Lynparza (olaparib 150 mg) Dailymed
Generic: olaparib is used for the treatment of Ovarian Neoplasms


Go PRO for all pill images
Recent Major Changes Section
Indications and Usage (1.3 )                                                                         9/2023
Dosage and Administration (2.1 )                                                                 9/2023
Indications and Usage (1.8 )                                                                         5/2023
Dosage and Administration (2 )                                                                    5/2023
Warnings and Precautions, Myelodysplastic Syndrome/Acute Myeloid Leukemia (5.1 )                                                                                                            11/2023
1 Indications And Usage
Lynparza is a poly (ADP-ribose) polymerase (PARP) inhibitor indicated:
Ovarian cancer
‚ÄĘ for the maintenance treatment of adult patients with deleterious or suspected deleterious germline or somatic BRCA-mutated advanced epithelial ovarian, fallopian tube or primary peritoneal cancer who are in complete or partial response to first-line platinum-based chemotherapy. Select patients for therapy based on an FDA-approved companion diagnostic for Lynparza. (1.1 ,2.1 )‚ÄĘ in combination with bevacizumab for the maintenance treatment of adult patients with advanced epithelial ovarian, fallopian tube or primary peritoneal cancer who are in complete or partial response to first-line platinum-based chemotherapy and whose cancer is associated with homologous recombination deficiency (HRD)-positive status defined by either:
‚ÄĘ a deleterious or suspected deleterious BRCA mutation, and/or‚ÄĘ genomic instability.¬† Select patients for therapy based on an FDA-approved companion diagnostic for Lynparza. (1.2 ,2.1 )‚ÄĘ for the maintenance treatment of adult patients with deleterious or suspected deleterious germline or somatic BRCA-mutated recurrent epithelial ovarian, fallopian tube or primary peritoneal cancer, who are in complete or partial response to platinum-based chemotherapy. Select patients for therapy based on an FDA-approved companion diagnostic for Lynparza. (1.3 ,2.1 )
Breast cancer
‚ÄĘ for the adjuvant treatment of adult patients with deleterious or suspected deleterious gBRCAm human epidermal growth factor receptor 2 (HER2)-negative high risk early breast cancer who have been treated with neoadjuvant or adjuvant chemotherapy. Select patients for therapy based on an FDA-approved companion diagnostic for Lynparza. (1.4 ,2.1 )‚ÄĘ for the treatment of adult patients with deleterious or suspected deleterious gBRCAm, HER2-negative metastatic breast cancer who have been treated with chemotherapy in the neoadjuvant, adjuvant or metastatic setting. Patients with hormone receptor (HR)-positive breast cancer should have been treated with a prior endocrine therapy or be considered inappropriate for endocrine therapy. Select patients for therapy based on an FDA-approved companion diagnostic for Lynparza. (1.5 ,2.1 )
Pancreatic cancer
‚ÄĘ for the maintenance treatment of adult patients with deleterious or suspected deleterious gBRCAm metastatic pancreatic adenocarcinoma whose disease has not progressed on at least 16 weeks of a first-line platinum-based chemotherapy regimen. Select patients for therapy based on an FDA-approved companion diagnostic for Lynparza. (1.6 ,2.1 )
Prostate cancer
‚ÄĘ for the treatment of adult patients with deleterious or suspected deleterious germline or somatic homologous recombination repair (HRR) gene-mutated metastatic castration-resistant prostate cancer (mCRPC) who have progressed following prior treatment with enzalutamide or abiraterone. Select patients for therapy based on an FDA-approved companion diagnostic for Lynparza. (1.7 ,2.1 )‚ÄĘ in combination with abiraterone and prednisone or prednisolone for the treatment of adult patients with deleterious or suspected deleterious BRCA-mutated (BRCAm) metastatic castration-resistant prostate cancer (mCRPC). Select patients for therapy based on an FDA-approved companion diagnostic for Lynparza. (1.8 ,2.1 )1.1 First-Line Maintenance Treatment of -mutated Advanced Ovarian Cancer
Lynparza is indicated for the maintenance treatment of adult patients with deleterious or suspected deleterious germline or somatic BRCA-mutated advanced epithelial ovarian, fallopian tube or primary peritoneal cancer who are in complete or partial response to first-line platinum-based chemotherapy. Select patients for therapy based on an FDA-approved companion diagnostic for Lynparza [see Dosage and Administration (2.1) ].
1.2 First-line Maintenance Treatment of HRD-positive Advanced Ovarian Cancer in Combination with Bevacizumab
Lynparza is indicated in combination with bevacizumab for the maintenance treatment of adult patients with advanced epithelial ovarian, fallopian tube or primary peritoneal cancer who are in complete or partial response to first-line platinum-based chemotherapy and whose cancer is associated with homologous recombination deficiency (HRD)-positive status defined by either:
 
‚ÄĘ a deleterious or suspected deleterious BRCA mutation, and/or‚ÄĘ genomic instability.
Select patients for therapy based on an FDA-approved companion diagnostic for Lynparza [see Dosage and Administration (2.1)].
1.3 Maintenance Treatment of -mutated Recurrent Ovarian Cancer
Lynparza is indicated for the maintenance treatment of adult patients with deleterious or suspected deleterious germline or somatic BRCA-mutated recurrent epithelial ovarian, fallopian tube or primary peritoneal cancer, who are in complete or partial response to platinum-based chemotherapy. Select patients for therapy based on an FDA-approved companion diagnostic for Lynparza [see Dosage and Administration (2.1)].
1.4 Adjuvant Treatment of Germline -mutated HER2-negative High Risk Early Breast Cancer
Lynparza is indicated for the adjuvant treatment of adult patients with deleterious or suspected deleterious gBRCAm human epidermal growth factor receptor 2 (HER2)-negative high risk early breast cancer who have been treated with neoadjuvant or adjuvant chemotherapy. Select patients for therapy based on an FDA-approved companion diagnostic for Lynparza [see Dosage and Administration (2.1)].
1.5 Germline -mutated HER2-negative Metastatic Breast Cancer
Lynparza is indicated for the treatment of adult patients with deleterious or suspected deleterious gBRCAm, HER2-negative metastatic breast cancer, who have been treated with chemotherapy in the neoadjuvant, adjuvant, or metastatic setting. Patients with hormone receptor (HR)-positive breast cancer should have been treated with a prior endocrine therapy or be considered inappropriate for endocrine therapy. Select patients for therapy based on an FDA-approved companion diagnostic for Lynparza [see Dosage and Administration (2.1)].
1.6 First-Line Maintenance Treatment of Germline -mutated Metastatic Pancreatic Adenocarcinoma
Lynparza is indicated for the maintenance treatment of adult patients with deleterious or suspected deleterious gBRCAm metastatic pancreatic adenocarcinoma whose disease has not progressed on at least 16 weeks of a first-line platinum-based chemotherapy regimen. Select patients for therapy based on an FDA-approved companion diagnostic for Lynparza [see Dosage and Administration (2.1)].
1.7 HRR Gene-mutated Metastatic Castration-Resistant Prostate Cancer
Lynparza is indicated for the treatment of adult patients with deleterious or suspected deleterious germline or somatic homologous recombination repair (HRR) gene-mutated metastatic castration-resistant prostate cancer (mCRPC) who have progressed following prior treatment with enzalutamide or abiraterone. Select patients for therapy based on an FDA-approved companion diagnostic for Lynparza [see Dosage and Administration (2.1)].
1.8 Treatment of -mutated Metastatic Castration-Resistant Prostate Cancer in Combination with Abiraterone and Prednisone or Prednisolone
Lynparza is indicated in combination with abiraterone and prednisone or prednisolone for the treatment of adult patients with deleterious or suspected deleterious BRCA-mutated (BRCAm) metastatic castration-resistant prostate cancer (mCRPC). Select patients for therapy based on an FDA-approved companion diagnostic for Lynparza [seeDosage and Administration (2.1)].
2 Dosage And Administration
‚ÄĘ Recommended dosage is 300 mg taken orally twice daily with or without food. See Full Prescribing Information for the recommended duration.(2.2) ‚ÄĘ Patients receiving Lynparza for mCRPC should also receive a gonadotropin-releasing hormone (GnRH) analog concurrently or should have had bilateral orchiectomy.(2.2) ‚ÄĘ For moderate renal impairment (CLcr 31-50 mL/min), reduce Lynparza dosage to 200 mg orally twice daily.(2.5) 2.1 Patient Selection
Information on FDA-approved tests for the detection of genetic mutations is available at http://www.fda.gov/companiondiagnostics .
Select patients for treatment with Lynparza based on the presence of deleterious or suspected deleterious HRR gene mutations, including BRCA mutations, or genomic instability based on the indication, biomarker, and sample type (Table 1).
Table 1 Biomarker Testing for Patient Selection Where testing fails or tissue sample is unavailable/insufficient, or when germline testing is negative, consider using an alternative test, if available.
Indication
Biomarker
Sample type
Tumor
Blood
Plasma
(ctDNA)
First-line maintenance treatment of germline or somatic BRCAm advanced ovarian cancer
BRCA1m, BRCA2m
X
X
First-line maintenance treatment of HRD-positive advanced ovarian cancer in combination with bevacizumab
BRCA1m, BRCA2m and/or genomic instability
X
Maintenance treatment of germline or somatic BRCAm recurrent ovarian cancer
BRCA1m, BRCA2m
X
X
Adjuvant treatment of gBRCAm HER2-negative high risk early breast cancer
gBRCA1m, gBRCA2m
X
gBRCAm HER2-negative metastatic breast cancer
gBRCA1m, gBRCA2m
X
First-line maintenance treatment of germline BRCA-mutated metastatic pancreatic adenocarcinoma
gBRCA1m, gBRCA2m
X
Germline or somatic HRR gene-mutated metastatic castration-resistant prostate cancer
ATMm, BRCA1m, BRCA2m, BARD1m, BRIP1m, CDK12m, CHEK1m, CHEK2m, FANCLm, PALB2m, RAD51Bm, RAD51Cm, RAD51Dm, RAD54Lm
X
gBRCA1m, gBRCA2m
X
ATMm, BRCA1m, BRCA2m
X
BRCA-mutated metastatic castration-resistant prostate cancer in combination with abiraterone and prednisone or prednisolone
BRCA1m, BRCA2m
X
X
X
2.2 Recommended Dosage
The recommended dosage of Lynparza is 300 mg taken orally twice daily, with or without food.
If a patient misses a dose of Lynparza, instruct patient to take their next dose at its scheduled time.
Instruct patients to swallow tablets whole. Do not chew, crush, dissolve, or divide tablet.
First-Line Maintenance Treatment of BRCA-mutated Advanced Ovarian Cancer
Continue treatment until disease progression, unacceptable toxicity, or completion of 2 years of treatment. Patients with a complete response (no radiological evidence of disease) at 2 years should stop treatment. Patients with evidence of disease at 2 years, who in the opinion of the treating healthcare provider can derive further benefit from continuous treatment, can be treated beyond 2 years.
First-Line Maintenance Treatment of HRD-positive Advanced Ovarian Cancer in Combination with Bevacizumab
Continue Lynparza treatment until disease progression, unacceptable toxicity, or completion of 2 years of treatment. Patients with a complete response (no radiological evidence of disease) at 2 years should stop treatment. Patients with evidence of disease at 2 years, who in the opinion of the treating healthcare provider can derive further benefit from continuous Lynparza treatment, can be treated beyond 2 years.
When used with Lynparza, the recommended dose of bevacizumab is 15 mg/kg every three weeks. Bevacizumab should be given for a total of 15 months including the period given with chemotherapy and given as maintenance. Refer to the Prescribing Information for bevacizumab when used in combination with Lynparza for more information.
Adjuvant Treatment of Germline BRCA-mutated HER2-negative High Risk Early Breast Cancer
Continue treatment for a total of 1 year, or until disease recurrence, or unacceptable toxicity, whichever occurs first. Patients receiving Lynparza for hormone receptor positive HER2-negative breast cancer should continue concurrent treatment with endocrine therapy as per current clinical practice guidelines.
Germline or Somatic BRCA-mutated Recurrent Ovarian Cancer, Germline BRCA-mutated HER2-negative Metastatic Breast Cancer, Germline BRCA-mutated Metastatic Pancreatic Adenocarcinoma, and HRR Gene-mutated Metastatic Castration-Resistant Prostate Cancer
Continue treatment until disease progression or unacceptable toxicity for:
 
‚ÄĘ Maintenance treatment of germline or somatic BRCA-mutated recurrent ovarian cancer. ‚ÄĘ Germline BRCA-mutated HER-2 negative metastatic breast cancer. ‚ÄĘ First-line maintenance treatment of germline BRCA-mutated metastatic pancreatic adenocarcinoma. ‚ÄĘ HRR gene-mutated metastatic castration-resistant prostate cancer.
BRCA-mutated Metastatic Castration-Resistant Prostate Cancer in Combination with Abiraterone and Prednisone or Prednisolone
Continue treatment until disease progression or unacceptable toxicity.
When used with Lynparza, the recommended dose of abiraterone is 1000 mg taken orally once daily. Abiraterone should be given in combination with prednisone or prednisolone 5 mg orally twice daily. Refer to the Prescribing Information for abiraterone for dosing information.
Patients with mCRPC should also receive a gonadotropin-releasing hormone (GnRH) analog concurrently or should have had bilateral orchiectomy.
2.3 Dosage Modifications for Adverse Reactions
To manage adverse reactions, consider interruption of treatment or dose reduction. The recommended dose reduction is 250 mg taken twice daily.
If a further dose reduction is required, then reduce to 200 mg taken twice daily.
2.4 Dosage Modifications for Concomitant Use with Strong or Moderate CYP3A Inhibitors
Avoid concomitant use of strong or moderate CYP3A inhibitors with Lynparza.
If concomitant use cannot be avoided, reduce Lynparza dosage to:
‚ÄĘ 100 mg twice daily when used concomitantly with a strong CYP3A inhibitor.‚ÄĘ 150 mg twice daily when used concomitantly with a moderate CYP3A inhibitor.
After the inhibitor has been discontinued for 3 to 5 elimination half-lives, resume the Lynparza dose taken prior to initiating the CYP3A inhibitor [see Drug Interactions (7.2) and Clinical Pharmacology (12.3)].
2.5 Dosage Modifications for Renal Impairment
Moderate Renal Impairment
In patients with moderate renal impairment (CLcr 31-50 mL/min), reduce the Lynparza dosage to 200 mg orally twice daily [see Use in Specific Populations (8.6) and Clinical Pharmacology (12.3)].
3 Dosage Forms And Strengths
Tablets:
‚ÄĘ 150 mg: green to green/grey, oval, bi-convex, film-coated, with debossment ‚ÄėOP150‚Äô on one side and plain on the reverse side.‚ÄĘ 100 mg: yellow to dark yellow, oval, bi-convex, film-coated, with debossment ‚ÄėOP100‚Äô on one side and plain on the reverse side.
Tablets: 150 mg, 100 mg(3)
4 Contraindications
None.
None.(4)
5 Warnings And Precautions
‚ÄĘ Myelodysplastic Syndrome/Acute Myeloid Leukemia (MDS/AML): Occurred in approximately 1.2% of patients with various BRCAm, gBRCAm, HRR gene-mutated or HRD-positive cancers exposed to Lynparza and the majority of events had a fatal outcome. Monitor patients for hematological toxicity at baseline and monthly thereafter. Discontinue if MDS/AML is confirmed.(5.1) ‚ÄĘ Pneumonitis: Occurred in 0.8% of patients exposed to Lynparza, and some cases were fatal. Interrupt treatment if pneumonitis is suspected. Discontinue if pneumonitis is confirmed.(5.2) ‚ÄĘ Venous thromboembolism (VTE), including severe or fatal pulmonary embolism (PE), occurred in patients treated with Lynparza. VTE occurred in 8% of patients with mCRPC. Monitor patients for signs and symptoms of VTE and PE and treat as medically appropriate. (5.3 )‚ÄĘ Embryo-Fetal Toxicity: Can cause fetal harm. Advise of the potential risk to a fetus and to use effective contraception. (5.4 ,8.1 ,8.3 )¬† 5.1 Myelodysplastic Syndrome/Acute Myeloid Leukemia
Myelodysplastic syndrome (MDS)/Acute Myeloid Leukemia (AML) has occurred in patients treated with Lynparza and some cases were fatal.
In clinical studies, among 2219 patients with various BRCAm, gBRCAm, HRR gene-mutated or HRD-positive cancers who received Lynparza as a single agent or as part of combination regimen, consistent with approved indications, the cumulative incidence of MDS/AML was approximately 1.2% (26/2219) [see Adverse Reactions (6.1)]. Of these, 54% (14/26) had a fatal outcome. The median duration of therapy with Lynparza in patients who developed MDS/AML was approximately 2 years (range: < 6 months to > 4 years). All of these patients had received previous chemotherapy with platinum agents and/or other DNA damaging agents including radiotherapy.
In SOLO1, patients with newly diagnosed advanced BRCAm ovarian cancer, the incidence of MDS/AML was 1.9% (5/260) in patients who received Lynparza and 0.8% (1/130) in patients who received placebo based on an updated analysis. In PAOLA-1, of patients with newly diagnosed advanced ovarian cancer with HRD-positive status, the incidence of MDS/AML was 1.6% (4/255) in patients who received Lynparza and 2.3% (3/131) in the control arm.
In SOLO2, patients with BRCAm platinum-sensitive relapsed ovarian cancer, the incidence of MDS/AML was 8% (15/195) in patients who received Lynparza and 4% (4/99) in patients who received placebo. The duration of Lynparza treatment prior to the diagnosis of MDS/AML ranged from 0.6 years to 4.5 years.
