month after receiving the last dose.
8.3 Females and Males of Reproductive Potential
ZEJULA can cause fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)].
Pregnancy Testing
Verify the pregnancy status of females of reproductive potential prior to initiating treatment with ZEJULA.
Contraception
Females: Advise females of reproductive potential to use effective contraception during treatment with ZEJULA and for 6Â months following the last dose.
Infertility
Males: Based on animal studies, ZEJULA may impair fertility in males of reproductive potential [see Nonclinical Toxicology (13.1)].
8.4 Pediatric Use
The safety and effectiveness of ZEJULA have not been established in pediatric patients.
8.5 Geriatric Use
In PRIMA, 39% of patients were aged 65 years or older and 10% were aged 75 years or older. In NOVA, 35% of patients were aged 65 years or older and 8% were aged 75 years or older. No overall differences in safety and effectiveness of ZEJULA were observed between these patients and younger patients but greater sensitivity of some older individuals cannot be ruled out.
8.6 Renal Impairment
No dose adjustment is necessary for patients with mild (CLcr: 60 to 89 mL/min) to moderate (CLcr: 30 to 59 mL/min) renal impairment. The degree of renal impairment was determined by creatinine clearance as estimated by the Cockcroft-Gault equation. The safety of ZEJULA in patients with severe renal impairment or end-stage renal disease undergoing hemodialysis is unknown.
8.7 Hepatic Impairment
For patients with moderate hepatic impairment, reduce the starting dosage of niraparib to 200 mg once daily [see Dosage and Administration (2.4)]. Niraparib exposure increased in patients with moderate hepatic impairment [total bilirubin ≥1.5 x upper level of normal (ULN) to 3.0 x ULN and any aspartate transaminase (AST) level]. Monitor patients for hematologic toxicity and reduce the dose further, if needed [see Dosage and Administration (2.3)].
For patients with mild hepatic impairment (total bilirubin <1.5 x ULN and any AST level or bilirubin ≤ULN and AST >ULN), no dose adjustment is needed.
The recommended dose of ZEJULA has not been established for patients with severe hepatic impairment (total bilirubin >3.0 x ULN and any AST level) [see Clinical Pharmacology (12.3)].
11 Description
Niraparib is an orally available poly (ADP-ribose) polymerase (PARP) inhibitor.
The chemical name for niraparib tosylate monohydrate is 2-{4-[(3S)-piperidin-3-yl]phenyl}-2H-indazole 7-carboxamide 4-methylbenzenesulfonate hydrate (1:1:1). The molecular formula is C26H30N4O5S and it has a molecular weight of 510.61Â amu. The molecular structure is shown below:
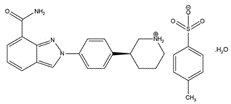
Niraparib tosylate monohydrate is a white to off-white, non-hygroscopic crystalline solid. Niraparib solubility is pH independent below the pKa of 9.95, with an aqueous free base solubility of 0.7 mg/mL to 1.1 mg/mL across the physiological pH range.
Each ZEJULA tablet contains 159.3 mg, 318.7 mg, or 478.0 mg of niraparib tosylate monohydrate equivalent to 100 mg, 200 mg, or 300 mg, respectively, of niraparib free base as the active ingredient. The inactive ingredients in the core tablet are crospovidone, lactose monohydrate, magnesium stearate, microcrystalline cellulose, povidone, and silicon dioxide. The film-coating consists of Opadry II Gray (100 mg), Opadry II Blue (200Â mg), or Opadry II Green (300 mg).
12 Clinical Pharmacology
12.1 Mechanism of Action
Niraparib is an inhibitor of PARP enzymes, including PARP-1 and PARP-2, that play a role in DNA repair. In vitro studies have shown that niraparib-induced cytotoxicity may involve inhibition of PARP enzymatic activity and increased formation of PARP-DNA complexes resulting in DNA damage, apoptosis, and cell death. Increased niraparib‑induced cytotoxicity was observed in tumor cell lines with or without deficiencies in BRCA1/2. Niraparib decreased tumor growth in mouse xenograft models of human cancer cell lines with deficiencies in BRCA1/2 and in human patient-derived xenograft tumor models with homologous recombination deficiency (HRD) that had either mutated or wild-type BRCA1/2.
12.2 Pharmacodynamics
The pharmacodynamic response of niraparib has not been characterized.
Hypertension and Cardiovascular Effects
Niraparib has the potential to cause effects on pulse rate and blood pressure in patients receiving the recommended dose, which may be related to pharmacological inhibition of the dopamine transporter (DAT), norepinephrine transporter (NET), and serotonin transporter (SERT) [see Nonclinical Toxicology (13.2)].
