Voriconazole (voriconazole 50 mg) Dailymed
Generic: voriconazole is used for the treatment of Aspergillosis Candidiasis Pregnancy Fusariosis
IMPRINT: V50
SHAPE: round
COLOR: white

Go PRO for all pill images
Recent Major Changes Section
Warnings and Precautions, Photosensitivity (5.6 )                                                             10/2022
1 Indications And Usage
Voriconazole tablets are an azole antifungal indicated for the treatment of adults and pediatric patients 2 years of age and older with:
- Invasive aspergillosis (
1.1 )- Candidemia in non-neutropenics and other deep tissue Candida infections (
1.2 )- Esophageal candidiasis (
1.3 )- Serious fungal infections caused by Scedosporium apiospermum and Fusarium species including Fusarium solani, in patients intolerant of, or refractory to, other therapy (
1.4 ) 1.1 Invasive Aspergillosis
Voriconazole tablets are indicated in adults and pediatric patients (2 years of age and older) for the treatment of invasive aspergillosis (IA). In clinical trials, the majority of isolates recovered were Aspergillus fumigatus. There was a small number of cases of culture-proven disease due to species of Aspergillus other than A. fumigatus [see Clinical Studies (14.1, 14.5) and Microbiology (12.4)].
1.2 Candidemia in Non-neutropenic Patients and Other Deep Tissue Infections
Voriconazoletablets are indicated in adults and pediatric patients (2 years of age and older) for the treatment of candidemia in non-neutropenic patients and the following Candida infections: disseminated infections in skin and infections in abdomen, kidney, bladder wall, and wounds [see Clinical Studies (14.2, 14.5) and Microbiology (12.4)].
1.3 Esophageal Candidiasis
Voriconazoletablets are indicated in adults and pediatric patients (2 years of age and older) for the treatment of esophageal candidiasis (EC) in adults and pediatric patients 2 years of age and older [see Clinical Studies ( 14.3 , 14.5 ) and Microbiology ( 12.4 )].
1.4 Scedosporiosis and Fusariosis
Voriconazole tablets are indicated for the treatment of serious fungal infections caused by Scedosporium apiospermum (asexual form of Pseudallescheria boydii) and Fusarium spp. including Fusarium solani, in adults and pediatric patients (2 years of age and older) intolerant of, or refractory to, other therapy[see Clinical Studies (14.4) and Microbiology (12.4)].
1.5 Usage
Specimens for fungal culture and other relevant laboratory studies (including histopathology) should be obtained prior to therapy to isolate and identify causative organism(s). Therapy may be instituted before the results of the cultures and other laboratory studies are known. However, once these results become available, antifungal therapy should be adjusted accordingly.
2 Dosage And Administration
  
- Dosage in Adults (
2.3 )
 Infection    Loading dose    Maintenance Dose        Intravenous  infusion    Intravenous  infusion    Oral    Invasive Aspergillosis    6 mg/kg every 12 hours for the first 24 hours   4 mg/kg every  12 hours   200 mg  every  12 hours   Candidemia in nonneutropenics and other deep tissue Candida infections    3-4 mg/kg every  12 hours   200 mg  every  12 hours   Scedosporiosis and Fusariosis    4 mg/kg every  12 hours   200 mg  every  12 hours   Esophageal  Candidiasis    Not Evaluated   Not  Evaluated   200 mg every 12 hours 
 
- Adult patients weighing less than 40 kg: oral maintenance dose 100 mg or 150 mg every 12 hours
- Hepatic Impairment: Use half the maintenance dose in adult patients with mild to moderate hepatic impairment (Child-Pugh Class A and B) (
2.5 )- Renal Impairment: Avoid intravenous administration in adult patients with moderate to severe renal impairment (creatinine clearance <50 mL/min) (
2.6 )- Dosage in Pediatric Patients 2 years of age and older (
2.4 )- For pediatric patients 2 to less than 12 years of age and 12 to 14 years of age weighing less than 50 kg see Table below.