Do not start Lynparza until patients have recovered from hematological toxicity caused by previous chemotherapy (‚ȧ Grade 1). Monitor complete blood count for cytopenia at baseline and monthly thereafter for clinically significant changes during treatment. For prolonged hematological toxicities, interrupt Lynparza and monitor blood counts weekly until recovery. If the levels have not recovered to Grade 1 or less after 4 weeks, refer the patient to a hematologist for further investigations, including bone marrow analysis and blood sample for cytogenetics. If MDS/AML is confirmed, discontinue Lynparza.
5.2 Pneumonitis
In clinical studies enrolling 2901 patients with various cancers who received Lynparza as a single agent [see Adverse Reactions (6.1)], the incidence of pneumonitis, including fatal cases, was 0.8% (24/2901). If patients present with new or worsening respiratory symptoms such as dyspnea, cough and fever, or a radiological abnormality occurs, interrupt Lynparza treatment and promptly assess the source of the symptoms. If pneumonitis is confirmed, discontinue Lynparza treatment and treat the patient appropriately.
5.3 Venous Thromboembolism
Venous thromboembolism (VTE), including severe or fatal pulmonary embolism (PE), occurred in patients treated with Lynparza [see Adverse Reactions (6.1)].
In the combined data of two randomized, placebo-controlled clinical studies (PROfound and PROpel) in patients with metastatic castration-resistant prostate cancer (N=1180), VTE occurred in 8% of patients who received Lynparza, including pulmonary embolism in 6%. In the control arms, VTE occurred in 2.5% including pulmonary embolism in 1.5%.
Monitor patients for clinical signs and symptoms of venous thrombosis and pulmonary embolism and treat as medically appropriate, which may include long-term anticoagulation as clinically indicated.
5.4 Embryo-Fetal Toxicity
Lynparza can cause fetal harm when administered to a pregnant woman based on its mechanism of action and findings in animals. In an animal reproduction study, administration of olaparib to pregnant rats during the period of organogenesis caused teratogenicity and embryo-fetal toxicity at exposures below those in patients receiving the recommended human dose of 300 mg twice daily. Apprise pregnant women of the potential hazard to a fetus and the potential risk for loss of the pregnancy. Advise females of reproductive potential to use effective contraception during treatment and for 6 months following the last dose of Lynparza. Based on findings from genetic toxicity and animal reproduction studies, advise male patients with female partners of reproductive potential or who are pregnant to use effective contraception during treatment and for 3 months following the last dose of Lynparza [see Use in Specific Populations (8.1, 8.3)].
6 Adverse Reactions
The following adverse reactions are discussed elsewhere in the labeling:
‚ÄĘ Myelodysplastic Syndrome/Acute Myeloid Leukemia [see Warnings and Precautions (5.1)]‚ÄĘ Pneumonitis [see Warnings and Precautions (5.2)]‚ÄĘ Venous Thromboembolism [see Warnings and Precautions (5.3) ]
Most common adverse reactions (‚Č•10%) in clinical trials:
‚ÄĘ as a single agent were nausea, fatigue (including asthenia), anemia, vomiting, diarrhea, decreased appetite, headache, dysgeusia, cough, neutropenia, dyspnea, dizziness, dyspepsia, leukopenia, and thrombocytopenia.(6.1) ‚ÄĘ in combination with bevacizumab were nausea, fatigue (including asthenia), anemia, lymphopenia, vomiting, diarrhea, neutropenia, leukopenia, urinary tract infection, and headache. (6.1 )‚ÄĘ in combination with abiraterone and prednisone or prednisolone were anemia, fatigue, nausea, diarrhea, decreased appetite, lymphopenia, dizziness, and abdominal pain. (6.1 )
To report SUSPECTED ADVERSE REACTIONS, contact AstraZeneca at 1-800-236-9933 or FDA at 1-800-FDA-1088 orwww.fda.gov/medwatch.
6.1 Clinical Trial Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Unless otherwise specified, the data described in the WARNINGS AND PRECAUTIONS reflect exposure to Lynparza as a single agent in 2901 patients; 2135 patients with exposure to 300 mg twice daily tablet dose including five controlled, randomized, trials (SOLO-1, SOLO-2, OlympiAD, POLO, and PROfound) and to 400 mg twice daily capsule dose in 766 patients in other trials that were pooled to conduct safety analyses. In addition to the 2901 patients, certain subsections in the WARNINGS AND PRECAUTIONS include adverse reactions observed with exposure to Lynparza with abiraterone (n=398) in PROpel. All patients with metastatic castration resistant prostate cancer received concomitant ADT or previous bilateral orchiectomy.
In the pooled safety population, 56% of patients were exposed for 6 months or longer and 28% were exposed for greater than one year in the Lynparza group.
In this pooled safety population, the most common adverse reactions in ‚Č•10% of patients were nausea (60%), fatigue (55%), anemia (36%), vomiting (32%), diarrhea (24%), decreased appetite (22%), headache (16%), dysgeusia (15%), cough (15%), neutropenia (14%), dyspnea (14%), dizziness (12%), dyspepsia (12%), leukopenia (11%), and thrombocytopenia (10%).
First-Line Maintenance Treatment of BRCA-mutated Advanced Ovarian Cancer
SOLO-1
The safety of Lynparza for the maintenance treatment of patients with BRCA-mutated advanced ovarian cancer following first-line treatment with platinum-based chemotherapy was investigated in SOLO-1 [see Clinical Studies (14.1)]. Patients received Lynparza tablets 300 mg orally twice daily (n=260) or placebo (n=130) until disease progression or unacceptable toxicity. The median duration of study treatment was 25 months for patients who received Lynparza and 14 months for patients who received placebo.
Among patients who received Lynparza, dose interruptions due to an adverse reaction of any grade occurred in 52% and dose reductions due to an adverse reaction occurred in 28%. The most frequent adverse reactions leading to dose interruption or reduction of Lynparza were anemia (23%), nausea (14%), and vomiting (10%). Discontinuation due to adverse reactions occurred in 12% of patients receiving Lynparza. The most frequent adverse reactions that led to discontinuation of Lynparza were fatigue (3.1%), anemia (2.3%), and nausea (2.3%).
Tables 2 and 3 summarize adverse reactions and laboratory abnormalities in SOLO-1.
Table 2 Adverse Reactions Graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE), version 4.0. in SOLO-1 (‚Č•10% of Patients Who Received Lynparza)
Adverse Reaction
Lynparza tablets
n=260
Placebo
n=130
All Grades
(%)
Grades
3 ‚Äď 4 (%)
All
Grades(%)
Grades
3 ‚Äď 4 (%)
Gastrointestinal Disorders
     Nausea
77
1
38
0
     Abdominal painIncludes abdominal pain, abdominal pain lower, abdominal pain upper, abdominal distension, abdominal discomfort, and abdominal tenderness.
45
2
35
1
     Vomiting
40
0
15
1
     DiarrheaIncludes colitis, diarrhea, and gastroenteritis.
37
3
26
0
     Constipation
28
0
19
0
     Dyspepsia
17
0
12
0
     StomatitisIncludes stomatitis, aphthous ulcer, and mouth ulceration.
11
0
2
0
General Disorders and Administration Site Conditions
     FatigueIncludes asthenia, fatigue, lethargy, and malaise.
67
4
42
2
Blood and Lymphatic System Disorders
     Anemia
38
21
9
2
     NeutropeniaIncludes neutropenia and febrile neutropenia.
17
6
7
3
     LeukopeniaIncludes leukopenia and white blood cell count decreased.
13
3
8
0
     ThrombocytopeniaIncludes platelet count decreased and thrombocytopenia.
11
1
4
2
Infections and Infestations
     Upper respiratory tract     infection/ influenza/nasopharyngitis/bronchitis
28
0
23
0
     UTIIncludes urosepsis, urinary tract infection, urinary tract pain, and pyuria.
13
1
7
0
Nervous System Disorders
     Dysgeusia
26
0
4
0
     Dizziness
20
0
15
1
Metabolism and Nutrition Disorders
     Decreased appetite
20
0
10
0
Respiratory, Thoracic and Mediastinal Disorders
     DyspneaIncludes dyspnea and dyspnea exertional.
15
0
6
0
Clinically relevant adverse reactions that occurred in <10% of patients receiving Lynparza were increased blood creatinine (8%), lymphopenia (6%), VTE (3%), hypersensitivity (2%), MDS/AML (1.9%), dermatitis (1%), and increased mean cell volume (0.4%).
Table 3 Laboratory Abnormalities Reported in ‚Č•25% of Patients in SOLO-1
Laboratory
ParameterPatients were allowed to enter clinical studies with laboratory values of CTCAE Grade 1.
Lynparza tablets
nThis number represents the safety population. The derived values in the table are based on the total number of evaluable patients for each laboratory parameter. =260
Placebo
n=130
Grades 1-4
(%)
Grades 3-4
(%)
Grades 1-4
(%)
Grades 3-4
(%)
Decrease in hemoglobin
87
19
63
2
Increase in mean corpuscular volume
87
-
43
-
Decrease in leukocytes
70
7
52
1
Decrease in lymphocytes
67
14
29
5
Decrease in absolute neutrophil count
51
9
38
6
Decrease in platelets
35
1
20
2
Increase in serum creatinine
34
0
18
0
First-line Maintenance Treatment of HRD-positive Advanced Ovarian Cancer in Combination with Bevacizumab
PAOLA-1
The safety of Lynparza in combination with bevacizumab for the maintenance treatment of patients with advanced ovarian cancer following first-line treatment containing platinum-based chemotherapy and bevacizumab was investigated in PAOLA-1 [see Clinical Studies (14.2)]. This study was a placebo-controlled, double-blind study in which 802 patients received either Lynparza 300 mg BID in combination with bevacizumab (n=535) or placebo in combination with bevacizumab (n=267) until disease progression or unacceptable toxicity. The median duration of treatment with Lynparza was 17.3 months and 11 months for bevacizumab post-randomization on the Lynparza/bevacizumab arm.
Fatal adverse reactions occurred in 1 patient due to concurrent pneumonia and aplastic anemia. Serious adverse reactions occurred in 31% of patients who received Lynparza/bevacizumab. Serious adverse reactions in >5% of patients included hypertension (19%) and anemia (17%).
Dose interruptions due to an adverse reaction of any grade occurred in 54% of patients receiving Lynparza/bevacizumab and dose reductions due to an adverse reaction occurred in 41% of patients who received Lynparza/bevacizumab.
The most frequent adverse reactions leading to dose interruption in the Lynparza/bevacizumab arm were anemia (21%), nausea (7%), vomiting (3%), and fatigue (3%), and the most frequent adverse reactions leading to reduction in the Lynparza/bevacizumab arm were anemia (19%), nausea (7%), and fatigue (4%).
Discontinuation due to adverse reactions occurred in 20% of patients receiving Lynparza/bevacizumab. Specific adverse reactions that most frequently led to discontinuation in patients treated with Lynparza/bevacizumab were anemia (4%) and nausea (3%).
The most common adverse reactions (‚Č• 10%) for patients receiving Lynparza/bevacizumab irrespective of the frequency compared with the placebo/bevacizumab arm were nausea (53%), fatigue (including asthenia) (53%), anemia (41%), lymphopenia (24%), vomiting (22%), diarrhea (18%), neutropenia (18%), leukopenia (18%), urinary tract infection (15%), and headache (14%).
Tables 4 and 5 summarize adverse reactions and laboratory abnormalities in PAOLA-1, respectively.
Table 4 Adverse Reactions Graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE), version 4.0. Occurring in ‚Č•10% of Patients Treated with Lynparza/bevacizumab in PAOLA-1 and at ‚Č•5% Frequency Compared to the Placebo/bevacizumab Arm
Adverse Reactions
Lynparza/bevacizumab
n=535
Placebo/bevacizumab n=267
Grades 1-4
(%)
Grades 3-4
(%)
Grades 1-4
(%)
Grades 3-4
(%)
General Disorders and Administration Site Conditions
     Fatigue (including asthenia)Includes asthenia and fatigue.
53
5
32
1.5
Gastrointestinal Disorders
     Nausea
53
2.4
22
0.7
     Vomiting
22
1.7
11
1.9
Blood and Lymphatic Disorders
     AnemiaIncludes anemia, anemia macrocytic, erythropenia, haematocrit decreased, haemoglobin decreased, normochromic anemia, normochromic normocytic anemia, normocytic anemia, and red blood cell count decreased.
41
17
10
0.4
     LymphopeniaIncludes B-lymphocyte count decreased, lymphocyte count decreased, lymphopenia, and T-lymphocyte count decreased.
24
7
9
1.1
     LeukopeniaIncludes leukopenia and white blood cell count decreased.
18
1.9
10
1.5
Clinically relevant adverse reactions that occurred in <10% of patients receiving Lynparza/bevacizumab were dysgeusia (8%), dyspnea (8%), stomatitis (5%), dyspepsia (4.3%), erythema (3%), dizziness (2.6%), hypersensitivity (1.7%), and MDS/AML (0.7%).
Venous thromboembolism occurred more commonly in patients receiving Lynparza/bevacizumab (5%) than in those receiving placebo/bevacizumab (1.9%).
Table 5 Laboratory Abnormalities Reported in ‚Č•25% of Patients in PAOLA-1 Reported within 30 days of the last dose.
Laboratory ParameterPatients were allowed to enter clinical studies with laboratory values of CTCAE Grade 1.
Lynparza/bevacizumab n=535
Placebo/bevacizumab nThis number represents the safety population. The derived values in the table are based on the total number of evaluable patients for each laboratory parameter. =267
Grades 1-4
(%)
Grades 3-4
(%)
Grades 1-4
(%)
Grades 3-4
(%)
Decrease in hemoglobin
79
13
55
0.4
Decrease in lymphocytes
63
10
42
3.0
Increase in serum creatinine
61
0.4
36
0.4
Decrease in leukocytes
59
3.4
45
2.2
Decrease in absolute neutrophil count
35
7
30
3.7
Decrease in platelets
35
2.4
28
0.4
Maintenance Treatment of BRCA-mutated Recurrent Ovarian Cancer
SOLO-2
The safety of Lynparza for the maintenance treatment of patients with platinum sensitive gBRCAm ovarian cancer was investigated in SOLO-2 [see Clinical Studies (14.3)]. Patients received Lynparza tablets 300 mg orally twice daily (n=195) or placebo (n=99) until disease progression or unacceptable toxicity. The median duration of study treatment was 19.4 months for patients who received Lynparza and 5.6 months for patients who received placebo.
Among patients who received Lynparza, dose interruptions due to an adverse reaction of any grade occurred in 45% and dose reductions due to an adverse reaction occurred in 27%. The most frequent adverse reactions leading to dose interruption or reduction of Lynparza were anemia (22%), neutropenia (9%), and fatigue/asthenia (8%). Discontinuation due to an adverse reaction occurred in 11% of patients receiving Lynparza.
Tables 6 and 7 summarize adverse reactions and laboratory abnormalities in SOLO-2.
Table 6 Adverse Reactions Graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE), version 4.0. in SOLO-2 (‚Č•20% of Patients Who Received Lynparza)
Adverse Reaction
Lynparza tablets
n=195
Placebo
n=99
Grades 1-4
(%)
Grades 3-4
(%)
Grades 1-4
(%)
Grades 3-4
(%)
Gastrointestinal Disorders
    Nausea
76
3
33
0
    Vomiting
37
3
19
1
    Diarrhea
33
2
22
0
    StomatitisRepresents grouped term consisting of abscess oral, aphthous ulcer, gingival abscess, gingival disorder, gingival pain, gingivitis, mouth ulceration, mucosal infection, mucosal inflammation, oral candidiasis, oral discomfort, oral herpes, oral infection, oral mucosal erythema, oral pain, oropharyngeal discomfort, and oropharyngeal pain.
20
1
16
0
General Disorders and Administration Site Conditions
    Fatigue including asthenia
66
4
39
2
Blood and Lymphatic Disorders
    AnemiaRepresents grouped term consisting of anemia, hematocrit decreased, hemoglobin decreased, iron deficiency, mean cell volume increased, and red blood cell count decreased.
44
20
9
2
Infections and Infestations
    Nasopharyngitis/URI/sinusitis/ rhinitis/influenza
36
0
29
0
Musculoskeletal and Connective Tissue Disorders
    Arthralgia/myalgia
30
0
28
0
Nervous System Disorders
    Dysgeusia
27
0
7
0
    Headache
26
1
14
0
Metabolism and Nutrition Disorders
    Decreased appetite
22
0
11
0
Clinically relevant adverse reactions that occurred in <20% of patients receiving Lynparza were neutropenia (19%), cough (18%), leukopenia (16%), hypomagnesemia (14%), thrombocytopenia (14%), dizziness (13%), dyspepsia (11%), increased creatinine (11%), MDS/AML (8%), edema (8%), rash (6%), VTE (5%), and lymphopenia (1%).
Table 7 Laboratory Abnormalities Reported in ‚Č•25% of Patients in SOLO-2
Laboratory ParameterPatients were allowed to enter clinical studies with laboratory values of CTCAE Grade 1.
Lynparza tablets
nThis number represents the safety population. The derived values in the table are based on the total number of evaluable patients for each laboratory parameter. =195
Placebo
n=99
Grades 1-4
(%)
Grades 3-4
(%)
Grades 1-4
(%)
Grades 3-4
(%)
Increase in mean corpuscular volumeRepresents the proportion of subjects whose mean corpuscular volume was > upper limit of normal (ULN).