In the PRIMA study, mean pulse rate and blood pressure increased over baseline in the niraparib arm relative to the placebo arm at most on-study assessments. Mean greatest increases from baseline in pulse rate on treatment were 22.4 and 14.0 beats/min in the niraparib and placebo arms, respectively. Mean greatest increases from baseline in systolic blood pressure on treatment were 24.4 and 19.6Â mmHg in the niraparib and placebo arms, respectively. Mean greatest increases from baseline in diastolic blood pressure on treatment were 15.9 and 13.9 mmHg in the niraparib and placebo arms, respectively.
In the NOVA study, mean pulse rate and blood pressure increased over baseline in the niraparib arm relative to the placebo arm at all on-study assessments. Mean greatest increases from baseline in pulse rate on treatment were 24.1 and 15.8 beats/min in the niraparib and placebo arms, respectively. Mean greatest increases from baseline in systolic blood pressure on treatment were 24.5 and 18.3Â mmHg in the niraparib and placebo arms, respectively. Mean greatest increases from baseline in diastolic blood pressure on treatment were 16.5 and 11.6 mmHg in the niraparib and placebo arms, respectively.
Cardiac Electrophysiology
The potential for QTc prolongation with niraparib was evaluated in a randomized, placebo‑controlled trial in patients with cancer (367 patients on niraparib and 179 patients on placebo). No large changes in the mean QTc interval (>20 ms) were detected in the trial following the treatment of niraparib 300 mg once daily.
12.3 Pharmacokinetics
Following a single-dose administration of 300 mg niraparib, the mean (±SD) peak plasma concentration (Cmax) was 804 (±403) ng/mL. The exposure (Cmax and AUC) of niraparib increased in a dose-proportional manner with daily doses ranging from 30 mg (0.1 times the approved recommended dose) to 400 mg (1.3 times the approved recommended dose). The accumulation ratio of niraparib exposure following 21 days of repeated daily doses was approximately 2-fold for doses ranging from 30 to 400 mg.
Absorption
The absolute bioavailability of niraparib is approximately 73%. Following oral administration of niraparib, peak plasma concentration, Cmax, is reached within 3 hours.
Food Effect: Following a high-fat meal (800 to 1,000 calories with approximately 50% of total caloric content of the meal from fat) in patients with solid tumors, the Cmax of niraparib tablets increased by 11% and AUCinf increased by 28%, as compared with fasted conditions.
Distribution
Niraparib is 83.0% bound to human plasma proteins. The average (±SD) apparent volume of distribution (Vd/F) was 1,220 (±1,114) L. In a population pharmacokinetic analysis, the Vd/F of niraparib was 1,074 L in patients with cancer.
Elimination
Following multiple daily doses of 300 mg of niraparib, the mean half-life (t1/2) is 36 hours. In a population pharmacokinetic analysis, the apparent total clearance (CL/F) of niraparib was 16.2Â L/h in patients with cancer.
Metabolism: Niraparib is metabolized by carboxylesterases (CEs) to form a major inactive metabolite, which subsequently undergoes glucuronidation.
Excretion: Following administration of a single oral 300-mg dose of radio-labeled niraparib, the average percent recovery of the administered dose over 21 days was 47.5% (range: 33.4% to 60.2%) in urine and 38.8% (range: 28.3% to 47.0%) in feces. In pooled samples collected over 6 days, unchanged niraparib accounted for 11% and 19% of the administered dose recovered in urine and feces, respectively.
Specific Populations
Age (18 to 65 years), race/ethnicity, and mild to moderate renal impairment (CLcr ≥30 to 90 mL/min) had no clinically significant effect on the pharmacokinetics of niraparib.
The effect of severe renal impairment (CLcr <30 mL/min) or end-stage renal disease undergoing hemodialysis on the pharmacokinetics of niraparib is unknown.
Patients with Hepatic Impairment: Mild hepatic impairment (total bilirubin <1.5 x ULN and any AST level or bilirubin ≤ ULN and AST > ULN) had no clinically significant effect on the pharmacokinetics of niraparib.
In a trial of patients with moderate hepatic impairment (total bilirubin ≥1.5 x ULN to 3.0 x ULN and any AST level) (n = 8), niraparib AUCinf was 1.56 (90% CI: 1.06 to 2.30) times higher compared with patients with normal hepatic function (n = 9) following administration of a single 300-mg dose. Niraparib dosage reduction is recommended for patients with moderate hepatic impairment [see Dosage and Administration (2.4)]. Moderate hepatic impairment did not have an effect on niraparib Cmax or on niraparib protein binding.