 Infection    Loading dose    Maintenance Dose        Intravenous  infusion    Intravenous  infusion    Oral    Invasive Aspergillosis    9 mg/kg every12 hours for the first 24 hours   8 mg/kg every 12 hours after the first 24 hours   9 mg/kg every 12 hours (maximum dose of 350 mg every 12 hours)   Candidemia in nonneutropenics and other deep tissue Candida infections    Scedosporiosis and Fusariosis    Esophageal  Candidiasis    Not Evaluated   4 mg/kg every 12 hours   9 mg/kg every 12 hours (maximum dose of 350 mg every 12 hours) 
- For pediatric patients aged 12 to 14 years weighing greater than or equal to 50 kg and those aged 15 years and older regardless of body weight use adult dosage. (
2.4 )- Dosage adjustment of voriconazole tablets in pediatric patients with renal or hepatic impairment has not been established (
2.5 ,2.6 )    2.1 Important Administration Instructions for Use in All Patients
Administer voriconazole tablets  at least one hour before or after a meal.
2.3 Recommended Dosing Regimen in Adults
Invasive aspergillosis and serious fungal infections due to Fusarium spp. and Scedosporium apiospermum
See Table 1. Therapy must be initiated with the specified loading dose regimen of intravenous voriconazole on Day 1 followed by the recommended maintenance dose (RMD) regimen. Intravenous treatment should be continued for at least 7 days. Once the patient has clinically improved and can tolerate medication given by mouth, the oral tablet form of voriconazole  may be utilized. The recommended oral maintenance dose of 200 mg achieves a voriconazole exposure similar to 3 mg/kg intravenously; a 300 mg oral dose achieves an exposure similar to 4 mg/kg intravenously [see Clinical Pharmacology (12.3)].
Candidemia in non-neutropenic patients and other deep tissue Candida infections
See Table 1. Patients should be treated for at least 14 days following resolution of symptoms or following last positive culture, whichever is longer.
Esophageal Candidiasis
See Table 1. Patients should be treated for a minimum of 14 days and for at least 7 days following resolution of symptoms.
Table 1:Recommended Dosing Regimen (Adults) a¬†¬†¬†¬†¬†¬†¬†¬†¬† Increase dose when voriconazole tablets are co-administered with phenytoin or efavirenz (7); Decrease dose in patients with hepatic impairment (2.5) b¬†¬†¬†¬†¬†¬†¬†¬†¬† In healthy volunteer studies, the 200 mg oral every 12 hours dose provided an exposure (AUCŌĄ) similar to a 3 mg/kg intravenous infusion every 12 hours dose; the 300 mg oral every 12 hours dose provided an exposure (AUCŌĄ) similar to a 4 mg/kg intravenous infusion every 12 hours dose (12). c¬†¬†¬†¬†¬†¬†¬†¬†¬† Adult patients who weigh less than 40 kg should receive half of the oral maintenance dose. d¬†¬†¬†¬†¬†¬†¬†¬†¬† In a clinical study of IA, the median duration of intravenous voriconazole therapy was 10 days (range 2 to 85 days). The median duration of oral voriconazole therapy was 76 days (range 2 to 232 days) (14.1). e¬†¬†¬†¬†¬†¬†¬†¬†¬† In clinical trials, patients with candidemia received 3 mg/kg intravenous infusion every 12 hours as primary therapy, while patients with other deep tissue Candida infections received 4 mg/kg every 12 hours as salvage therapy. Appropriate dose should be based on the severity and nature of the infection. f¬†¬†¬†¬†¬†¬†¬†¬†¬†¬† Not evaluated in patients with EC. Infection Loading Dose Maintenance Dosea,b ¬† Intravenous infusion Intravenous infusion Oralc Invasive Aspergillosisd 6 mg/kg every 12 hours for the first 24 hours 4 mg/kg every 12 hours 200 mg¬†every 12 hours Candidemia in nonneutropenic patients and other deep tissue Candida infections 6 mg/kg every 12 hours for the first 24 hours 3-4 mg/kg¬†every 12 hourse 200 mg¬†every 12 hours Esophageal Candidiasis Not Evaluatedf Not Evaluatedf 200 mg every 12 hours Scedosporiosis and Fusariosis 6 mg/kg every 12 hours for the first 24 hours 4 mg/kg every 12 hours 200 mg every 12 hours
Method for Adjusting the Dosing Regimen in Adults
If patient’s response is inadequate, the oral maintenance dose may be increased from 200 mg every 12 hours (similar to 3 mg/kg intravenously every 12 hours) to 300 mg every 12 hours (similar to 4 mg/kg intravenously every 12 hours). For adult patients weighing less than 40 kg, the oral maintenance dose may be increased from 100 mg every 12 hours to 150 mg every 12 hours. If patient is unable to tolerate 300 mg orally every 12 hours, reduce the oral maintenance dose by 50 mg steps to a minimum of 200 mg every 12 hours (or to 100 mg every 12 hours for adult patients weighing less than 40 kg).