89
-
52
-
Decrease in hemoglobin
83
17
69
0
Decrease in leukocytes
69
5
48
1
Decrease in lymphocytes
67
11
37
1
Decrease in absolute neutrophil count
51
7
34
3
Increase in serum creatinine
44
0
29
0
Decrease in platelets
42
2
22
1
Adjuvant Treatment of germline BRCA-mutated HER2-negative High Risk Early Breast Cancer
OlympiA
The safety of Lynparza as monotherapy for the adjuvant treatment of patients with gBRCA-mutated HER2-negative high risk early breast cancer was investigated in OlympiA [see Clinical Studies (14.4) ]. This study was a randomized, double-blind, multi-center study in which patients received either Lynparza tablets 300 mg orally twice daily (n=911) or placebo (n=904) for a total of 1 year, or until disease recurrence, or unacceptable toxicity. The median duration of treatment was 1 year in both arms.
Dose interruptions due to an adverse reaction of any grade occurred in 31% of patients receiving Lynparza; dose reductions due to an adverse reaction occurred in 23% of patients receiving Lynparza. The most frequent adverse reactions leading to dose interruption of Lynparza were anemia (11%), neutropenia (6%), nausea (5%), leukopenia (3.5%), fatigue (3%), and vomiting (2.9%) and the most frequent adverse reactions leading to dose reduction of Lynparza were anemia (8%), nausea (4.7%), neutropenia (4.2%), fatigue (3.3%), leukopenia (1.8%), and vomiting (1.5%). Discontinuation due to adverse reactions occurred in 10% of patients receiving Lynparza. The adverse reactions that most frequently led to discontinuation of Lynparza were nausea (2%), anemia (1.8%), and fatigue (1.3%).
Tables 8 and 9 summarize the adverse reactions and laboratory abnormalities, respectively, in patients in OlympiA.
Table 8 Adverse Reactions Graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE), version 4.03 in OlympiA (‚Č• 10% of Patients Who Received Lynparza)
Adverse Reactions
Lynparza tablets
n=911
Placebo
n=904
Grades 1-4
(%)
Grades 3-4
(%)
Grades 1-4
(%)
Grades 3-4
(%)
Gastrointestinal Disorders
   Nausea
57
0.8
23
0
   Vomiting
23
0.7
8
0
   Diarrhea
18
0.3
14
0.3
   StomatitisIncludes aphthous ulcer, mouth ulceration, and stomatitis.
10
0.1
4.5
0
General Disorders and Administration Site Conditions
   Fatigue (including asthenia)
42
1.8
28
0.7
Blood and Lymphatic Disorders
   AnemiaIncludes anemia, anemia macrocytic, erythropenia, hematocrit decreased, hemoglobin decreased, normochromic anemia, normochromic normocytic anemia, normocytic anemia, and red blood cell count decreased.
24
9
3.9
0.3
   LeukopeniaIncludes leukopenia and white blood cell count decreased.
17
3
6
0.3
   NeutropeniaIncludes agranulocytosis, febrile neutropenia, granulocyte count decreased, granulocytopenia, idiopathic neutropenia, neutropenia, neutropenic infection, neutropenic sepsis, and neutrophil count decreased.
16
5
7
0.8
Nervous System Disorders
   Headache
20
0.2
17
0.1
   DysgeusiaIncludes dysgeusia and taste disorder.
12
0
4.8
0
   Dizziness
11
0.1
7
0.1
Metabolism and Nutrition Disorders
   Decreased appetite
13
0.2
6
0
Clinically relevant adverse reactions that occurred in <10% of patients receiving Lynparza were cough (9.2%), lymphopenia (7%), dyspepsia (6%), upper abdominal pain (4.9%), rash (4.9%), dyspnea (4.2%), thrombocytopenia (4.2%), increase in creatinine (2%), hypersensitivity (0.9%), VTE (0.5%), dermatitis (0.5%), increase in mean corpuscular volume (0.2%), and MDS/AML (0.2%).
Table 9 Laboratory Abnormalities Reported in ‚Č•25% of Patients in OlympiA
Laboratory ParameterPatients were allowed to enter clinical studies with laboratory values of CTCAE Grade 1.
Lynparza tablets
nThis number represents the safety population. The derived values in the table are based on the total number of evaluable patients for each laboratory parameter. = 911
Placebo
n=904
Grades 1-4
(%)
Grades 3-4
(%)
Grades 1-4
(%)
Grades 3-4
(%)
Decrease in lymphocytes
77
13
59
3.7
Increase in mean corpuscular volumeRepresents the proportion of subjects whose mean corpuscular volume was > ULN.
67
0
4.8
0
Decrease in hemoglobin
65
8
31
0.9
Decrease in leukocytes
64
5
42
0.7
Decrease in absolute neutrophil count
39
7
27
1.1
Germline BRCA-mutated HER2-negative Metastatic Breast Cancer
OlympiAD
The safety of Lynparza was evaluated in gBRCAm patients with HER2-negative metastatic breast cancer who had previously received up to two lines of chemotherapy for the treatment of metastatic disease in OlympiAD [see Clinical Studies (14.5) ]. Patients received either Lynparza tablets 300 mg orally twice daily (n=205) or a chemotherapy (capecitabine, eribulin, or vinorelbine) of the healthcare provider’s choice (n=91) until disease progression or unacceptable toxicity. The median duration of study treatment was 8.2 months in patients who received Lynparza and 3.4 months in patients who received chemotherapy.
Among patients who received Lynparza, dose interruptions due to an adverse reaction of any grade occurred in 35% and dose reductions due to an adverse reaction occurred in 25%. Discontinuation due to an adverse reaction occurred in 5% of patients receiving Lynparza.
Tables 10 and 11 summarize the adverse reactions and laboratory abnormalities in OlympiAD.
Table 10 Adverse Reactions Graded according to NCI CTCAE v4.0. in OlympiAD (‚Č•20% of Patients Who Received Lynparza)
Adverse Reaction
Lynparza tablets
n=205
Chemotherapy
n=91
Grades 1-4
(%)
Grades 3-4
(%)
Grades 1-4
(%)
Grades 3-4
(%)
Gastrointestinal Disorders
     Nausea
58
0
35
1
     Vomiting
30
0
15
1
     Diarrhea
21
1
22
0
Blood and Lymphatic Disorders
     AnemiaRepresents grouped terms consisting of anemia (anemia erythropenia, hematocrit decreased, hemoglobin decreased, and red blood cell count decreased).
40
16
26
4
     NeutropeniaRepresents grouped terms consisting of neutropenia (febrile neutropenia, granulocyte count decreased, granulocytopenia, neutropenia, neutropenic infection, neutropenic sepsis, and neutrophil count decreased).
27
9
50
26
     LeukopeniaRepresents grouped terms consisting of leukopenia (leukopenia and white blood cell count decreased).
25
5
31
13
General Disorders and Administration Site Conditions
     Fatigue (including asthenia)
37
4
36
1
Infections and Infestations
     Respiratory tract infectionRepresents grouped terms consisting of bronchitis, influenza, lower respiratory tract infection, nasopharyngitis, pharyngitis, respiratory tract infection, rhinitis, sinusitis, upper respiratory tract infection, and upper respiratory tract infection bacterial.
27
1
22
0
Nervous System Disorders
     Headache
20
1
15
2
Clinically relevant adverse reactions that occurred in <20% of patients receiving Lynparza were cough (18%), decreased appetite (16%), thrombocytopenia (11%), dysgeusia (9%), lymphopenia (8%), dyspepsia (8%), dizziness (7%), stomatitis (7%), upper abdominal pain (7%), rash (5%), increase in serum creatinine (3%), dermatitis (1%), and VTE (1%).
Table 11 Laboratory Abnormalities Reported in ‚Č•25% of Patients in OlympiAD
Laboratory ParameterPatients were allowed to enter clinical studies with laboratory values of CTCAE Grade 1.
Lynparza tablets
nThis number represents the safety population. The derived values in the table are based on the total number of evaluable patients for each laboratory parameter. = 205
Chemotherapy
n= 91
Grades 1-4
(%)
Grades 3-4
(%)
Grades 1-4
(%)
Grades 3-4
(%)
Decrease in hemoglobin
82
17
66
3
Decrease in lymphocytes
73
21
63
3
Decrease in leukocytes
71
8
70
23
Increase in mean corpuscular volumeRepresents the proportion of subjects whose mean corpuscular volume was > ULN.
71
-
33
-
Decrease in absolute neutrophil count
46
11
65
38
Decrease in platelets
33
3
28
0
First-line Maintenance Treatment of Germline BRCA-mutated Metastatic Pancreatic Adenocarcinoma
POLO
The safety of Lynparza as maintenance treatment of germline BRCA-mutated metastatic pancreatic adenocarcinoma following first-line treatment with platinum-based chemotherapy was evaluated in POLO [see Clinical Studies (14.6) ]. Patients received Lynparza tablets 300 mg orally twice daily (n=90) or placebo (n=61) until disease progression or unacceptable toxicity. Among patients receiving Lynparza, 34% were exposed for 6 months or longer and 25% were exposed for greater than one year.
Among patients who received Lynparza, dosage interruptions due to an adverse reaction of any grade occurred in 35% and dosage reductions due to an adverse reaction occurred in 17%. The most frequent adverse reactions leading to dosage interruption or reduction in patients who received Lynparza were anemia (11%), vomiting (5%), abdominal pain (4%), asthenia (3%), and fatigue (2%). Discontinuation due to adverse reactions occurred in 6% of patients receiving Lynparza. The most frequent adverse reaction that led to discontinuation of Lynparza was fatigue (2.2%).
Tables 12 and 13 summarize the adverse reactions and laboratory abnormalities in patients in POLO.
Table 12 Adverse Reactions Graded according to NCI CTCAE, version 4.0. in POLO (Occurring in ‚Č•10% of Patients who Received Lynparza)Adverse Reaction Lynparza tablets (n=91) This number represents the safety population. The derived values in the table are based on the total number of evaluable patients for each laboratory parameter. Placebo (n=60) All Grades (%) Grades 3 ‚Äď 4 (%) All Grades (%) Grades 3 ‚Äď 4 (%)
General Disorders and Administration Site Conditions
     FatigueIncludes asthenia and fatigue.
60
5
35
2
Gastrointestinal Disorders
     Nausea
45
0
23
2
     Abdominal painIncludes abdominal pain, abdominal pain upper, and abdominal pain lower.
34
2
37
5
     Diarrhea
29
0
15
0
     Constipation
23
0
10
0
     Vomiting
20
1
15
2
     StomatitisIncludes stomatitis and mouth ulceration.
10
0
5
0
Blood and Lymphatic System Disorders
     Anemia
27
11
17
3
     ThrombocytopeniaIncludes platelets count decreased and thrombocytopenia.
14
3
7
0
     NeutropeniaIncludes neutropenia, febrile neutropenia, and neutrophil count decreased.
12
4
8
3
Metabolism and Nutrition Disorders
     Decreased appetite
25
3
7
0
Musculoskeletal and Connective Tissue Disorders
     Back pain
19
0
17
2
     Arthralgia
15
1
10
0
Skin and Subcutaneous Tissue Disorder
     RashIncludes rash erythematous, rash macular, and rash maculo-papular.
15
0
5
0
Respiratory, Thoracic and Mediastinal Disorders
     DyspneaIncludes dyspnea and dyspnea exertional.
13
0
5
2
Infections and Infestations
     Nasopharyngitis
12
0
3
0
Nervous System Disorders
     Dysgeusia
11
0
5
0
Clinically relevant adverse reactions that occurred in <10% of patients receiving Lynparza were cough (9%), abdominal pain upper (7%), blood creatinine increased (7%), dizziness (7%), headache (7%), dyspepsia (5%), leukopenia (5%), VTE (3%), hypersensitivity (2%), and lymphopenia (2%).
Table 13 Laboratory Abnormalities Reported in ‚Č•25% of Patients in POLO
Laboratory
ParameterPatients were allowed to enter POLO with hemoglobin ‚Č•9 g/dL (CTCAE Grade 2) and other laboratory values of CTCAE Grade 1.
Lynparza tablets
nThis number represents the safety population. The derived values in the table are based on the total number of evaluable patients for each laboratory parameter. =91
Placebo
n=60
Grades 1-4
(%)
Grades 3-4
(%)
Grades 1-4 (%)
Grades 3-4
(%)
Increase in serum creatinine
99
2
85
0
Decrease in hemoglobin
86
11
65
0
Increase in mean corpuscular volumeRepresents the proportion of subjects whose mean corpuscular volume was > ULN.
71
-
30
-
Decrease in lymphocytes
61
9
27
0
Decrease in platelets
56
2
39
0
Decrease in leukocytes
50
3
23
0
Decrease in absolute neutrophil count
25
3
10
0
HRR Gene-mutated Metastatic Castration-Resistant Prostate Cancer
PROfound
The safety of Lynparza as monotherapy was evaluated in patients with mCRPC and HRR gene mutations who have progressed following prior treatment with enzalutamide or abiraterone in PROfound [see Clinical Studies (14.7) ]. This study was a randomized, open-label, multi-center study in which 386 patients received either Lynparza tablets 300 mg orally twice daily (n=256) or investigator’s choice of enzalutamide or abiraterone acetate (n=130) until disease progression or unacceptable toxicity. Among patients receiving Lynparza, 62% were exposed for 6 months or longer and 20% were exposed for greater than one year.
Fatal adverse reactions occurred in 4% of patients treated with Lynparza. These included pneumonia (1.2%), cardiopulmonary failure (0.4%), aspiration pneumonia (0.4%), intestinal diverticulum (0.4%), septic shock (0.4%), Budd-Chiari Syndrome (0.4%), sudden death (0.4%), and acute cardiac failure (0.4%).
Serious adverse reactions occurred in 36% of patients receiving Lynparza. The most frequent serious adverse reactions (‚Č•2%) were anemia (9%), pneumonia (4%), pulmonary embolism (2%), fatigue/asthenia (2%), and urinary tract infection (2%).
Dose interruptions due to an adverse reaction of any grade occurred in 45% of patients receiving Lynparza; dose reductions due to an adverse reaction occurred in 22% of Lynparza patients. The most frequent adverse reactions leading to dose interruption of Lynparza were anemia (25%) and thrombocytopenia (6%) and the most frequent adverse reaction leading to reduction of Lynparza was anemia (16%). Discontinuation due to adverse reactions occurred in 18% of Lynparza. The adverse reaction that most frequently led to discontinuation of Lynparza was anemia (7%).
Tables 14 and 15 summarize the adverse reactions and laboratory abnormalities, respectively, in patients in PROfound.
Table 14 Adverse Reactions Graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE), version 4.03. Reported in ‚Č•10% of Patients in PROfound
Adverse Reactions
Lynparza tablets
n=256
 
Enzalutamide or abiraterone
n=130
 
Grades 1-4
(%)
Grades 3-4
(%)
Grades 1-4
(%)
Grades 3-4
(%)
Blood and lymphatic disorders
    AnemiaIncludes anemia and hemoglobin decreased.
46
21
15
5
    ThrombocytopeniaIncludes platelet count decreased and thrombocytopenia.
12
4
3
0
Gastrointestinal disorders
    Nausea
41
1
19
0
    Diarrhea
21
1
7
0
    Vomiting
18
2
12
1
General disorders and administration site conditions
    Fatigue (including asthenia)
41
3
32
5
Metabolism and nutrition disorders
    Decreased appetite
30
1
18
1
Respiratory, thoracic, and mediastinal disorders
    Cough
11
0
2
0
    Dyspnea
10
2
3
0
Clinically relevant adverse reactions that occurred in <10% of patients receiving Lynparza were neutropenia (9%), VTE (7%), dizziness (7%), dysgeusia (7%), dyspepsia (7%), headache (6%), pneumonia (5%), stomatitis (5%), rash (4%), blood creatinine increase (4%), pneumonitis (2%), upper abdominal pain (2%), and hypersensitivity (1%).
Table 15 Laboratory Abnormalities Reported in ‚Č•25% of Patients in PROfound
LaboratoryParameterPatients were allowed to enter clinical studies with laboratory values of CTCAE Grade 1.
Lynparza tablets
nThis number represents the safety population. The derived values in the table are based on the total number of evaluable patients for each laboratory parameter. = 256
Enzalutamide or abiraterone
n=130
Grades 1-4
(%)
Grades 3-4
(%)
Grades 1-4
(%)
Grades 3-4
(%)
Decrease in hemoglobin
98
13
73
4
Decrease in lymphocytes
62
23
34
13
Decrease in leukocytes
53
4
21
0
Decrease in absolute neutrophil count
34
3
9
0
Treatment of BRCA-mutated Metastatic Castration-Resistant Prostate Cancer in Combination with Abiraterone and Prednisone or Prednisolone
PROpel
The safety of Lynparza in combination with abiraterone and prednisone or prednisolone for the treatment of patients in the first-line mCRPC setting was investigated in PROpel [see Clinical Studies (14.8)]. Patients were randomized to receive either Lynparza tablets 300 mg orally twice daily plus abiraterone tablets 1000 mg once daily (Lynparza/abiraterone) (n=398), or placebo plus abiraterone 1000 mg once daily (placebo/abiraterone) (n=396) until disease progression or unacceptable toxicity. Patients in both arms also received either prednisone or prednisolone 5 mg twice daily.
Fatal adverse reactions occurred in 6% of patients, including COVID-19 (3%) and pneumonias (0.5%).