The effect of severe hepatic impairment (total bilirubin >3.0 x ULN and any AST level) on the pharmacokinetics of niraparib is unknown.
Drug Interaction Studies
No clinical drug interaction studies have been performed with ZEJULA.
In Vitro Studies: Inhibition of Cytochrome P450 (CYP) Enzymes: Neither niraparib nor the major primary metabolite M1 is an inhibitor of CYP1A2, CYP2B6, CYP2C8, CYP2C9, CYP2C19, CYP2D6, and CYP3A4.
 Induction of CYP Enzymes: Neither niraparib nor M1 is a CYP3A4 inducer. Niraparib weakly induces CYP1A2 in vitro. Substrate of CYP Enzymes: Niraparib is a substrate of CEs and the resulting M1 is further metabolized through the formation of glucuronides in vivo. Inhibition of Uridine 5'-Diphospho-Glucuronosyltransferases (UGTs): Niraparib did not exhibit inhibitory effect against the UGT isoforms (UGT1A1, UGT1A4, UGT1A9, and UGT2B7) up to 200 microM in vitro. Therefore, the potential for a clinically relevant inhibition of UGTs by niraparib is minimal. Inhibition of Transporter Systems: Niraparib is a weak inhibitor of breast cancer resistance protein (BCRP) but does not inhibit P-glycoprotein (P-gp), bile salt export pump (BSEP), or multidrug resistance-associated protein 2 (MRP2).
Niraparib is an inhibitor of multidrug and toxin extrusion (MATE) 1 and 2 with IC50 of 0.18 microM and ≤0.14 microM, respectively. Increased plasma concentrations of coadministered drugs that are substrates of these transporters (e.g., metformin) cannot be excluded.
The M1 metabolite is not an inhibitor of P-gp, BCRP, BSEP, MRP2, or MATE1 or 2. Neither niraparib nor M1 is an inhibitor of organic anion transporting polypeptide (OATP)1B1, OATP1B3, organic cation transporter (OCT1)1, organic anion transporter (OAT)1, OAT3, or OCT2.
 Substrate of Transporter Systems: Niraparib is a substrate of P-gp and BCRP. Niraparib is not a substrate of BSEP, MRP2, or MATE1 or 2. The M1 metabolite is not a substrate of P-gp, BCRP, BSEP, or MRP2. However, M1 is a substrate of MATE1 and 2. Neither niraparib nor M1 is a substrate of OATP1B1, OATP1B3, OCT1, OAT1, OAT3, or OCT2.
13 Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies have not been conducted with niraparib.
Niraparib was clastogenic in an in vitro mammalian chromosomal aberration assay and in an in vivo rat bone marrow micronucleus assay. This clastogenicity is consistent with genomic instability resulting from the primary pharmacology of niraparib and indicates potential for genotoxicity in humans. Niraparib was not mutagenic in a bacterial reverse mutation assay (Ames) test.
Fertility studies in animals have not been conducted with niraparib. In repeat-dose oral toxicity studies, niraparib was administered daily for up to 3 months’ duration in rats and dogs. Reduced sperm, spermatids, and germ cells in epididymides and testes were observed at doses ≥10 mg/kg and ≥1.5 mg/kg in rats and dogs, respectively. These dose levels resulted in systemic exposures approximately 0.3 and 0.012 times, respectively, the human exposure (AUC0-24h) at the recommended dose of 300 mg daily. There was a trend toward reversibility of these findings 4 weeks after dosing was stopped.
13.2 Animal Toxicology and/or Pharmacology
In vitro, niraparib bound to DAT, NET, and SERT and inhibited uptake of norepinephrine and dopamine in cells with IC50 values that were lower than the Cmin at steady-state in patients receiving the recommended dose. Niraparib has the potential to cause effects in patients related to inhibition of these transporters (e.g., cardiovascular, central nervous system).
Intravenous administration of niraparib to vagotomized dogs over 30 minutes at 1, 3, and 10 mg/kg resulted in an increased range of arterial pressures of 13% to 20%, 18% to 27%, and 19% to 25%, respectively, and increased range of heart rates of 2% to 11%, 4% to 17%, and 12% to 21%, respectively, above pre-dose levels. The unbound plasma concentrations of niraparib in dogs at these dose levels were approximately 0.5, 1.5, and 5.8 times the unbound Cmax at steady‑state in patients receiving the recommended dose.