If patient is unable to tolerate 4 mg/kg intravenously every 12 hours, reduce the intravenous maintenance dose to 3 mg/kg every 12 hours.
2.4 Recommended Dosing Regimen in Pediatric Patients
The recommended dosing regimen for pediatric patients 2 to less than 12 years of age and 12 to 14 years of age with body weight less than 50 kg is shown in Table 2. For pediatric patients 12 to 14 years of age with a body weight greater than or equal to 50 kg and those 15 years of age and above regardless of body weight, administer the adult dosing regimen of voriconazole tablets[see Dosage and Administration (2.3)].
Table 2: Recommended Dosing Regimen for Pediatric Patients 2 to less than 12 years of age and 12 to 14 years of age with body weight less than 50 kg^ ^ Based on a population pharmacokinetic analysis in 112 immunocompromised pediatric patients aged 2 to less than 12 years of age and 26 immunocompromised pediatric patients aged 12 to less than 17 years of age. * In the Phase 3 clinical trials, patients with IA received intravenous (IV) treatment for at least 6 weeks and up to a maximum of 12 weeks. Patients received IV treatment for at least the first 7 days of therapy and then could be switched to oral voriconazole therapy.   † Study treatment for primary or salvage invasive candidiasis and candidemia (ICC) or EC consisted of intravenous voriconazole, with an option to switch to oral therapy after at least 5 days of IV therapy, based on subjects meeting switch criteria. For subjects with primary or salvage ICC, voriconazole was administered for at least 14 days after the last positive culture. A maximum of 42 days of treatment was permitted. Patients with primary or salvage EC were treated for at least 7 days after the resolution of clinical signs and symptoms. A maximum of 42 days of treatment was permitted.      Loading Dose    Maintenance Dose        Intravenous  infusion    Intravenous  infusion    Oral    Invasive Aspergillosis*    9 mg/kg every  12 hours for the first 24 hours   8 mg/kg every  12 hours after the first 24 hours   9 mg/kg every 12 hours (maximum dose of 350 mg every 12 hours)   Candidemia in nonneutropenics  and other deep tissue Candida infections †    Scedosporiosis and Fusariosis    Esophageal Candidiasis†    Not Evaluated   4 mg/kg every  12 hours   9 mg/kg every  12 hours (maximum dose of 350 mg every 12 hours) 
Initiate therapy with an intravenous infusion regimen. Consider an oral regimen only after there is a significant clinical improvement. Note that an 8 mg/kg intravenous dose will provide voriconazole exposure approximately 2-fold higher than a 9 mg/kg oral dose. 
The oral dose recommendation for children is based on studies in which voriconazole was administered as the powder for oral suspension formulation. Bioequivalence between the voriconazole powder for oral suspension and voriconazole tablets has not been investigated in a pediatric population.
Oral bioavailability may be limited in pediatric patients 2 to 12 years with malabsorption and very low body weight for age. In that case, intravenous voriconazole administration is recommended.
Method for Adjusting the Dosing Regimen in Pediatric Patients
Pediatric Patients 2 to less than 12 years of age and 12 to 14 years of age with body weight less than 50 kg
If patient response is inadequate and the patient is able to tolerate the initial intravenous maintenance dose, the maintenance dose may be increased by 1 mg/kg steps. If patient response is inadequate and the patient is able to tolerate the oral maintenance dose, the dose may be increased by 1 mg/kg steps or 50 mg steps to a maximum of 350 mg every 12 hours. If patients are unable to tolerate the initial intravenous maintenance dose, reduce the dose by 1 mg/kg steps. If patients are unable to tolerate the oral maintenance dose, reduce the dose by 1 mg/kg or 50 mg steps.