Serious adverse reactions occurred in 39% of patients. Serious adverse reactions reported in > 2% of patients included anemia (6%), COVID-19 (6%), pneumonia (4.5%), pulmonary embolism (3.5%), and urinary tract infection (3%).
Permanent discontinuation of Lynparza due to adverse reactions occurred in 16% of patients treated in the Lynparza with abiraterone arm. The most common adverse reactions which resulted in permanent discontinuation of Lynparza were anemia (4.3%) and pneumonia (1.5%).
Dosage interruption of Lynparza due to adverse reactions occurred in 48% of patients treated in the Lynparza with abiraterone arm. The most common (>2%) adverse reactions requiring dosage interruption of Lynparza were anemia (16%), COVID-19 (6%) fatigue (3.5%), nausea (2.8%), pulmonary embolism (2.3%), and diarrhea (2.3%).
Dose reduction of Lynparza due to adverse reactions occurred in 21% of patients treated in the Lynparza with abiraterone arm. The most common (>2%) adverse reactions requiring dosage reductions of Lynparza were anemia (11%) and fatigue (2.5%).
The most common adverse reactions (‚Č•10%) in patients who received Lynparza/abiraterone were anemia (48%), fatigue (38%), nausea (30%), diarrhea (19%), decreased appetite (16%), lymphopenia (14%), abdominal pain (13%), and dizziness (14%).
Tables 16 and 17 summarize adverse reactions and laboratory abnormalities in PROpel, respectively.
Table 16 Adverse Reactions (‚Č•10%) in Patients Who Received Lynparza (with a Difference of ‚Č•5% Compared to Placebo) in PROpel
Adverse ReactionsGraded according to the National Cancer Institute Common Terminology Criteria for Adverse Events (NCI CTCAE), version 4.03.
Lynparza/abirateronen=398
Placebo/abirateronen=396
Grades 1-4
(%)
Grades 3-4
(%)
Grades 1-4
(%)
Grades 3-4
(%)
Blood and Lymphatic Disorders
         AnemiaIncludes anemia, anemia macrocytic, and red blood cell count decreased
48
16
18
3.3
         LymphopeniaIncludes lymphocyte count decreased and lymphopenia
14
5
6
1.8
General Disorders and Administration Site Conditions
         Fatigue (including asthenia)
38
2.3
30
1.5
Gastrointestinal Disorders
         Nausea
30
0.3
14
0.3
         Diarrhea
19
1
10
0.3
         Abdominal painIncludes abdominal discomfort, abdominal pain, abdominal pain upper, and abdominal pain lower
13
0
7
0.5
Metabolism and nutrition disorders
         Decreased appetite
16
1
7
0
Nervous System Disorders
         DizzinessIncludes dizziness and vertigo.
14
0.3
7
0
Clinically relevant adverse reactions that occurred in <10% for patients receiving Lynparza plus abiraterone were headache (9%), VTE (8%), rash (7%), dysgeusia (6%), acute kidney injury (3%), and stomatitis (2.5%).
Table 17 Selected Laboratory Abnormalities Reported in ‚Č•20% of Patients in PROpel
Laboratory Parameter
Lynparza/abirateronen=398This number represents the safety population. The derived values in the table are based on the total number of evaluable patients for each laboratory parameter.
Placebo/abiraterone n=396
Grades 1-4
(%)
Grades 3-4
(%)
Grades 1-4
(%)
Grades 3-4
(%)
Decrease in hemoglobin
97
12
81
1.3
Decrease in lymphocytes
70
23
49
11
Decrease in platelets
23
1.2
20
0.3
Decrease in absolute neutrophil count
23
5
6
0
6.2 Postmarketing Experience
The following adverse reactions have been identified during post-approval use of Lynparza. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Immune System Disorders: Hypersensitivity including angioedema.
Skin and subcutaneous tissue disorders: Erythema nodosum, rash, dermatitis.
7 Drug Interactions
‚ÄĘ Strong or moderate CYP3A inhibitors: Avoid concomitant use. If concomitant use cannot be avoided, reduce Lynparza dosage. (2.4 ,7.2 ,12.3 )‚ÄĘ Strong or moderate CYP3A inducers: Avoid concomitant use. (7.2 ,12.3 )7.1 Use with Anticancer Agents
Clinical studies of Lynparza with other myelosuppressive anticancer agents, including DNA damaging agents, indicate a potentiation and prolongation of myelosuppressive toxicity.
7.2 Effect of Other Drugs on Lynparza
Strong and Moderate CYP3A Inhibitors
Coadministration of CYP3A inhibitors can increase olaparib concentrations, which may increase the risk for adverse reactions [see Clinical Pharmacology (12.3)]. Avoid coadministration of strong or moderate CYP3A inhibitors. If the strong or moderate inhibitor must be coadministered, reduce the dose of Lynparza [see Dosage and Administration (2.4)].
Strong and Moderate CYP3A Inducers
Concomitant use with a strong or moderate CYP3A inducer decreased olaparib exposure, which may reduce Lynparza efficacy [see Clinical Pharmacology (12.3)]. Avoid coadministration of strong or moderate CYP3A inducers.
8 Use In Specific Populations
Lactation: Advise women not to breastfeed.(8.2)
8.1 Pregnancy
Risk Summary
Based on findings in animals and its mechanism of action [see Clinical Pharmacology (12.1)], Lynparza can cause fetal harm when administered to a pregnant woman. There are no available data on Lynparza use in pregnant women to inform the drug-associated risk. In an animal reproduction study, the administration of olaparib to pregnant rats during the period of organogenesis caused teratogenicity and embryo-fetal toxicity at exposures below those in patients receiving the recommended human dose of 300 mg twice daily (see Data). Apprise pregnant women of the potential hazard to the fetus and the potential risk for loss of the pregnancy.
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. The estimated background risk in the U.S. general population of major birth defects is 2-4%; and the risk for spontaneous abortion is approximately 15-20% in clinically recognized pregnancies.
Data
Animal Data
In a fertility and early embryonic development study in female rats, olaparib was administered orally for 14 days before mating through to Day 6 of pregnancy, which resulted in increased post-implantation loss at a dose level of 15 mg/kg/day (with maternal systemic exposures approximately 7% of the human exposure (AUC0-24h) at the recommended dose).
In an embryo-fetal development study, pregnant rats received oral doses of 0.05 and 0.5 mg/kg/day olaparib during the period of organogenesis. A dose of 0.5 mg/kg/day (with maternal systemic exposures approximately 0.18% of human exposure (AUC0-24h) at the recommended dose) caused embryo-fetal toxicities including increased post-implantation loss and major malformations of the eyes (anophthalmia, microphthalmia), vertebrae/ribs (extra rib or ossification center; fused or absent neural arches, ribs, and sternebrae), skull (fused exoccipital), and diaphragm (hernia). Additional abnormalities or variants included incomplete or absent ossification (vertebrae/sternebrae, ribs, limbs) and other findings in the vertebrae/sternebrae, pelvic girdle, lung, thymus, liver, ureter, and umbilical artery. Some findings noted above in the eyes, ribs, and ureter were observed at a dose of 0.05 mg/kg/day olaparib at lower incidence.
8.2 Lactation
Risk Summary
No data are available regarding the presence of olaparib in human milk, or on its effects on the breastfed infant or on milk production. Because of the potential for serious adverse reactions in the breastfed infants from Lynparza, advise a lactating woman not to breastfeed during treatment with Lynparza and for one month after receiving the last dose.
8.3 Females and Males of Reproductive Potential
Lynparza can cause fetal harm when administered to a pregnant woman [see Use in Specific Populations (8.1)].
Pregnancy Testing
Verify pregnancy status in females of reproductive potential prior to initiating treatment with Lynparza.
Contraception
Females
Advise females of reproductive potential to use effective contraception during treatment with Lynparza and for 6 months following the last dose.
Males
Based on findings in genetic toxicity and animal reproduction studies, advise male patients with female partners of reproductive potential or who are pregnant to use effective contraception during treatment and for 3 months following the last dose of Lynparza. Advise male patients not to donate sperm during therapy and for 3 months following the last dose of Lynparza [see Use in Specific Populations (8.1) and Nonclinical Toxicology (13.1)].
8.4 Pediatric Use
Safety and effectiveness of Lynparza have not been established in pediatric patients.
8.5 Geriatric Use
Of the 2901 patients with advanced solid tumors who received Lynparza as a single agent, 680 (23%) patients were aged ‚Č•65 years, and this included 206 (7%) patients who were aged ‚Č•75 years. Thirteen (0.4%) patients were aged ‚Č•85 years.
Of the 535 patients with advanced solid tumors who received Lynparza tablets 300 mg orally twice daily in combination with bevacizumab (PAOLA-1), 204 (38%) patients were aged ‚Č•65 years, and this included 31 (6%) patients who were aged ‚Č•75 years.
Of the 398 patients with advanced solid tumors who received Lynparza tablets 300 mg orally twice daily in combination with abiraterone and prednisone or prednisolone (PROpel), 268 (67%) patients were aged ‚Č•65 years, and this included 95 (24%) patients who were aged ‚Č•75 years.
No overall differences in the safety or effectiveness of Lynparza were observed between these patients and younger patients.
8.6 Renal Impairment
No dosage modification is recommended in patients with mild renal impairment (CLcr 51 to 80 mL/min estimated by Cockcroft-Gault). Reduce Lynparza dosage to 200 mg twice daily in patients with moderate renal impairment (CLcr 31 to 50 mL/min) [see Dosage and Administration (2.5)]. There are no data in patients with severe renal impairment or end-stage disease (CLcr ‚ȧ30 mL/min) [see Clinical Pharmacology (12.3)].
8.7 Hepatic Impairment
No adjustment to the starting dose is required in patients with mild or moderate hepatic impairment (Child-Pugh classification A and B). There are no data in patients with severe hepatic impairment (Child-Pugh classification C) [see Clinical Pharmacology (12.3)].
11 Description
Olaparib is a poly (ADP-ribose) polymerase (PARP) inhibitor. The chemical name is 4-[(3-{[4-(cyclopropylcarbonyl)piperazin-1-yl]carbonyl}-4-fluorophenyl)methyl]phthalazin-1(2H)-one. The empirical molecular formula for Lynparza is C24H23FN4O3 and the relative molecular mass is 434.46. It has the following chemical structure:
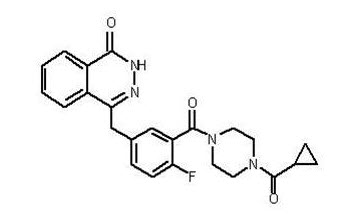
Olaparib is a crystalline solid, is non-chiral and shows pH-independent low solubility across the physiological pH range.
Lynparza (olaparib) tablets for oral use contain 100 mg or 150 mg of olaparib. Inactive ingredients in the tablet core are copovidone, mannitol, colloidal silicon dioxide, and sodium stearyl fumarate. The tablet coating consists of hypromellose, polyethylene glycol 400, titanium dioxide, ferric oxide yellow, and ferrosoferric oxide (150 mg tablet only).
12 Clinical Pharmacology
12.1 Mechanism of Action
Olaparib is an inhibitor of poly (ADP-ribose) polymerase (PARP) enzymes, including PARP1, PARP2, and PARP3. PARP enzymes are involved in normal cellular functions, such as DNA transcription and DNA repair. Olaparib has been shown to inhibit growth of select tumor cell lines in vitro and decrease tumor growth in mouse xenograft models of human cancer, both as monotherapy or following platinum-based chemotherapy. Increased cytotoxicity and anti-tumor activity following treatment with olaparib were noted in cell lines and mouse tumor models with deficiencies in BRCA1/2, ATM, or other genes involved in the homologous recombination repair (HRR) of DNA damage and correlated with platinum response. In vitro studies have shown that olaparib-induced cytotoxicity may involve inhibition of PARP enzymatic activity and increased formation of PARP-DNA complexes, resulting in DNA damage and cancer cell death. In prostate cancer models, PARP1 has been shown to contribute to androgen receptor (AR) activity regulation; the combination of olaparib and AR inhibition resulted in cytotoxicity in vitro and anti-tumor activity in mouse xenograft models.
12.2 Pharmacodynamics
Cardiac Electrophysiology
The effect of olaparib on cardiac repolarization was assessed in 119 patients following a single dose of 300 mg and in 109 patients following multiple dosing of 300 mg twice daily. No clinically relevant effect of olaparib on QT interval was observed.
12.3 Pharmacokinetics
The area under the curve (AUC) of olaparib increases approximately proportionally following administration of single doses of 25 mg to 450 mg (0.08 to 1.5 times the recommended dose) and maximal concentrations (Cmax) increased slightly less than proportionally for the same dose range. Olaparib showed time-dependent pharmacokinetics and an AUC mean accumulation ratio of 1.8 is observed at steady state following a dose of 300 mg twice daily.
The mean (CV%) olaparib Cmax is 5.4 őľg/mL (32%) and AUC is 39.2 őľg*h/mL (44%) following a single 300 mg dose. The mean steady state olaparib Cmax and AUC is 7.6 őľg/mL (35%) and 49.2 őľg*h/mL (44%), following a dose of 300 mg twice daily.
Absorption
Following oral administration of olaparib, the median time to peak plasma concentration is 1.5 hours.
Effect of Food
Co-administration of a high fat and high calorie meal (800-1000 kcal, 50% of the calorie content made up from fat) with olaparib slowed the rate (tmax delayed by 2.5 hours) of absorption, but did not significantly alter the extent of olaparib absorption (mean AUC increased by approximately 8%).
Distribution
The mean (¬Ī standard deviation) apparent volume of distribution of olaparib is 158 ¬Ī 136 L following a single 300 mg dose of Lynparza. The protein binding of olaparib is approximately 82% in vitro.
Elimination
The mean (¬Ī standard deviation) terminal plasma half-life of olaparib is 14.9 ¬Ī 8.2 hours and the apparent plasma clearance is 7.4 ¬Ī 3.9 L/h following a single 300 mg dose of Lynparza.
Metabolism
Olaparib is metabolized by cytochrome P450 (CYP) 3A in vitro.
Following an oral dose of radiolabeled olaparib to female patients, unchanged olaparib accounted for 70% of the circulating radioactivity in plasma. It was extensively metabolized with unchanged drug accounting for 15% and 6% of radioactivity in urine and feces, respectively. The majority of the metabolism is attributable to oxidation reactions with a number of the components produced undergoing subsequent glucuronide or sulfate conjugation.
Excretion
Following a single dose of radiolabeled olaparib, 86% of the dosed radioactivity was recovered within a 7-day collection period, 44% via the urine and 42% via the feces. The majority of the material was excreted as metabolites.
Specific Populations
Patients with Renal Impairment
In a renal impairment trial, the mean AUC increased by 24% and Cmax by 15%, when olaparib was dosed in patients with mild renal impairment (CLcr=51-80 mL/min defined by the Cockcroft-Gault equation; n=13) and by 44% and 26%, respectively, when olaparib was dosed in patients with moderate renal impairment (CLcr=31-50 mL/min; n=13), compared to those with normal renal function (CLcr ‚Č•81 mL/min; n=12). There was no evidence of a relationship between the extent of plasma protein binding of olaparib and creatinine clearance. There are no data in patients with severe renal impairment or end-stage renal disease (CLcr ‚ȧ30 mL/min).
Patients with Hepatic Impairment
In a hepatic impairment trial, the mean AUC increased by 15% and the mean Cmax increased by 13% when olaparib was dosed in patients with mild hepatic impairment (Child-Pugh classification A; n=10) and the mean AUC increased by 8% and the mean Cmax decreased by 13% when olaparib was dosed in patients with moderate hepatic impairment (Child-Pugh classification B; n=8), compared to patients with normal hepatic function (n=13). Hepatic impairment has no effect on the protein binding of olaparib and, therefore, total plasma exposure was representative of free drug. There are no data in patients with severe hepatic impairment (Child-Pugh classification C).
Drug Interaction Studies
Clinical Studies
CYP3A Inhibitors: Concomitant use of itraconazole (strong CYP3A inhibitor) increased olaparib Cmax by 42% and AUC by 170%. Concomitant use of fluconazole (moderate CYP3A inhibitor) is predicted to increase olaparib Cmax by 14% and AUC by 121%.
CYP3A Inducers: Concomitant use of rifampicin (strong CYP3A inducer) decreased olaparib Cmax by 71% and AUC by 87%. Concomitant use of efavirenz (moderate CYP3A inducer) is predicted to decrease olaparib Cmax by 31% and AUC by 60%.
In vitro Studies
CYP Enzymes: Olaparib is both an inhibitor and inducer of CYP3A and an inducer of CYP2B6. Olaparib is predicted to be a weak CYP3A inhibitor in humans.
UGT Enzymes: Olaparib is an inhibitor of UGT1A1.
Transporters: Olaparib is an inhibitor of BCRP, OATP1B1, OCT1, OCT2, OAT3, MATE1, and MATE2K. Olaparib is a substrate and inhibitor of the efflux transporter P-gp. The potential for olaparib to induce P-gp has not been evaluated.
13 Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies have not been conducted with olaparib.
Olaparib was clastogenic in an in vitro chromosomal aberration assay in mammalian Chinese hamster ovary (CHO) cells and in an in vivo rat bone marrow micronucleus assay. This clastogenicity is consistent with genomic instability resulting from the primary pharmacology of olaparib and indicates potential for genotoxicity in humans. Olaparib was not mutagenic in a bacterial reverse mutation (Ames) test.