In addition, niraparib crossed the blood-brain barrier in rats and monkeys following oral administration. The cerebrospinal fluid:plasma Cmax ratios of niraparib administered at 10Â mg/kg orally to 2 rhesus monkeys were 0.10 and 0.52.
14 Clinical Studies
14.1 First-Line Maintenance Treatment of Advanced Ovarian Cancer
PRIMA (NCT02655016) was a double-blind, placebo-controlled trial in which patients (N = 733) in complete or partial response to first-line platinum-based chemotherapy were randomized 2:1 to ZEJULA or matched placebo. Initially, the patients received a starting dosage of 300 mg once daily regardless of body weight or platelet count. The study was amended to include a starting dose of 200 mg for patients weighing <77 kg (<170 lbs) OR with a platelet count of <150,000/mcL or 300 mg for patients weighing ≥77 kg (≥170 lbs) AND who had a platelet count ≥150,000/mcL.
Patients were randomized post-completion of first-line platinum-based chemotherapy plus surgery. Randomization was stratified by best response during the front-line platinum regimen (complete response vs. partial response), neoadjuvant chemotherapy (NACT) (yes vs. no), and HRD status (positive vs. negative or not determined). HRD status was determined using the FDA-approved Myriad myChoice CDx assay. HRD positive status included either tumor BRCA mutant (tBRCAm) or a genomic instability score (GIS) ≥42.
The major efficacy outcome measure, progression-free survival (PFS), was determined by blinded independent central review (BICR) per Response Evaluation Criteria in Solid Tumors (RECIST) version 1.1. In some cases, criteria other than RECIST, such as clinical signs and symptoms and increasing CA-125, were also applied. Overall survival was an additional efficacy outcome measure. PFS testing was performed hierarchically: first in the homologous recombination (HR)-deficient (HRD positive) population, then in the overall population. The median age of 62 ranged from 32 to 85 years among patients randomized with ZEJULA and 33 to 88 years among patients randomized with placebo. Eighty-nine percent of all patients were White. Sixty-nine percent of patients randomized with ZEJULA and 71% of patients randomized with placebo had an Eastern Cooperative Oncology Group Performance Status (ECOG PS) of 0 at study baseline. Approximately 45% of patients were enrolled in the U.S. or Canada. In the overall population, 65% of patients had stage III disease and 35% had stage IV disease. Sixty‑seven percent of the patients received NACT. Sixty-nine percent of the patients had a complete response to the first-line platinum-based chemotherapy. Approximately 35% (n = 258) of patients received a starting dose of 200 or 300 mg depending on baseline body weight and platelet count. Among those patients, 186 patients received a starting dose of 200 mg.
PRIMA demonstrated a statistically significant improvement in PFS for patients randomized to ZEJULA as compared with placebo in the HR-deficient and overall population (Table 10, Figure 1, and Figure 2).
Table 10. Efficacy Results – PRIMA (determined by BICRa) BICR = Blinded Independent Central Review, HR = Homologous Recombination, NE = not estimable. a Efficacy analysis was based on blinded independent central review. b Based on a stratified Cox proportional hazards model. c Based on a stratified log-rank test.
HR-Deficient Population
Overall Population
ZEJULA
(n = 247)
Placebo
(n = 126)
ZEJULA
(n = 487)
Placebo
(n = 246)
Progression-free survival events, n (%)
81
(33)
73
(58)
232
(48)
155
(63)
Progression-free survival median in months (95%Â CI)
21.9
(19.3, NE)
10.4
(8.1, 12.1)
13.8
(11.5, 14.9)
8.2
(7.3, 8.5)
Hazard ratiob (95% CI)
0.43
(0.31, 0.59)
0.62
(0.50, 0.76)
P valuec
<0.0001
<0.0001
In exploratory subgroup analyses of patients who were administered a starting dose of ZEJULA or matched placebo based on baseline weight or platelet count, the hazard ratio for PFS was 0.39 (95% CI: 0.22, 0.72) in the HR-deficient subgroup (n = 130) and 0.68 (95% CI: 0.48, 0.97) in the overall population (n = 258).