In a fertility study, female rats received oral olaparib at doses of 0.05, 0.5, and 15 mg/kg/day for at least 14 days before mating through the first week of pregnancy. There were no adverse effects on mating and fertility rates at doses up to 15 mg/kg/day (maternal systemic exposures approximately 7% of the human exposure (AUC0-24h) at the recommended dose).
In a male fertility study, olaparib had no effect on mating and fertility in rats at oral doses up to 40 mg/kg/day following at least 70 days of olaparib treatment (with systemic exposures of approximately 5% of the human exposure (AUC0-24h) at the recommended dose).
14 Clinical Studies
14.1 First-Line Maintenance Treatment of -mutated Advanced Ovarian Cancer
The efficacy of Lynparza was evaluated in SOLO-1 (NCT01844986), a randomized (2:1), double-blind, placebo-controlled, multi-center trial in patients with BRCA-mutated advanced ovarian, fallopian tube, or primary peritoneal cancer following first-line platinum-based chemotherapy. Patients were randomized to receive Lynparza tablets 300 mg orally twice daily or placebo. Treatment was continued for up to 2 years or until disease progression or unacceptable toxicity; however, patients with evidence of disease at 2 years, who in the opinion of the treating healthcare provider could derive further benefit from continuous treatment, could be treated beyond 2 years. Randomization was stratified by response to first-line platinum-based chemotherapy (complete or partial response). The major efficacy outcome was investigator-assessed progression-free survival (PFS) evaluated according to Response Evaluation Criteria in Solid Tumors (RECIST), version 1.1.
A total of 391 patients were randomized, 260 to Lynparza and 131 to placebo. The median age of patients treated with Lynparza was 53 years (range: 29 to 82) and 53 years (range: 31 to 84) among patients on placebo. The ECOG performance status (PS) was 0 in 77% of patients receiving Lynparza and 80% of patients receiving placebo. Of all patients, 82% were White, 36% were enrolled in the U.S. or Canada, and 82% were in complete response to their most recent platinum-based regimen. The majority of patients (n=389) had germline BRCA mutation (gBRCAm), and 2 patients had somatic BRCAm (sBRCAm).
Of the 391 patients randomized in SOLO-1, 386 were retrospectively or prospectively tested with a Myriad BRACAnalysis test and 383 patients were confirmed to have deleterious or suspected deleterious gBRCAm status; 253 were randomized to the Lynparza arm and 130 to the placebo arm. Two out of 391 patients randomized in SOLO-1 were confirmed to have sBRCAm based on an investigational Foundation Medicine tissue test.
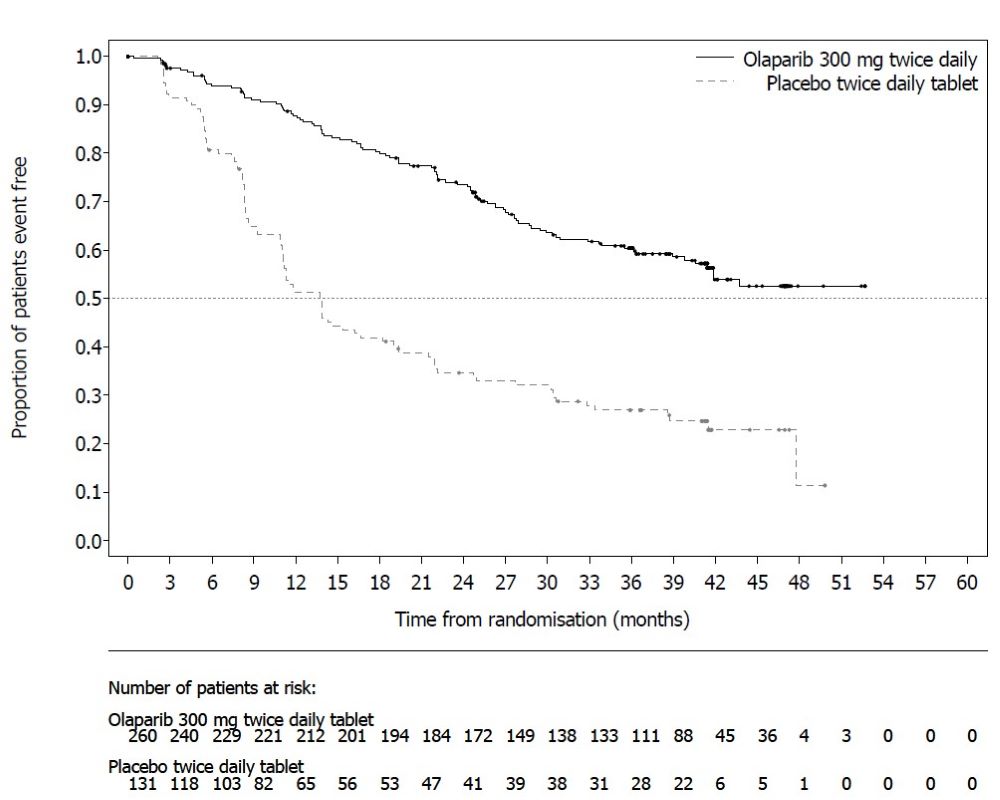
SOLO-1 demonstrated a statistically significant improvement in investigator-assessed PFS for Lynparza compared to placebo. Results from a blinded independent review were consistent. At the time of the analysis of PFS, overall survival (OS) data were not mature (21% of patients had died). Efficacy results are presented in Table 18 and Figure 1.
Table 18 Efficacy Results - SOLO-1 (Investigator Assessment)
Lynparza tablets
(n=260)
Placebo
(n=131)
Progression-Free SurvivalMedian follow-up of 41 months in both treatment arms.
     Number of events (%)
102 (39%)
96 (73%)
     Median, months
NR
13.8
     Hazard ratioA value <1 favors Lynparza. Hazard ratio from a Cox proportional hazards model including response to previous platinum chemotherapy (complete response versus partial response) as a covariate. (95% CI)
0.30 (0.23, 0.41)
     p-valueThe p-value is derived from a stratified log-rank test.
<0.0001
NR not reached; CI Confidence Interval.
Figure 1 Kaplan-Meier Curves of Investigator-Assessed Progression-Free Survival - SOLO-1
14.2 First-line Maintenance Treatment of HRD-positive Advanced Ovarian Cancer in Combination with Bevacizumab
PAOLA-1 (NCT02477644) was a randomized, double-blind, placebo-controlled, multi-center trial that compared the efficacy of Lynparza in combination with bevacizumab versus placebo/bevacizumab for the maintenance treatment of advanced high-grade epithelial ovarian cancer, fallopian tube or primary peritoneal cancer following first-line platinum-based chemotherapy and bevacizumab. Randomization was stratified by first-line treatment outcome (timing and outcome of cytoreductive surgery and response to platinum-based chemotherapy) and tBRCAm status, determined by prospective local testing. All available clinical samples were retrospectively tested with Myriad myChoice¬ģ CDx. Patients were required to have no evidence of disease (NED) due to complete surgical resection, or who were in complete response (CR), or partial response (PR) following completion of first-line platinum-containing chemotherapy and bevacizumab. Patients were randomized (2:1) to receive Lynparza tablets 300 mg orally twice daily in combination with bevacizumab (n=537) 15 mg/kg every three weeks or placebo/bevacizumab (n=269). Patients continued bevacizumab in the maintenance setting and started treatment with Lynparza after a minimum of 3 weeks and up to a maximum of 9 weeks following completion of their last dose of chemotherapy. Lynparza treatment was continued for up to 2 years or until progression of the underlying disease or unacceptable toxicity. Patients who in the opinion of the treating physician could derive further benefit from continuous treatment could be treated beyond 2 years. Treatment with bevacizumab was for a total of up to 15 months, including the period given with chemotherapy and given as maintenance.
The major efficacy outcome measure was investigator-assessed PFS evaluated according to RECIST, version 1.1. An additional efficacy endpoint was overall survival (OS).
A statistically significant difference in PFS was observed in the intent-to-treat (ITT) population. Planned exploratory analyses of PFS and OS were conducted in patients with known HRD status. The PFS hazard ratio (HR) for patients with HRD-negative tumors (277/806; 34%) was 1.00 (95% CI: 0.75, 1.34) and the OS HR was 1.18 (95% CI: 0.87, 1.60) indicating that the clinical benefit was primarily attributed to the results seen in the HRD-positive population. Efficacy results in patients with HRD-positive tumors are summarized in Table 19, Figure 2, and Figure 3.
Among the 387 patients (48%) with HRD-positive tumors identified post-randomization using the Myriad myChoice¬ģ HRD Plus tumor test the median age was 58 years in both arms (range 32-82). Ovarian cancer was the primary tumor type in 87% of patients in both arms. Ninety five percent (95%) were serous histological type. The ECOG performance score was 0 in 75% of patients and 1 in 24% of patients. All patients had received first-line platinum-based therapy and bevacizumab. First-line treatment outcomes at screening indicated that patients had no evidence of disease with complete macroscopic resection at initial debulking surgery (36%), no evidence of disease/CR with complete macroscopic resection at interval debulking surgery (29%, both arms), no evidence of disease/CR in patients who had either incomplete resection (at initial or interval debulking surgery) or no debulking surgery (16%, both arms) and patients with a partial response (19%, both arms). Sixty-two (62%) of patients in the Lynparza/bevacizumab arm and 58% of patients in placebo/bevacizumab arm had tumors with a deleterious BRCA mutation. Patients were not restricted by the surgical outcome with 67% having complete cytoreduction at initial or interval debulking surgery and 33% having residual macroscopic disease.
Table 19 Efficacy Results - PAOLA-1 (HRD-positive status, Investigator Assessment)
Lynparza/bevacizumab
(n=255)
Placebo/bevacizumab
(n=132)
Progression-Free SurvivalResults from a blinded independent review of PFS were consistent with those from investigator-assessed PFS
     Number of events (%)
87 (34%)
92 (70%)
     Median, months
37.2
17.7
     Hazard ratioThe analysis was performed using an unstratified Cox proportional hazards model. (95% CI)
0.33 (0.25, 0.45)
Overall SurvivalBased on final OS subgroup analysis.
     Number of events (%)
93 (36%)
69 (52%)
     Median, months
75.2
57.3
     Hazard ratio† (95% CI)
0.62 (0.45, 0.85)
CI Confidence interval.
Figure 2 Kaplan-Meier Curves of Investigator-Assessed Progression-Free Survival ‚Äď PAOLA-1 (HRD-positive status)
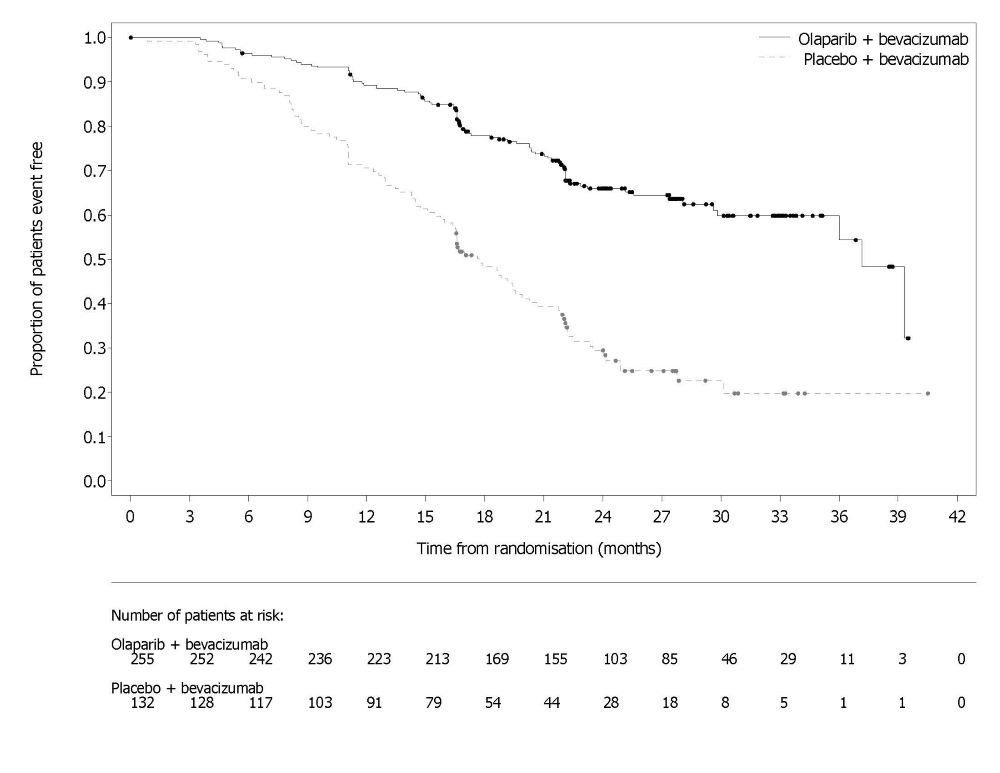
Figure 3 Kaplan-Meier Curves of Overall Survival ‚Äď PAOLA-1 (HRD-positive status)*
*             Based on final OS subgroup analysis.
14.3 Maintenance Treatment of -mutated Recurrent Ovarian Cancer
The efficacy of Lynparza was evaluated in SOLO-2 (NCT01874353), a randomized (2:1) double-blind, placebo-controlled trial in patients with BRCAm ovarian, fallopian tube, or primary peritoneal cancer who were in response to platinum-based therapy. Patients were randomized to Lynparza tablets 300 mg orally twice daily or placebo until unacceptable toxicity or progressive disease. Randomization was stratified by response to last platinum chemotherapy (complete versus partial) and time to disease progression in the penultimate platinum-based chemotherapy prior to enrollment (6-12 months versus >12 months). All patients had received at least two prior platinum-containing regimens and were in response (complete or partial) to their most recent platinum-based regimen. The major efficacy outcome measure was investigator-assessed PFS evaluated according to RECIST, version 1.1. An additional efficacy outcome measure was OS.
A total of 295 patients were randomized, 196 to Lynparza and 99 to placebo. The median age of patients treated with Lynparza was 56 years (range: 28 to 83) and 56 years (range: 39 to 78) among patients treated with placebo. The ECOG PS was 0 in 83% of patients receiving Lynparza and 78% of patients receiving placebo. Of all patients, 89% were White, 17% were enrolled in the U.S. or Canada, 47% were in complete response to their most recent platinum-based regimen, and 40% had a progression-free interval of 6-12 months since their penultimate platinum regimen. Prior bevacizumab therapy was reported for 17% of those treated with Lynparza and 20% of those receiving placebo. Approximately 44% of patients on the Lynparza arm and 37% on placebo had received three or more lines of platinum-based treatment. All (100%) patients enrolled had gBRCA mutations.
All patients had a deleterious or suspected deleterious germline BRCA mutation as detected either by a local test (n=236) or central Myriad CLIA test (n=59), subsequently confirmed by BRACAnalysis CDx¬ģ (n=286).
SOLO-2 demonstrated a statistically significant improvement in investigator-assessed PFS in patients randomized to Lynparza as compared with placebo. Results from a blinded independent review were consistent. The final analysis of OS did not reach statistical significance. Efficacy results are presented in Table 20 and Figures 4 and 5.
Table 20 Efficacy Results - SOLO-2 (Investigator Assessment) Lynparza tablets(n=196) Placebo(n=99)
Progression-Free Survival
       Number of events (%)
107 (55%)
80 (81%)
     Median, months
19.1
5.5
     Hazard ratioHazard ratio from a Cox proportional hazards model including response to last platinum chemotherapy (complete response versus partial response) and time to disease progression in the penultimate platinum-based chemotherapy prior to enrollment (6-12 month versus >12 months) as covariates. (95% CI)
0.30 (0.22, 0.41)
      p-valueThe p-value is derived from a stratified log-rank test.
<0.0001
Overall Survival
Number of events (%)
116 (59)
65 (66)
     Median, months
51.7
38.8
     Hazard ratio(95% CI)
0.74 (0.54, 1.00)
      p-value
0.0537
Figure 4 Kaplan-Meier Curves of Investigator-Assessed Progression-Free Survival ‚Äď SOLO-2
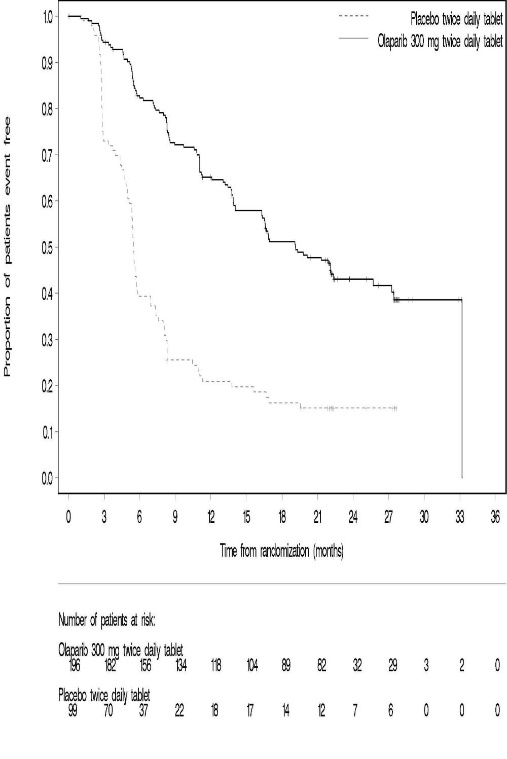
Figure 5 Kaplan-Meier Curves of Overall Survival ‚Äď SOLO-2
14.4 Adjuvant Treatment of Germline -mutated HER2-negative High Risk Early Breast Cancer
The efficacy of Lynparza was evaluated in OlympiA (NCT02032823), a randomized (1:1), double-blind, placebo-controlled, international study in patients with gBRCAm HER2-negative high risk early breast cancer who had completed definitive local treatment and neoadjuvant or adjuvant chemotherapy. Patients were randomized to receive Lynparza tablets 300 mg orally twice daily or placebo. Treatment was continued for up to 1 year, or until disease recurrence, or unacceptable toxicity. Patients were required to have completed at least 6 cycles of neoadjuvant or adjuvant chemotherapy containing anthracyclines, taxanes or both. Prior platinum for previous cancer (e.g., ovarian) or as adjuvant or neoadjuvant treatment for breast cancer was allowed. Patients with high-risk early breast cancer were defined as follows:
‚ÄĘ patients who received prior neoadjuvant chemotherapy: patients with either triple negative breast cancer (TNBC) or hormone receptor positive breast cancer must have had residual invasive cancer in the breast and/or the resected lymph nodes (non-pathologic complete response) at the time of surgery. Additionally, patients with hormone receptor positive breast cancer must have had a score of ‚Č•3 based on pre-treatment clinical and post-treatment pathologic stage (CPS), estrogen receptor (ER) status, and histologic grade as shown in Table 21.