Figure 1. Progression-Free Survival – PRIMA Patients with HR-Deficient Tumors (Intent-to-Treat Population, N = 373)
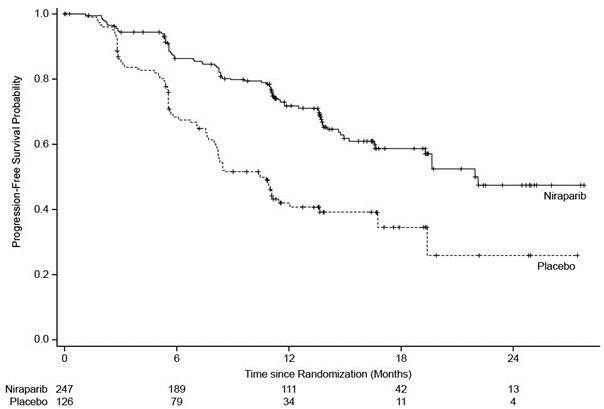
Figure 2. Progression-Free Survival – PRIMA Overall Population (Intent-to-Treat Population, N = 733)
At the time of the PFS analysis, overall survival data were immature, with 11% deaths in the overall population.
14.2 Maintenance Treatment of Recurrent Germline -mutated Ovarian Cancer
NOVA (NCT01847274) was a double-blind, placebo-controlled trial in which patients (NÂ =Â 553) with platinum-sensitive recurrent epithelial ovarian, fallopian tube, or primary peritoneal cancer were randomized 2:1 to ZEJULA 300 mg orally daily or matched placebo within 8 weeks of the last therapy. Treatment was continued until disease progression or unacceptable toxicity. All patients had received at least 2 prior platinum-containing regimens and were in response (complete or partial) to their most recent platinum-based regimen.
Randomization was stratified by time to progression after the penultimate platinum therapy (6 to <12 months and ≥12 months), use of bevacizumab in conjunction with the penultimate or last platinum regimen (yes/no), and best response during the most recent platinum regimen (complete response and partial response). Eligible patients were assigned to 1 of 2 cohorts based on the results of germline BRCA testing with Myriad BRACAnalysis CDx. Patients with deleterious or suspected deleterious germline BRCA mutations (gBRCAmut) were assigned to the germline BRCA-mutated (gBRCAmut) cohort (n = 203), and those without germline BRCA mutations were assigned to the non-gBRCAmut cohort (n = 350). The efficacy results are based on the gBRCAmut cohort only.
The major efficacy outcome measure, PFS, was determined primarily by central independent assessment per RECIST version 1.1. In some cases, criteria other than RECIST, such as clinical signs and symptoms and increasing CA-125, were also applied. Overall survival (OS) was an additional outcome measure.
For the gBRCAmut cohort, the median age of patients was 57 years among patients treated with ZEJULA and 58 years among patients treated with placebo. Eighty-eight percent of all patients were White. Sixty-six percent of patients receiving ZEJULA and 74% of patients receiving placebo had an ECOG PS of 0 at study baseline. Approximately 40% of patients were enrolled in the U.S. or Canada, and 51% of all patients were in complete response to most recent platinum‑based regimen, with 39% on both arms with an interval of 6 to 12 months since the penultimate platinum regimen. Twenty-four percent of those treated with ZEJULA and 26% treated with placebo had received prior bevacizumab therapy. Approximately 50% of patients had 3 or more lines of treatment.
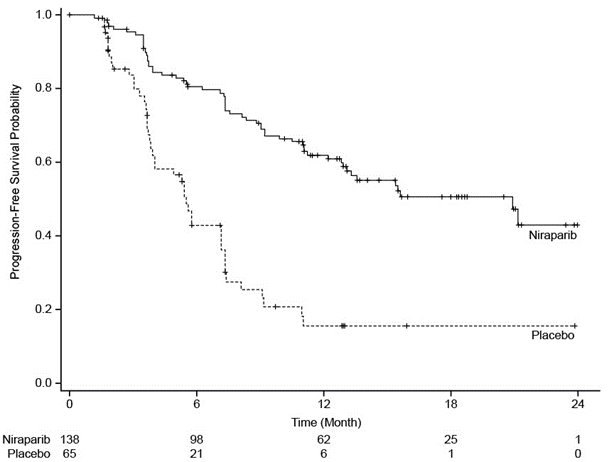
The trial demonstrated a statistically significant improvement in PFS for patients randomized to ZEJULA as compared with placebo in the gBRCAmut cohort (Table 11 and Figure 3).
Table 11. Efficacy Results – NOVA gBRCAmut Cohort (IRC Assessmenta) IRC = Independent Review Committee, gBRCAmut = germline BRCA-mutated, NR = not reached. a Efficacy analysis was based on blinded central independent radiologic and clinical oncology review committee. b Based on a stratified Cox proportional hazards model. c Based on a stratified log-rank test.
ZEJULA
(n = 138)
Placebo
(n = 65)
Progression-free survival median in months (95% CI)
21.0
(12.9, NR)
5.5
(3.8, 7.2)
Hazard ratiob (95%Â CI)
0.26
(0.17, 0.41)
P valuec
<0.0001
Figure 3. Progression-Free Survival – NOVA gBRCAmut Cohort Based on IRC Assessment (N = 203)
gBRCAmut = germline BRCA-mutated, IRC = Independent Review Committee.
A final OS analysis was conducted after 154 events were observed. Exploratory OS results showed a HR of 0.85 (95% CI: 0.61, 1.20) in the gBRCAmut cohort with a median OS of 40.9 months (95% CI: 34.9, 52.9) for patients treated with ZEJULA and 38.1 months (95% CI: 27.6, 47.3) for patients on placebo.
16 How Supplied/storage And Handling
ZEJULA is available as oval-shaped, film-coated tablets containing 100 mg, 200 mg, or 300 mg of niraparib free base.
ZEJULA 100-mg tablets are gray, debossed with “100” on one side and “Zejula” on the other side. Bottle of 30 tablets (NDC 0173-0909-13).
ZEJULA 200-mg tablets are blue, debossed with “200” on one side and “Zejula” on the other side. Bottle of 30 tablets (NDC 0173-0912-13).
ZEJULA 300-mg tablets are green, debossed with “300” on one side and “Zejula” on the other side. Bottle of 30 tablets (NDC 0173-0915-13).
Store and dispense in the original bottle. Store at 20°C to 25°C (68°F to 77°F); excursions are permitted between 15°C to 30°C (59°F to 86°F) [see USP Controlled Room Temperature].
17 Patient Counseling Information
Advise the patient to read the FDA-approved patient labeling (Patient Information).
Myelodysplastic Syndrome/Acute Myeloid Leukemia
Advise patients to contact their healthcare provider if they experience weakness, feeling tired, fever, weight loss, frequent infections, bruising, bleeding easily, breathlessness, blood in urine or stool, and/or laboratory findings of low blood cell counts or a need for blood transfusions. This may be a sign of hematological toxicity or MDS or AML, which has been reported in patients treated with ZEJULA [see Warnings and Precautions (5.1)].
Bone Marrow Suppression
Advise patients that periodic monitoring of their blood counts is required. Advise patients to contact their healthcare provider for new onset of bleeding, fever, or symptoms of infection [see Warnings and Precautions (5.2)].
Hypertension and Cardiovascular Effects
Advise patients to undergo blood pressure and heart rate monitoring at least weekly for the first 2 months, then monthly for the first year of treatment and periodically thereafter. Advise patients to contact their healthcare provider if blood pressure is elevated [see Warnings and Precautions (5.3)].
Posterior Reversible Encephalopathy Syndrome
Inform patients that they are at risk of developing posterior reversible encephalopathy syndrome (PRES) that can present with signs and symptoms including seizure, headaches, altered mental status, or vision changes. Advise patients to contact their healthcare provider if they develop any of these signs or symptoms [see Warnings and Precautions (5.4)].
Dosing Instructions
Inform patients on how to take ZEJULA [see Dosage and Administration (2.2)]. ZEJULA should be taken once daily. Instruct patients that if they miss a dose of ZEJULA not to take an extra dose to make up for the one that they missed. They should take their next dose at the regularly scheduled time. Each tablet should be swallowed whole. ZEJULA may be taken with or without food. Bedtime administration may be a potential method for managing nausea.
Embryo-Fetal Toxicity
Advise females to inform their healthcare provider if they are pregnant or become pregnant. Inform female patients of the risk to a fetus and potential loss of the pregnancy [see Warnings and Precautions (5.5), Use in Specific Populations (8.1)].
Contraception
Advise females of reproductive potential to use effective contraception during treatment with ZEJULA and for 6 months after receiving the last dose [see Use in Specific Populations (8.3)].
Lactation
Advise patients not to breastfeed while taking ZEJULA and for 1 month after the last dose [see Use in Specific Populations (8.2)].
Trademarks are owned by or licensed to the GSK group of companies. Opadry is a trademark owned by or licensed to its respective owner and is not owned by or licensed to the GSK group of companies. The maker of Opadry is not affiliated with and does not endorse the GSK group of companies or its products.
Manufactured for
GlaxoSmithKline
Durham, NC 27701
©2024 GSK group of companies or its licensor.
ZJT:3PI
Spl Patient Package Insert Section
Patient Information
ZEJULA (zuh-JOO-luh)
(niraparib)
tablets
What is the most important information I should know about ZEJULA?