Table 21 Early Breast Cancer Stage, Receptor Status, and Grade Scoring Requirements for Study Enrollment Total score of ‚Č•3 required for patients with hormone receptor positive breast cancer. Stage/feature Points
Clinical Stage
(pre-treatment)
I/IIA
0
IIB/IIIA
1
IIIB/IIIC
2
Pathologic Stage (post-treatment)
0/I
0
IIA/IIB/IIIA/IIIB
1
IIIC
2
Receptor status
ER positive
0
ER negative 
1
Nuclear grade
Nuclear grade 1-2
0
Nuclear grade 3
1
‚ÄĘ patients who received prior adjuvant chemotherapy: patients with TNBC must have had node positive disease or node negative disease with a ‚Č•2cm primary tumor; patients with hormone receptor positive, HER2-negative breast cancer must have had ‚Č•4 pathologically confirmed positive lymph nodes.
Randomization was stratified by hormone receptor status (hormone receptor positive versus triple negative), by prior neoadjuvant versus adjuvant chemotherapy, and by prior platinum use for breast cancer (yes versus no).
The major efficacy outcome measure was invasive disease free survival (IDFS), defined as the time from randomization to date of first recurrence, where recurrence is defined as invasive loco-regional, distant recurrence, contralateral invasive breast cancer, new cancer or death from any cause. An additional efficacy outcome measure was OS.
A total of 1836 patients were randomized, 921 to Lynparza and 915 to placebo. Demographic and baseline characteristics were well balanced between arms. The median age was 42 years. Sixty-seven percent (67%) of patients were White, 29% were Asian, and 3% were Black. Three percent (3%) of patients were Hispanic or Latino. Two patients (0.2%) in the Lynparza arm and four patients (0.4%) in the placebo arm were male. Sixty-one percent (61%) of patients were pre-menopausal. Eighty-nine percent (89%) of patients were ECOG performance status 0 and 11% ECOG PS 1. Eighty-two percent (82%) of patients had TNBC and 18% had hormone receptor-positive disease. Fifty percent (50%) of patients had received prior neoadjuvant and 50% received prior adjuvant chemotherapy. Ninety-four percent (94%) of patients received anthracycline and taxane chemotherapy. Twenty-six (26%) of patients overall had received prior platinum for breast cancer. Ninety percent (90%) of patients with hormone receptor positive breast cancer received concurrent endocrine therapy.
Patients enrolled based on local gBRCA test results provided a sample for retrospective confirmatory central testing with BRACAnalysis¬ģ. Out of 1836 patients enrolled into OlympiA, 1623 were confirmed as gBRCAm by Myriad BRACAnalysis¬ģ, either prospectively or retrospectively.
A statistically significant improvement in IDFS and OS was demonstrated in patients in the Lynparza arm compared with the placebo arm. Efficacy data for OlympiA (FAS) are presented in Table 22 and Figures 6 and 7.
Table 22 Efficacy Results ‚Äď OlympiA
Lynparza tablets
(N=921)
Placebo
(N=915)
Invasive Disease Free Survival (IDFS)Data from the pre-specified interim analysis (86% of the number of events for the planned final analysis).
Number of events (%)
106 (12)
178 (20)
Hazard ratio (95% CI)Based on the stratified Cox's Proportional Hazards Model.
0.58 (0.46, 0.74)
p‚ÄĎvalue (2‚ÄĎsided)p-value from a stratified log-rank test. Compared with the allocated alpha of 0.005 for IDFS and 0.015 for OS.
< 0.0001
3-year event-free rate, % (95% CI)Percentage are calculated using Kaplan-Meier estimates.
86 (82.8, 88.4)
77 (73.7, 80.1)
Overall SurvivalData from the pre-specified second interim analysis of OS (at ~330 IDFS events).
Number of events (%)
75 (8)
109 (12)
Hazard ratio (95% CI)
0.68 (0.50, 0.91)
p‚ÄĎvalue (2-sided)
0.0091
3-year event-free rate, % (95% CI)
93 (90.8, 94.4)
89 (86.7, 91)
CI = confidence interval.
Figure 6 Kaplan-Meier Curves of IDFS ‚Äď OlympiA
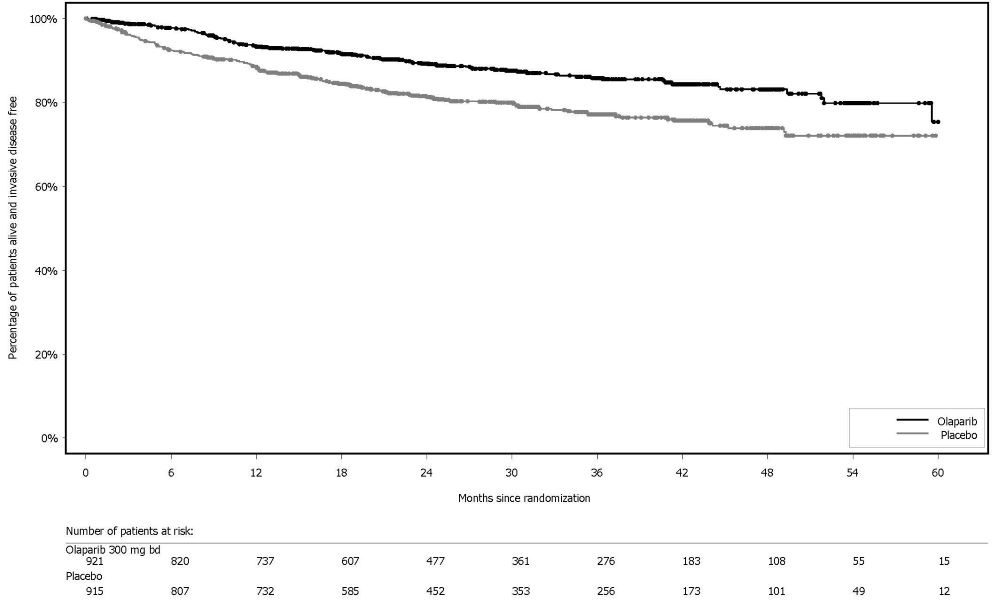
Figure 7 Kaplan-Meier Curves of OS ‚Äď OlympiA
14.5 Treatment of Germline -mutated HER2-negative Metastatic Breast Cancer
The efficacy of Lynparza was evaluated in OlympiAD (NCT02000622), an open-label randomized (2:1) study in patients with gBRCAm HER2-negative metastatic breast cancer. Patients were required to have received treatment with an anthracycline (unless contraindicated) and a taxane, in the neoadjuvant, adjuvant or metastatic setting. Patients with hormone receptor-positive disease must have progressed on at least 1 endocrine therapy (adjuvant or metastatic), or have disease that the treating healthcare provider believed to be inappropriate for endocrine therapy. Patients with prior platinum therapy were required to have no evidence of disease progress during platinum treatment. No prior treatment with a PARP inhibitor was permitted. Patients were randomized to Lynparza tablets 300 mg orally twice daily or healthcare provider’s choice of chemotherapy (capecitabine, eribulin, or vinorelbine, at standard doses) until progression or unacceptable toxicity. Randomization was stratified by prior use of chemotherapy for metastatic disease (yes vs no), hormone receptor status (hormone receptor positive vs triple negative), and previous use of platinum-based chemotherapy (yes vs no). The major efficacy outcome measure was PFS assessed by blinded independent central review (BICR) using RECIST version 1.1.
A total of 302 patients were randomized, 205 to Lynparza and 97 to chemotherapy. Among the 205 patients treated with Lynparza, the median age was 44 years (range: 22 to 76), 65% were White, 4% were males and all the patients had an ECOG PS of 0 or 1. Approximately 50% of patients had triple-negative tumors and 50% had estrogen receptor and/or progesterone receptor positive tumors and the proportions were balanced across treatment arms. Patients in each treatment arm had received a median of 1 prior chemotherapy regimen for metastatic disease; approximately 30% had not received a prior chemotherapy regimen for metastatic breast cancer. Twenty-one percent of patients in the Lynparza arm and 14% in the chemotherapy arm had received platinum therapy for metastatic disease. Seven percent of patients in each treatment arm had received platinum therapy for localized disease.
Of the 302 patients randomized onto OlympiAD, 299 were tested with the BRACAnalysis CDx¬ģ and 297 were confirmed to have deleterious or suspected deleterious gBRCAm status; 202 were randomized to the Lynparza arm and 95 to the healthcare provider‚Äôs choice of chemotherapy arm.
A statistically significant improvement in PFS was demonstrated for the Lynparza arm compared to the chemotherapy arm. Efficacy data for OlympiAD are displayed in Table 23 and Figure 8. Consistent results were observed across patient subgroups defined by study stratification factors. An exploratory analysis of investigator-assessed PFS was consistent with the BICR-assessed PFS results.
Table 23 Efficacy Results - OlympiAD (BICR-assessed)
Lynparza tablets
(n=205)
Chemotherapy
(n=97)
Progression-Free Survival
     Number of events (%)
163 (80%)
71 (73%)
     Median, months
7.0
4.2
     Hazard ratio (95% CI)Hazard ratio is derived from a stratified log-rank test, stratified by ER, PgR negative versus ER and or PgR positive and prior chemotherapy (yes versus no).
0.58 (0.43, 0.80)
     p-valueFor PFS, p-value (2-sided) was compared to 0.05.
0.0009
Patients with Measurable Disease
n=167
n=66
     Objective Response Rate (95% CI)Response based on confirmed responses. The confirmed complete response rate was 7.8% for Lynparza compared to 1.5% for chemotherapy arm.
52% (44, 60)
23% (13, 35)
Overall Survival
     Number of events (%)
130 (63%)
62 (64%)
     Median, months
19.3
17.1
     Hazard ratio (95% CI)
0.90 (0.66, 1.23)
Figure 8 Kaplan-Meier Curves of Progression-Free Survival - OlympiAD
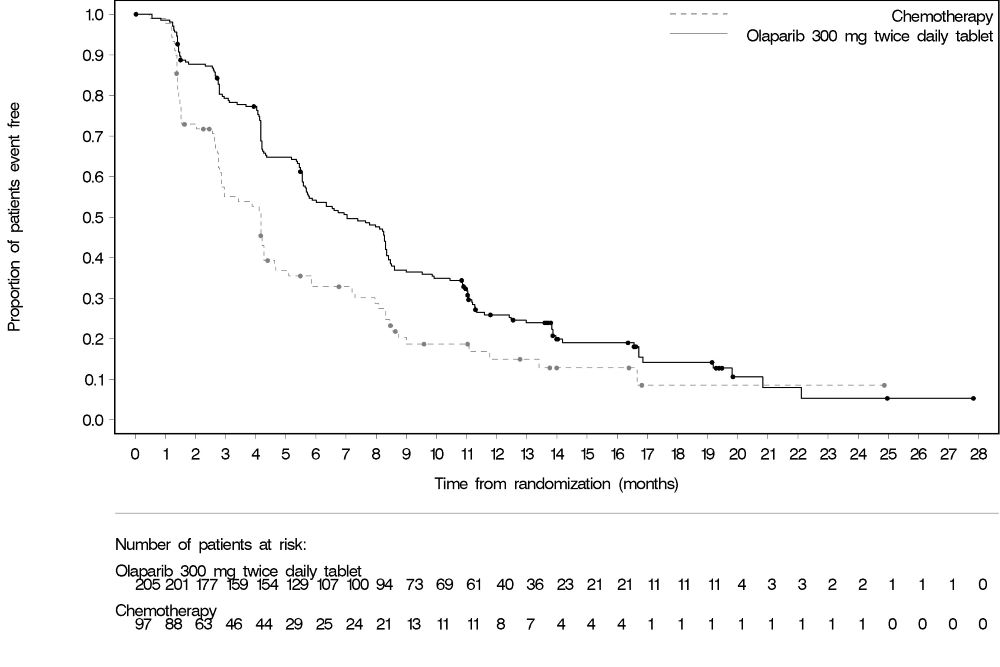
14.6 First-Line Maintenance Treatment of Germline -mutated Metastatic Pancreatic Adenocarcinoma
The efficacy of Lynparza was evaluated in POLO (NCT02184195), a randomized (3:2), double-blind placebo-controlled, multi-center trial. Patients were required to have metastatic pancreatic adenocarcinoma with a deleterious or suspected deleterious germline BRCA mutation (gBRCAm) and absence of disease progression after receipt of first-line platinum-based chemotherapy for at least 16 weeks. Patients were randomized to receive Lynparza tablets 300 mg orally twice daily or placebo until disease progression or unacceptable toxicity. The major efficacy outcome measure was PFS by BICR using RECIST, version 1.1 modified to assess patients with clinical complete response at entry who were assessed as having no evidence of disease unless they had progressed based on the appearance of new lesions. Additional efficacy outcome measures were OS and ORR.
A total of 154 patients were randomized, 92 to Lynparza and 62 to placebo. The median age was 57 years (range 36 to 84); 54% were male; 92% were White, 4% were Asian, and 3% were Black; baseline ECOG PS was 0 (67%) or 1 (31%). The median time from initiation of first-line platinum-based chemotherapy to randomization was 5.8 months (range 3.4 to 33.4 months). Seventy-five percent (75%) of patients received FOLFIRINOX with a median of 9 cycles (range 4-61), 8% received FOLFOX or XELOX, 4% received GEMOX, and 3% received gemcitabine plus cisplatin; 49% achieved a complete or partial response to platinum-based chemotherapy.
All patients had a deleterious or suspected deleterious germline BRCA-mutation as detected by the Myriad BRACAnalysis ¬ģ or BRACAnalysis CDx¬ģ at a central laboratory only (n=106), local BRCA test only (n=4), or both local and central testing (n=44). Among the 150 patients with central test results, 30% had a mutation in BRCA1; 69% had a mutation in BRCA2; and 1 patient (1%) had mutations in both BRCA1 and BRCA2.
POLO demonstrated a statistically significant improvement in BICR-assessed PFS in patients randomized to Lynparza as compared with placebo. The final analysis of OS did not reach statistical significance. Efficacy results of POLO are provided in Table 24 and Figure 9.
Table 24 Efficacy Results - POLO (BICR-assessed)
Lynparza tablets
(n=92)
Placebo
(n=62)
Progression-Free Survival
     Number of events (%)Number of events: Progression - Lynparza 55, placebo 44; death before BICR-documented progression - Lynparza 5, placebo 0.
60 (65)
44 (71)
     Median, months (95% CI)
7.4 (4.1, 11.0)
3.8 (3.5, 4.9)
     Hazard ratioHazard ratio, 95% CI, and p-value calculated from a log-rank test. A hazard ratio <1 favors Lynparza. (95% CI)
0.53 (0.35, 0.81)
     p-value
0.0035
Overall Survival
     Number of events (%)
61 (66)
47 (76)
     Median, months (95% CI)
19.0 (15.3, 26.3)
19.2 (14.3, 26.1)
     Hazard ratio† (95% CI)
0.83 (0.56, 1.22)
     p-value
0.3487
Patients with Measurable Disease
n=78
n=52
Objective Response Rate (95% CI)
23% (14, 34)
12% (4, 23)
     Complete response (%)
2 (2.6)
0
     Partial response (%)
16 (21)
6 (12)
Duration of Response (DOR)
     Median time in months (95% CI)
25 (15, NC)
4 (2, NC)
NC Not calculable.