ZEJULA may cause serious side effects including:
• Bone marrow problems called myelodysplastic syndrome (MDS) or a type of cancer of the blood called acute myeloid leukemia (AML). Some people who have ovarian cancer and who have received previous treatment with chemotherapy or certain other medicines for their cancer have developed MDS or AML during treatment with ZEJULA. MDS or AML may lead to death. If you develop MDS or AML, your healthcare provider will stop treatment with ZEJULA. Symptoms of low blood cell counts (low red blood cells, low white blood cells, and low platelets) are common during treatment with ZEJULA, but can be a sign of serious bone marrow problems, including MDS or AML. Symptoms may include:
o weaknesso feeling tiredo weight losso frequent infections
o fevero shortness of breatho blood in urine or stoolo bruising or bleeding more easily
 Your healthcare provider will do blood tests to check your blood cell counts:o before treatment with ZEJULA.o weekly for the first month of treatment with ZEJULA.o every month for the next 11 months, then as needed during treatment with ZEJULA.• High blood pressure. High blood pressure is common during treatment with ZEJULA and can become serious. Your healthcare provider will check your blood pressure and heart rate at least weekly for the first 2 months, then monthly for the first year and as needed thereafter during your treatment with ZEJULA.• Posterior reversible encephalopathy syndrome (PRES). PRES is a condition that affects the brain and may happen during treatment with ZEJULA. If you have headache, vision changes, confusion, or seizure with or without high blood pressure, please contact your healthcare provider.
See “What are the possible side effects of ZEJULA?” for more information about side effects.
What is ZEJULA?
ZEJULA is a prescription medicine used for the:
• maintenance treatment of adults with advanced ovarian cancer, fallopian tube cancer, or primary peritoneal cancer. ZEJULA is used after the cancer has responded (complete or partial response) to treatment with platinum-based chemotherapy.• maintenance treatment of adults with ovarian cancer, fallopian tube cancer, or primary peritoneal cancer with a certain type of inherited (germline) abnormal BRCA gene that comes back. ZEJULA is used after the cancer has responded (complete or partial response) to treatment with platinum-based chemotherapy. Your healthcare provider will perform a test to make sure that ZEJULA is right for you.
It is not known if ZEJULA is safe and effective in children.
Before taking ZEJULA, tell your healthcare provider about all of your medical conditions, including if you:
• have heart problems.• have liver problems.• have high blood pressure.• are pregnant or plan to become pregnant. ZEJULA can harm your unborn baby and may cause loss of pregnancy (miscarriage).• If you are able to become pregnant, your healthcare provider should perform a pregnancy test before you start treatment with ZEJULA.• Females who are able to become pregnant should use effective birth control (contraception) during treatment with ZEJULA and for 6 months after the last dose of ZEJULA. Talk to your healthcare provider about birth control methods that may be right for you.• Tell your healthcare provider right away if you become pregnant.• are breastfeeding or plan to breastfeed. It is not known if ZEJULA passes into your breast milk. Do not breastfeed during treatment with ZEJULA and for 1 month after the last dose of ZEJULA. Talk to your healthcare provider about the best way to feed your baby during this time.
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements.
How should I take ZEJULA?
• Take ZEJULA exactly as your healthcare provider tells you to.• Take ZEJULA 1 time each day, at the same time each day.• ZEJULA may be taken with or without food.• ZEJULA tablets should be swallowed whole. Do not chew, crush, or split ZEJULA tablets before swallowing.• Taking ZEJULA at bedtime may help relieve any nausea symptoms you may have.• Do not stop taking ZEJULA without first talking with your healthcare provider.• If you miss a dose of ZEJULA, take your next dose at your scheduled time. Do not take an extra dose to make up for a missed dose.• If you vomit after taking a dose of ZEJULA, do not take an extra dose. Take your next dose at your scheduled time.• If you take too much ZEJULA, call your healthcare provider or go to the nearest hospital emergency room right away.
What are the possible side effects of ZEJULA?
ZEJULA may cause serious side effects, including:
• See “What is the most important information I should know about ZEJULA?”
The most common side effects of ZEJULA include:
• nausea• tiredness• constipation• pain in your muscles and back• pain in the stomach area• vomiting• loss of appetite• trouble sleeping• headache
• shortness of breath• rash• diarrhea• cough• dizziness• changes in the amount or color of your urine• urinary tract infection• low levels of magnesium in the blood
Your healthcare provider may change your dose, temporarily stop treatment, or permanently stop treatment with ZEJULA if you have certain side effects.