Figure 9 Kaplan-Meier Curves of BICR-Assessed Progression-Free Survival ‚Äď POLO
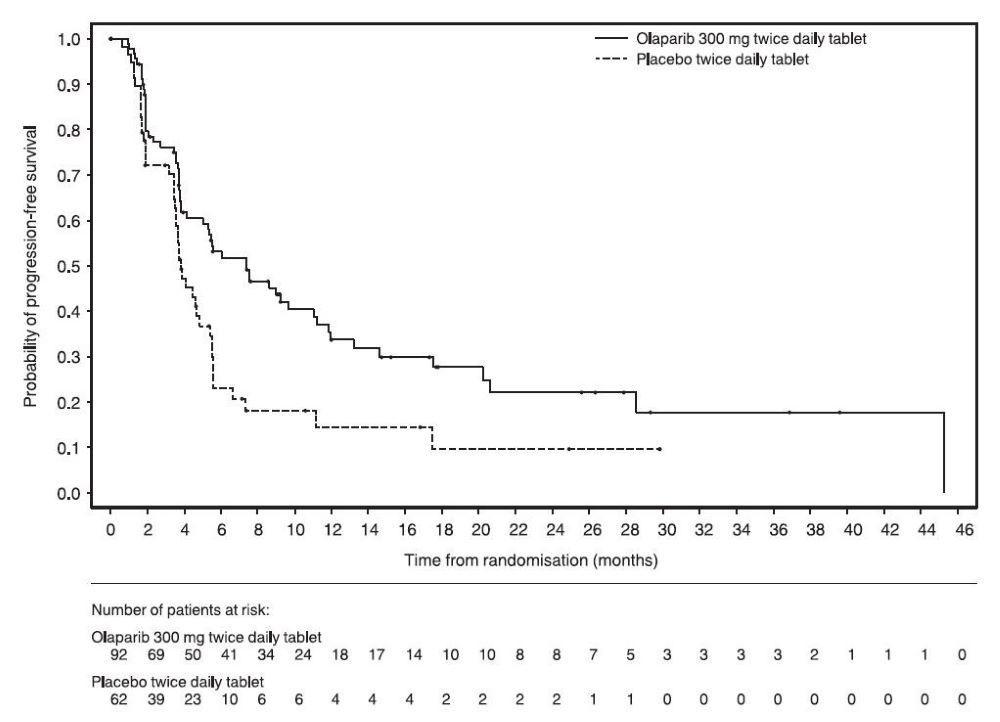
14.7 HRR Gene-mutated Metastatic Castration-Resistant Prostate Cancer
The efficacy of Lynparza was evaluated in PROfound (NCT02987543), randomized, open-label, multi-center trial that evaluated the efficacy of Lynparza 300 mg twice daily versus a comparator arm of investigator’s choice of enzalutamide or abiraterone acetate in men with metastatic castration-resistant prostate cancer (mCRPC). All patients received a GnRH analog or had prior bilateral orchiectomy. Patients needed to have progressed on prior enzalutamide or abiraterone for the treatment of metastatic prostate cancer and/or CRPC and have a tumor mutation in one of 15 genes involved in the homologous recombination repair (HRR) pathway.
Patients were divided into two cohorts based on HRR gene mutation status. Patients with mutations in either BRCA1, BRCA2, or ATM were randomized in Cohort A; patients with mutations among 12 other genes involved in the HRR pathway (BARD1, BRIP1, CDK12, CHEK1, CHEK2, FANCL, PALB2, PPP2R2A, RAD51B, RAD51C, RAD51D, or RAD54L) were randomized in Cohort B; patients with co-mutations (BRCA1, BRCA2, or ATM plus a Cohort B gene) were assigned to Cohort A. Although patients with PPP2R2A gene mutations were enrolled in the trial, Lynparza is not indicated for the treatment of patients with this gene mutation due to unfavorable risk-benefit. Patients were randomized (2:1), 256 to Lynparza arm and 131 to enzalutamide or abiraterone acetate arm; in Cohort A there were 245 (162 Lynparza arm and 83 in enzalutamide or abiraterone acetate arm) and in Cohort B there were 142 patients (94 in Lynparza arm and 48 in enzalutamide or abiraterone acetate arm). Randomization was stratified by prior receipt of taxane chemotherapy and presence of measurable disease by RECIST 1.1. Treatment was continued until objective radiological disease progression determined by BICR. Upon radiological progression confirmed by BICR, patients randomized to enzalutamide or abiraterone acetate were given the option to switch to Lynparza. Patients with HRR gene mutations were identified by tissue-based testing using the Foundation Medicine FoundationOne¬ģ clinical trial HRR assay performed at a central laboratory.
Determination of deleterious or suspected deleterious somatic or germline HRR mutation status in line with the FDA-approved mutation classification and testing criteria for the Foundation Medicine F1CDx tissue-based assay and assessment of the germline-BRCA status using the Myriad BRACAnalysis CDx blood-based assay was performed retrospectively. Representation of individual gene mutations by cohort is provided in Table 25. No patients were enrolled who had mutations in two of the 15 pre-specified HRR genes: FANCL and RAD51C.
Table 25 Frequency of Patients with HRR Mutations Enrolled in PROfound
HRR Mutation
Cohort A
N=245
n (%)
Cohort BThree patients with single BRCA2 or ATM gene mutations and 1 patient with co-occurring BRCA2+CDK12 gene mutations were incorrectly assigned to Cohort B.
N=142
n (%)
Single mutation
224 (91)
135 (95)
    BRCA2
127 (52)
1 (<1)
    ATM
84 (34)
2 (1)
    BRCA1
13 (5)
0
    CDK12
0
89 (63)
    CHEK2
0
12 (8)
    PPP2R2ALynparza is not indicated for patients with PPP2R2A mutations.
0
10 (7)
    RAD51B
0
5 (4)
    RAD54L
0
5 (4)
    PALB2
0
4 (3)
    BRIP1
0
3 (2)
    CHEK1
0
2 (1)
    BARD1
0
1 (<1)
    RAD51D
0
1 (<1)
Co-occurring mutationPatients with co-occurring mutations (BRCA1, BRCA2, or ATM plus a Cohort B gene) were assigned to Cohort A.
21 (9)
7 (5)
In Cohort A+B, the median age was 69 years (range: 47 to 91 years) in both arms; 69% were White, 29% were Asian, and 1% were Black. The ECOG performance score was 0 or 1 in most patients (95%) in both arms. In patients treated with Lynparza, the proportion of patients with RECIST 1.1 measurable disease at baseline was 58%, including 17% with lung and 10% with liver metastases, respectively. At randomization, 66% of patients had received prior taxane chemotherapy, 40% had received enzalutamide, 38% had received abiraterone acetate, and 20% had received both enzalutamide and abiraterone acetate. Patient characteristics were well-balanced between arms.
The major efficacy outcome of the study was radiological progression-free survival (rPFS) (Cohort A) as determined by BICR using RECIST version 1.1 and Prostate Cancer Clinical Trials Working Group 3 (PCWG3) (bone) criteria. Additional efficacy outcomes included confirmed objective response rate (ORR) (Cohort A), rPFS (combined Cohorts A+B) as assessed by BICR, and overall survival (OS) (Cohort A).
PROfound demonstrated a statistically significant improvement in BICR-assessed rPFS for Lynparza compared to investigator’s choice of enzalutamide or abiraterone acetate in Cohort A and Cohort A+B. In an exploratory analysis for patients in Cohort B, the median rPFS was 4.8 months for Lynparza vs 3.3 months for comparator with a HR of 0.88 (95% CI 0.58, 1.36). The major efficacy outcome was supported by a statistically significant improvement in ORR by BICR for patients with measurable disease at baseline in Cohort A. In Cohort B, ORR by BICR was 3.7% (95% CI 0.5, 12.7) in Lynparza treated patients and 8.3% (95% CI 1.0, 27.0) in patients treated with enzalutamide or abiraterone acetate.
The final analysis of overall survival (OS) demonstrated a statistically significant improvement in OS in patients randomized to Lynparza compared to patients in the enzalutamide or abiraterone acetate arm in Cohort A.
Efficacy results of PROfound are provided in Tables 26 and 27 and Figures 10 and 11.
Table 26 Efficacy Results - PROfound (BICR-assessed)
Cohort A
Cohort A+BAlthough 10 patients with PPP2R2A mutation were included in all analyses of Cohort A+B, Lynparza is not indicated for this population due to unfavorable risk-benefit.
Lynparza tablets(n=162)
Enzalutamide or Abiraterone acetate(n=83)
Lynparza tablets(n=256)
Enzalutamide or Abiraterone acetate(n=131)
Radiological Progression-Free Survival (rPFS)
     Number of events (%)
106 (65)
68 (82)
180 (70)
99 (76)
     Median (95% CI), in months
7.4 (6.2, 9.3)
3.6 (1.9, 3.7)
5.8 (5.5, 7.4)
3.5 (2.2, 3.7)
     Hazard ratio (95% CI)The HR and CI were calculated using a Cox proportional hazards model adjusted for prior taxane use and measurable disease. An HR <1 favors Lynparza 300 mg taken orally twice daily.
0.34 (0.25, 0.47)
0.49 (0.38, 0.63)
     p-valueThe analysis was performed using the log-rank test stratified by prior taxane use and measurable disease.
<0.0001
<0.0001
Confirmed ORR
Patients with measurable disease at baseline
n=84
n=43
-
-
    ORR, n (%)
28 (33)
1 (2)
-
-
    (95% CI)
(23, 45)
(0, 12)
-
-
    p-value
<0.0001
-
Overall Survival
n=162
n=83
-
-
    Number of events (%)
91 (56)
57 (69)
-
-
    Median (95% CI), in months
19.1 (17.4, 23.4)
14.7 (11.9, 18.8)
-
-
    Hazard ratio (95% CI)
0.69 (0.50, 0.97)
-
    p-value
0.0175
-
CI Confidence interval
Figure 10 Kaplan-Meier Curves of BICR-Assessed rPFS ‚Äď PROfound ‚Äď Cohort A
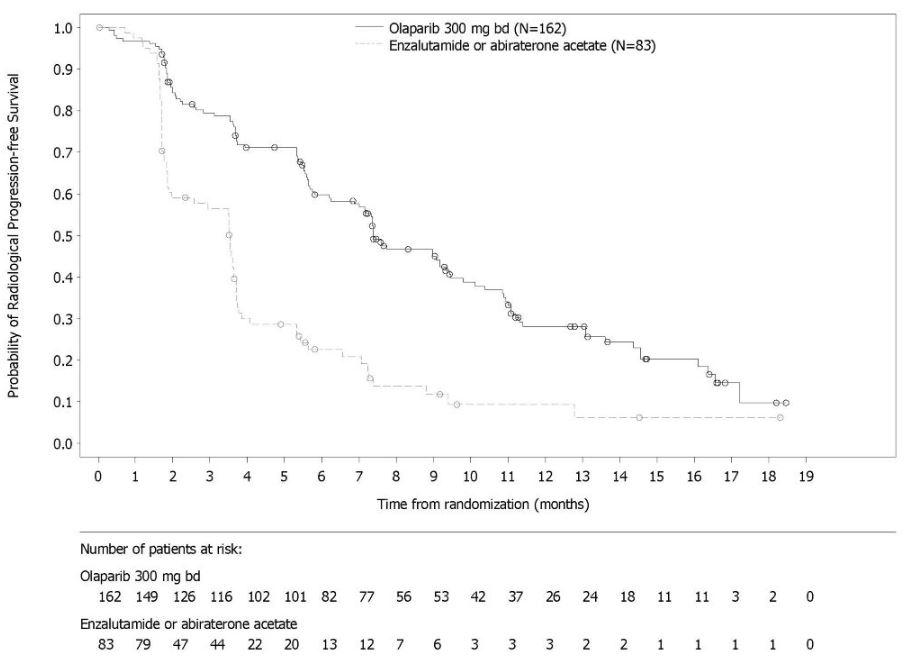
Consistent results were observed in exploratory analyses of rPFS for patients who received or did not receive prior taxane therapy and for those with germline-BRCA mutations identified using the Myriad BRACAnalysis CDx assay compared with those with BRCA mutations identified using the Foundation Medicine F1CDx assay.
Figure 11 Kaplan-Meier Curves of Overall Survival ‚Äď PROfound ‚Äď Cohort A
Response data by HRR mutations for patients in the Lynparza arm are presented in Table 27. In the comparator arm of Cohorts A and B, a total of three patients achieved a partial response, including one patient with an ATM mutation alone and 2 patients with co-occurring mutations (one with PALB2+PPP2R2A and one with CDK12+PALB2).
Table 27 Response Rate and Duration of Response by HRR Mutation in Patients with Measurable Disease at Baseline on the Lynparza Arm ‚Äď PROfound (BICR-assessed)
HRR mutationNo patients with FANCL or RAD51C enrolled. Three patients with PPP2R2A mutations had measurable disease, however, Lynparza is not indicated for patients with PPP2R2A mutation.
  Patients  (N=138)
  Confirmed ORRIn patients with a single BRCA2 mutation the median duration of response in the Lynparza arm (n=24) was 5.6 months (95% C.I: 5.5, 9.2). In the 3 responders with a single ATM mutation in the Lynparza arm, the duration of response ranged from 5.8+ to 9.0 months. In the 2 responders with a single CDK12 mutation in the Lynparza arm, the duration of response was 3.7 and 7.2 months. + denotes ongoing response.
  n (%)
  95% CI
Single mutation
BRCA2
  43
  24 (56)
  (40, 71)
ATM
  30
  3 (10)
  (2, 27)
CDK12
  34
  2 (6)
  (1, 20)
BRCA1
  6
  SD, PD (4), NE
  NA
CHEK2
  4
  SD (2), PD (2)
  NA
BRIP1
  2
  SD, PD
  NA
PALB2
  2
  SD, PD
  NA
CHEK1
  1
  PD
  NA
RAD51B
  1
  SD
  NA
RAD51D
  1
  PD
  NA
RAD54L
  1
  SD
  NA
Co-occurring mutations
BRCA2/CDK12
  2
  PR, SD
  NA
BRCA2/ATM
  2
  SD, SD
  NA
BRCA2/BARD1
  1
  PD
  NA
BRCA2/CHEK2
  1
  SD
  NA
CDK12/CHEK1
  1
  SD
  NA
CDK12/PALB2
  1
  PD
  NA
BRCA2/CDK12/CHEK2
  1
  PD
  NA
BRCA2/CHEK2/RAD51D
  1
  SD
  NA
  PR Partial response; SD Stable disease; PD Progressive disease; NE Not evaluable; NA Not applicable due to small numbers or lack of response.14.8 Treatment of BRCA-mutated Metastatic Castration-Resistant Prostate Cancer in Combination with Abiraterone and Prednisone or Prednisolone
The efficacy of Lynparza in the treatment of patients with mCRPC was investigated in PROpel (NCT03732820), a randomized, double-blind, placebo-controlled, multi-center study that compared the efficacy of Lynparza in combination with abiraterone with placebo plus abiraterone for patients with mCRPC. Patients (n=796) were randomized (1:1) to receive Lynparza tablets 300 mg orally twice daily in combination with abiraterone 1000 mg daily (n=399) compared with placebo plus abiraterone (n=397). All patients received either prednisone or prednisolone 5 mg twice daily, and a GnRH analog or prior bilateral orchiectomy. Patients with prior treatment with abiraterone were excluded. Prior docetaxel for localized or metastatic hormone-sensitive prostate cancer (mHSPC) was allowed. Randomization was stratified by metastases (bone only, visceral, or other) and docetaxel treatment at mHSPC stage (yes or no). Lynparza treatment was continued until objective radiological disease progression determined by investigator or unacceptable toxicity.
BRCA gene mutation (BRCAm) status was assessed after randomization and before primary analysis by both NGS-based tumor tissue and ctDNA tests. BRCAm classification criteria in line with the FDA approved assays were used to determine the deleterious and suspected deleterious somatic or germline mutation status of patients.
The major efficacy outcome measure was investigator-assessed rPFS evaluated according to RECIST, version 1.1 and Prostate Cancer Working Group (PCWG3) (bone) criteria. Overall survival (OS) was an additional efficacy outcome measure.
Of the 796 patients tested, 85 (11%) had BRCAm determined by either a positive ctDNA test (9%) or a tumor tissue test (6%). Among these 85 patients, the median age was 68 years (range 43 to 85), and 67% were 65 years or older; 72% were White, 22% Asian, and 2% Black or African American; 66% had ECOG performance status (PS) 0 and 34% had ECOG PS 1; 25% had prior docetaxel treatment for mHSPC; 53% had bone-only metastases, 15% had visceral metastases, and 32% had other metastases.
A statistically significant improvement in rPFS for Lynparza/abiraterone compared to placebo/abiraterone was observed in the intention to treat (ITT) population. In an exploratory analysis in the subgroup of 711 patients without an identified BRCAm, the rPFS hazard ratio was 0.77 (95% CI: 0.63, 0.96) and the OS hazard ratio was 0.92 (95% CI: 0.74, 1.14), indicating that the improvement in the ITT population was primarily attributed to the results seen in the subgroup of patients with BRCAm.
Results of an exploratory analysis in the subgroup of 85 patients on PROpel with BRCAm are summarized in Table 28 and Figure 12 and 13.
Results from the BICR assessment were consistent with the investigator-assessed rPFS results.
Table 28 Efficacy Results ‚Äď PROpel (Patients with BRCAm)
Lynparza/abiraterone
N = 47
Placebo/abiraterone
N = 38
Radiological Progression-Free Survival (rPFS)Investigator-assessed.
Events, n (%)
14 (30)
28 (74)
Median (95% CI), months
NR (NR, NR)
8 (6, 15)
Hazard ratio (95% CI)Calculated using an unstratified univariable Cox proportional hazards model.
0.24 (0.12, 0.45)
Overall Survival (OS)
Events, n (%)
13 (28)
25 (66)
Median (95% CI), months
NR (NR, NR)
23 (18, 34)
Hazard ratio (95% CI)
0.30 (0.15, 0.59)
NR: Not reached.