These are not all the possible side effects of ZEJULA.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store ZEJULA?
• Store ZEJULA at room temperature between 68°F to 77°F (20°C to 25°C).• Store ZEJULA tablets in the original bottle.
Keep ZEJULA and all medicines out of the reach of children.
General information about the safe and effective use of ZEJULA.
Medicines are sometimes prescribed for purposes other than those uled in a Patient Information leaflet. Do not use ZEJULA for a condition for which it was not prescribed. Do not give ZEJULA to other people, even if they have the same symptoms that you have. It may harm them. You can ask your healthcare provider or pharmacist for information about ZEJULA that is written for health professionals.
What are the ingredients in ZEJULA?
Active ingredient: niraparib.
Inactive ingredients:
Core tablet: crospovidone, lactose monohydrate, magnesium stearate, microcrystalline cellulose, povidone, and silicon dioxide.
Film-coating: Opadry II Gray (100 mg), Opadry II Blue (200 mg), or Opadry II Green (300 mg).
For more information about ZEJULA, call 1-888-825-5249.
Trademarks are owned by or licensed to the GSK group of companies. Opadry is a trademark owned by or licensed to its respective owner and is not owned by or licensed to the GSK group of companies. The maker of Opadry is not affiliated with and does not endorse the GSK group of companies or its products.
Manufactured for:
GlaxoSmithKline, Durham, NC 27701
©2023 GSK group of companies or its licensor.
ZJT:1PIL
This Patient Information has been approved by the U.S. Food and Drug Administration     Revised: 4/2023
Package Label.principal Display Panel
PRINCIPAL DISPLAY PANEL
NDC 0173-0909-13
Zejula
(niraparib)
tablets
100 mg
GSK
Rx only
30 tablets
Each 100-mg tablet is equivalent to 159.3 mg of niraparib tosylate monohydrate.
Store at 20°C to 25°C (68°F to 77°F); excursions are permitted between 15°C to 30°C (59°F to 86°F).[See USP Controlled Room Temperature]. Store in original package.
Do not accept if membrane seal under cap is missing or broken. See prescribing information for dosage information. Keep out of reach of children.
Trademarks owned or licensed by GSK.
Mfd for GSK, Durham, NC 27701
©2023 GSK or licensor.
Rev. 1/23
629735

Package Label.principal Display Panel
PRINCIPAL DISPLAY PANEL
NDC 0173-0912-13
Zejula
(niraparib)
tablets
200 mg
GSK
Rx only
30 tablets
Each 200-mg tablet is equivalent to 318.7 mg of niraparib tosylate monohydrate.
Store at 20°C to 25°C (68°F to 77°F); excursions are permitted between 15°C to 30°C (59°F to 86°F).[See USP Controlled Room Temperature]. Store in original package.
Do not accept if membrane seal under cap is missing or broken. See prescribing information for dosage information. Keep out of reach of children.
Trademarks owned or licensed by GSK.
Mfd for GSK, Durham, NC 27701
©2023 GSK or licensor.
Rev. 1/23
629736

Package Label.principal Display Panel
PRINCIPAL DISPLAY PANEL
NDC 0173-0915-13
Zejula
(niraparib)
Tablets
300 mg
GSK
Rx only
30 tablets
Each 300-mg tablet is equivalent to 478.0 mg of niraparib tosylate monohydrate.
Store at 20°C to 25°C (68°F to 77°F); excursions are permitted between 15°C to 30°C (59°F to 86°F).[See USP Controlled Room Temperature]. Store in original package.
Do not accept if membrane seal under cap is missing or broken. See prescribing information for dosage information. Keep out of reach of children.
Trademarks owned or licensed by GSK.
Mfd for GSK, Durham, NC 27701
©2023 GSK or licensor.
Rev. 1/23
629737

DISCLAIMER:
"This tool does not provide medical advice, and is for informational and educational purposes only, and is not a substitute for professional medical advice, treatment or diagnosis. Call your doctor to receive medical advice. If you think you may have a medical emergency, please dial 911."
"Do not rely on openFDA to make decisions regarding medical care. While we make every effort to ensure that data is accurate, you should assume all results are unvalidated. We may limit or otherwise restrict your access to the API in line with our Terms of Service."
"This product uses publicly available data from the U.S. National Library of Medicine (NLM), National Institutes of Health, Department of Health and Human Services; NLM is not responsible for the product and does not endorse or recommend this or any other product."
PillSync may earn a commission via links on our site