Figure 12 Kaplan-Meier Curves of rPFS ‚Äď PROpel (Patients with BRCAm, Investigator Assessment)
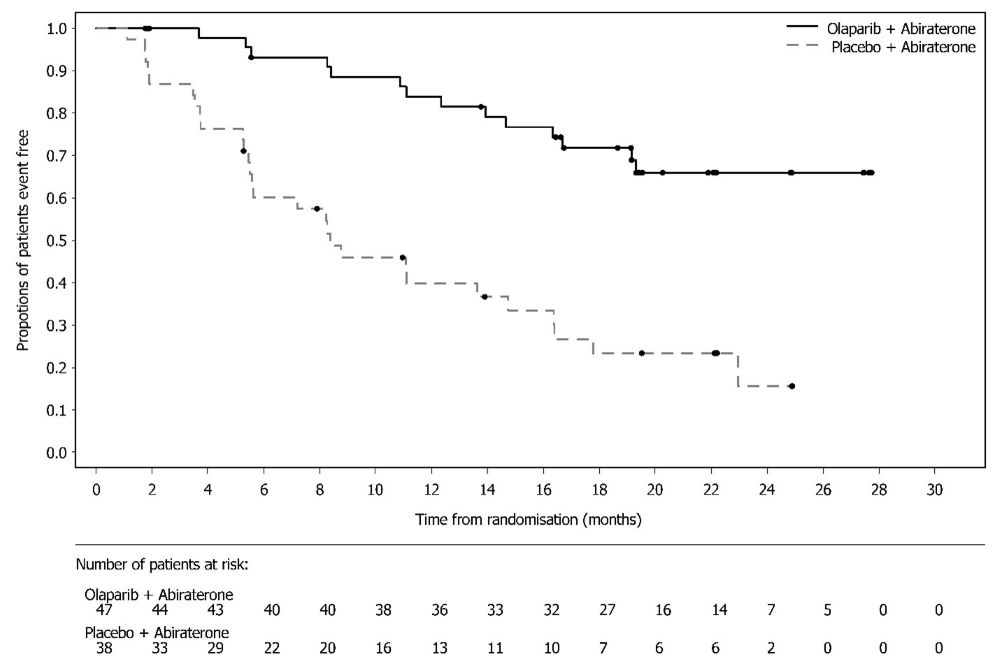
Figure 13 Kaplan-Meier Curves of OS ‚Äď PROpel (Patients with BRCAm)
16 How Supplied/storage And Handling
Lynparza is available as 150 mg and 100 mg tablets.
‚ÄĘ 150 mg tablets: green to green/grey, oval, bi-convex, film-coated tablet, with debossment ‚ÄėOP150‚Äô on one side and plain on the reverse, are available in:
‚ąė Bottles of 60 tablets (NDC 0310-0679-60) and‚ąė Bottles of 120 tablets (NDC 0310-0679-12).‚ÄĘ 100 mg tablets: yellow to dark yellow, oval, bi-convex, film-coated tablet, with debossment ‚ÄėOP100‚Äô on one side and plain on the reverse, are available in:
‚ąė Bottles of 60 tablets (NDC 0310-0668-60) and‚ąė Bottles of 120 tablets (NDC 0310-0668-12).
Store at 20¬ļC to 25¬ļC (68¬ļF to 77¬ļF), excursions permitted to 15¬ļC to 30¬ļC (59¬ļF to 86¬ļF) [see USP Controlled Room Temperature]. Store in original bottle to protect from moisture.
17 Patient Counseling Information
Advise the patient to read the FDA-approved patient labeling (Medication Guide).
MDS/AML
Advise patients to contact their healthcare provider if they experience weakness, feeling tired, fever, weight loss, frequent infections, bruising, bleeding easily, breathlessness, blood in urine or stool, and/or laboratory findings of low blood cell counts, or a need for blood transfusions. This may be a sign of hematological toxicity or a more serious uncommon bone marrow problem called ‚Äėmyelodysplastic syndrome‚Äô (MDS) or ‚Äėacute myeloid leukemia‚Äô (AML) which have been reported in patients treated with Lynparza [see Warnings and Precautions (5.1)].
Pneumonitis
Advise patients to contact their healthcare provider if they experience any new or worsening respiratory symptoms including shortness of breath, fever, cough, or wheezing [see Warnings and Precautions (5.2)].
Venous Thromboembolism
Advise patients to immediately report any signs or symptoms of thromboembolism such as pain or swelling in an extremity, shortness of breath, chest pain, tachypnea, and tachycardia [see Warnings and Precautions (5.3)].
Embryo-Fetal Toxicity
Inform pregnant women of the risk to a fetus and potential loss of the pregnancy. Advise females to inform their healthcare provider of known or suspected pregnancy [see Use in Specific Populations (8.1)].
Advise females of reproductive potential to use effective contraception during treatment with Lynparza and for 6 months after the last dose [see Use in Specific Populations (8.3)].
Advise male patients with female partners of reproductive potential or who are pregnant to use effective contraception during treatment and for 3 months after receiving the last dose of Lynparza. Advise male patients not to donate sperm during therapy and for 3 months following the last dose of Lynparza [see Warnings and Precautions (5.4) and Use in Specific Populations (8.3) ].
Lactation
Advise patients not to breastfeed while taking Lynparza and for one month after receiving the last dose [see Use in Specific Populations (8.2)].
Drug Interactions
Advise patients and caregivers to inform their healthcare provider of all concomitant medications, including prescription medicines, over-the-counter drugs, vitamins, and herbal products. Inform patients to avoid grapefruit, grapefruit juice, Seville oranges, and Seville orange juice while taking Lynparza [see Drug Interactions (7.2)].
Nausea/Vomiting
Advise patients that mild or moderate nausea and/or vomiting is very common in patients receiving Lynparza and that they should contact their healthcare provider who will advise on available antiemetic treatment options [see Adverse Reactions (6.1)].
Distributed by:
AstraZeneca Pharmaceuticals LP
Wilmington, DE 19850
© AstraZeneca 2023
Spl Medguide Section
Medication Guide
Lynparza¬ģ (Lin-par-zah)
(olaparib)
tablets
 
What is the most important information I should know about Lynparza?
Lynparza may cause serious side effects, including:
Bone marrow problems called Myelodysplastic Syndrome (MDS) or Acute Myeloid Leukemia (AML). Some people who have received previous treatment with chemotherapy, radiotherapy or certain other medicines for their cancer have developed MDS or AML during treatment with Lynparza. MDS or AML may lead to death. If you develop MDS or AML, your healthcare provider will stop treatment with Lynparza.
Symptoms of low blood cell counts are common during treatment with Lynparza, but can be a sign of serious bone marrow problems, including MDS or AML. Symptoms may include:
‚ÄĘ weakness‚ÄĘ weight loss‚ÄĘ fever‚ÄĘ frequent infections
‚ÄĘ blood in urine or stool‚ÄĘ shortness of breath‚ÄĘ feeling very tired‚ÄĘ bruising or bleeding more easily
Your healthcare provider will do blood tests to check your blood cell counts:
‚ÄĘ before treatment with Lynparza‚ÄĘ every month during treatment with Lynparza‚ÄĘ weekly if you have low blood cell counts that last a long time. Your healthcare provider may stop treatment with Lynparza until your blood cell counts improve.
Lung problems (pneumonitis). Tell your healthcare provider if you have any new or worsening symptoms of lung problems, including shortness of breath, fever, cough, or wheezing. Your healthcare provider may do a chest x-ray if you have any of these symptoms. Your healthcare provider may temporarily or completely stop treatment if you develop pneumonitis. Pneumonitis may lead to death.
Blood clots (Venous Thromboembolism). Some people may develop a blood clot in a deep vein, usually in the leg (venous thrombosis), or a clot in the lungs (pulmonary embolism) which may be severe or lead to death. Tell your healthcare provider right away if you have any symptoms such as pain or swelling in an extremity, shortness of breath, chest pain, breathing that is more rapid than normal (tachypnea), or heart beats faster than normal (tachycardia). Your healthcare provider will monitor you for these symptoms and may prescribe blood thinner medicine.
What is Lynparza?
Lynparza is a prescription medicine used to treat adults who have:
‚ÄĘ advanced ovarian cancer, fallopian tube cancer, or primary peritoneal cancer with a certain type of inherited (germline) or acquired (somatic) abnormal BRCA gene. Lynparza is used alone as maintenance treatment after the cancer has responded to your first treatment with platinum-based chemotherapy. Your healthcare provider will perform a test to make sure that Lynparza is right for you.‚ÄĘ advanced ovarian cancer, fallopian tube cancer or primary peritoneal cancer with a certain type of abnormal BRCA gene or a positive laboratory tumor test for genomic instability called HRD. Lynparza is used in combination with another anti-cancer medicine, bevacizumab, as maintenance treatment after the cancer has responded to your first treatment with platinum-based chemotherapy. Your healthcare provider will perform a test to make sure that Lynparza is right for you.‚ÄĘ ovarian cancer, fallopian tube cancer, or primary peritoneal cancer with a certain type of inherited (germline) or acquired (somatic) abnormal BRCA gene. Lynparza is used as maintenance treatment when the cancer has come back. Lynparza is used after the cancer has responded to treatment with platinum-based chemotherapy. Your healthcare provider will perform a test to make sure that Lynparza is right for you.‚ÄĘ human epidermal growth factor receptor 2 (HER2)-negative early breast cancer with a certain type of inherited (germline) abnormal BRCA gene. Lynparza is given after surgery (treatment after surgery is called adjuvant therapy). You should have received chemotherapy medicines before or after surgery to remove the tumor. Your healthcare provider will perform a test to make sure that Lynparza is right for you.‚ÄĘ a certain type of abnormal inherited BRCA gene, human epidermal growth factor receptor 2 (HER2)-negative breast cancer that has spread to other parts of the body (metastatic). You should have received chemotherapy medicines, either before or after your cancer has spread. If you have hormone receptor (HR)-positive disease, you should have been treated with hormonal therapy. Your healthcare provider will perform a test to make sure that Lynparza is right for you.‚ÄĘ metastatic pancreatic cancer with a certain type of abnormal inherited BRCA gene. Lynparza is used as maintenance treatment after your cancer has not progressed on at least 16 weeks of treatment with platinum-based chemotherapy. Your healthcare provider will perform a test to make sure that Lynparza is right for you.‚ÄĘ prostate cancer with certain inherited or acquired abnormal genes called homologous recombination repair (HRR genes). Lynparza is used when the cancer has spread to other parts of the body (metastatic), and no longer responds to a medical or surgical treatment that lowers testosterone, and has progressed after treatment with enzalutamide or abiraterone. Your healthcare provider will perform a test to make sure Lynparza is right for you.‚ÄĘ prostate cancer with a certain type of abnormal inherited or acquired BRCA gene that has spread to other parts of the body (metastatic) and no longer responds to a medical or surgical treatment that lowers testosterone. Lynparza is used in combination with another anti-cancer medicine, abiraterone, together with the steroid medicine, prednisone or prednisolone. Your healthcare provider will perform a test to make sure that Lynparza is right for you.
It is not known if Lynparza is safe and effective in children.
Before taking Lynparza, tell your healthcare provider about all of your medical conditions, including if you:
‚ÄĘ have lung or breathing problems‚ÄĘ have kidney problems‚ÄĘ are pregnant, become pregnant, or plan to become pregnant. Lynparza can harm your unborn baby and may cause loss of pregnancy (miscarriage).
‚ąė If you are able to become pregnant, your healthcare provider may do a pregnancy test before you start treatment with Lynparza.‚ąė Females who are able to become pregnant should use effective birth control (contraception) during treatment with Lynparza and for 6 months after the last dose of Lynparza. Talk to your healthcare provider about birth control methods that may be right for you. Tell your healthcare provider right away if you become pregnant or think you might be pregnant following treatment with Lynparza.‚ąė Males with female partners who are pregnant or able to become pregnant should use effective birth control (contraception) during treatment with Lynparza and for 3 months after the last dose of Lynparza.‚ąė Do not donate sperm during treatment with Lynparza and for 3 months after your last dose.‚ÄĘ are breastfeeding or plan to breastfeed. It is not known if Lynparza passes into your breast milk. Do not breastfeed during treatment with Lynparza and for 1 month after receiving the last dose of Lynparza. Talk to your healthcare provider about the best way to feed your baby during this time.
 
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements. Taking Lynparza and certain other medicines may affect how Lynparza works and may cause side effects.
How should I take Lynparza?
‚ÄĘ Take Lynparza tablets exactly as your healthcare provider tells you.‚ÄĘ Do not change your dose or stop taking Lynparza unless your healthcare provider tells you to. Your healthcare provider may temporarily stop treatment with Lynparza or change your dose of Lynparza if you experience side effects.‚ÄĘ Your healthcare provider will decide how long you stay on treatment.‚ÄĘ Take Lynparza by mouth 2 times a day with or without food.‚ÄĘ Each dose should be taken about 12 hours apart.‚ÄĘ Swallow Lynparza tablets whole. Do not chew, crush, dissolve, or divide the tablets.‚ÄĘ If you are taking Lynparza for early breast cancer and you have hormone receptor-positive disease, you should continue to take hormonal therapy during your treatment with Lynparza.‚ÄĘ If you are taking Lynparza for prostate cancer and you are receiving gonadotropin-releasing hormone (GnRH) analog therapy, you should continue with this treatment during your treatment with Lynparza unless you have had a surgery to lower the amount of testosterone in your body (surgical castration).‚ÄĘ If you miss a dose of Lynparza, take your next dose at your usual scheduled time. Do not take an extra dose to make up for a missed dose.‚ÄĘ If you take too much Lynparza, call your healthcare provider or go to the nearest hospital emergency room right away.
What should I avoid while taking Lynparza?
Avoid grapefruit, grapefruit juice, Seville oranges and Seville orange juice during treatment with Lynparza since they may increase the level of Lynparza in your blood.
What are the possible side effects of Lynparza?
Lynparza may cause serious side effects.
See ‚ÄúWhat is the most important information I should know about Lynparza?‚ÄĚ
The most common side effects of Lynparza when used alone are:
‚ÄĘ nausea or vomiting. Tell your healthcare provider if you get nausea or vomiting. Your healthcare provider may prescribe medicines to treat these symptoms.
‚ÄĘ tiredness or weakness‚ÄĘ low red blood cell counts‚ÄĘ diarrhea‚ÄĘ loss of appetite‚ÄĘ headache‚ÄĘ changes in the way food tastes
‚ÄĘ cough‚ÄĘ low white blood cell counts‚ÄĘ shortness of breath‚ÄĘ dizziness‚ÄĘ indigestion or heartburn‚ÄĘ low platelet counts
The most common side effects of Lynparza when used in combination with bevacizumab are:
‚ÄĘ nausea or vomiting. Tell your healthcare provider if you get nausea or vomiting. Your healthcare provider may prescribe medicines to treat these symptoms.‚ÄĘ tiredness or weakness‚ÄĘ low red blood cell counts‚ÄĘ low white blood cell counts‚ÄĘ diarrhea‚ÄĘ urinary tract infection‚ÄĘ headache
The most common side effects of Lynparza when used in combination with abiraterone are:
‚ÄĘ low red blood cell counts‚ÄĘ tiredness or weakness‚ÄĘ nausea or vomiting. Tell your healthcare provider if you get nausea or vomiting. Your healthcare provider may prescribe medicines to treat these symptoms.
‚ÄĘ diarrhea‚ÄĘ loss of appetite‚ÄĘ low white blood cell counts‚ÄĘ dizziness‚ÄĘ abdominal pain
These are not all of the possible side effects of Lynparza.
Call your healthcare provider for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store Lynparza?
‚ÄĘ Store Lynparza at room temperature, between 68¬įF to 77¬įF (20¬įC to 25¬įC).‚ÄĘ Store Lynparza in the original bottle to protect it from moisture.
Keep Lynparza and all medicines out of reach of children.
General information about the safe and effective use of Lynparza.
Medicines are sometimes prescribed for purposes other than those uled in a Medication Guide. Do not use Lynparza for a condition for which it was not prescribed. Do not give Lynparza to other people, even if they have the same symptoms you have. It may harm them.
You can ask your healthcare provider or pharmacist for information about Lynparza that is written for health professionals.
What are the ingredients in Lynparza?
Active ingredient: olaparib
Inactive ingredients:
Tablet contains: copovidone, mannitol, colloidal silicon dioxide and sodium stearyl fumarate
Tablet coating contains: hypromellose, polyethylene glycol 400, titanium dioxide, ferric oxide yellow and ferrosoferric oxide (150 mg tablet only)
Lynparza is a registered trademark of the AstraZeneca group of companies.
© AstraZeneca 2023
Distributed by:
AstraZeneca Pharmaceuticals LP
Wilmington, DE 19850
For more information, call 1-800-236-9933 or go to www.Lynparza.com.
This Medication Guide has been approved by the U.S. Food and Drug Administration.                          Revised: 11/2023
Package/label Principal Display Panel 100 Mg
NDC 0310-0668-60
Lynparza¬ģ
(olaparib) tablets
100 mg
Dispense accompanying
Medication Guide to each patient.
60 Tablets
Rx only
AstraZeneca
Package/label Principal Display Panel 150 Mg
NDC 0310-0679-12
Lynparza¬ģ
(olaparib) tablets
150 mg
Dispense accompanying
Medication Guide to each patient.
120 Tablets
Rx only
AstraZeneca

DISCLAIMER:
"This tool does not provide medical advice, and is for informational and educational purposes only, and is not a substitute for professional medical advice, treatment or diagnosis. Call your doctor to receive medical advice. If you think you may have a medical emergency, please dial 911."
"Do not rely on openFDA to make decisions regarding medical care. While we make every effort to ensure that data is accurate, you should assume all results are unvalidated. We may limit or otherwise restrict your access to the API in line with our Terms of Service."
"This product uses publicly available data from the U.S. National Library of Medicine (NLM), National Institutes of Health, Department of Health and Human Services; NLM is not responsible for the product and does not endorse or recommend this or any other product."
PillSync may earn a commission via links on our site