Pediatric patients 12 to 14 years of age weighing greater than or equal to 50 kg and 15 years of age and older regardless of body weight:
Use the optimal method for titrating dosage recommended for adults [see Dosage and Administration (2.3)].
2.5 Dosage Modifications in Patients With Hepatic Impairment
Adults
The maintenance dose of voriconazole should be reduced in adult patients with mild to moderate hepatic impairment, Child-Pugh Class A and B. There are no PK data to allow for dosage adjustment recommendations in patients with severe hepatic impairment (Child-Pugh Class C).
Duration of therapy should be based on the severity of the patient’s underlying disease, recovery from immunosuppression, and clinical response.
Adult patients with baseline liver function tests (ALT, AST) of up to 5 times the upper limit of normal (ULN) were included in the clinical program. Dose adjustments are not necessary for adult patients with this degree of abnormal liver function, but continued monitoring of liver function tests for further elevations is recommended [see Warnings and Precautions (5.1)].
It is recommended that the recommended voriconazole loading dose regimens be used, but that the maintenance dose be halved in adult patients with mild to moderate hepatic cirrhosis (Child-Pugh Class A and B) [see Clinical Pharmacology (12.3)].
Voriconazole has not been studied in adult patients with severe hepatic cirrhosis (Child-Pugh Class C) or in patients with chronic hepatitis B or chronic hepatitis C disease. Voriconazole has been associated with elevations in liver function tests and with clinical signs of liver damage, such as jaundice. Voriconazole tablets should only be used in patients with severe hepatic impairment if the benefit outweighs the potential risk. Patients with hepatic impairment must be carefully monitored for drug toxicity.
Pediatric Patients
 Dosage adjustment of voriconazole tablets in pediatric patients with hepatic impairment has not been established [see Use in Specific Populations (8.4)].
2.6 Dosage Modifications in Patients With Renal Impairment
Adult Patients
The pharmacokinetics of orally administered voriconazole tablets are not significantly affected by renal impairment. Therefore, no adjustment is necessary for oral dosing in patients with mild to severe renal impairment [see Clinical Pharmacology (12.3)].
In patients with moderate or severe renal impairment (creatinine clearance <50 mL/min) who are receiving an intravenous infusion of voriconazole, accumulation of the intravenous vehicle, SBECD, occurs. Oral voriconazole should be administered to these patients, unless an assessment of the benefit/risk to the patient justifies the use of intravenous voriconazole. Serum creatinine levels should be closely monitored in these patients, and, if increases occur, consideration should be given to changing to oral voriconazole therapy [see Warnings and Precautions (5.7)].
Voriconazole and the intravenous vehicle, SBECD, are dialyzable. A 4-hour hemodialysis session does not remove a sufficient amount of voriconazole to warrant dose adjustment [see Clinical Pharmacology (12.3)].
Pediatric Patients
Dosage adjustment of voriconazole tablets in pediatric patients with renal impairment has not been established [see Use in Specific Populations (8.4)].
2.7 Dosage Adjustment When Co-Administered With Phenytoin or Efavirenz
The maintenance dose of voriconazole should be increased when co-administered with phenytoin or efavirenz. Use the optimal method for titrating dosage [see Drug Interactions (7) and Dosage and Administration (2.3)].
3 Dosage Forms And Strengths
Tablets
Voriconazole 50 mg tablets; white, round-shaped, biconvex, film-coated tablets debossed with ‚ÄėV50‚Äô on one side and plain on other side. Voriconazole 200 mg tablets; white, oval-shaped, biconvex, film-coated tablets debossed with ‚ÄėV200‚Äô on one side and plain on other side.
- Tablets: 50 mg, 200 mg
4 Contraindications
- Voriconazole tablets are contraindicated in patients with known hypersensitivity to voriconazole or its excipients. There is no information regarding cross-sensitivity between voriconazole and other azole antifungal agents. Caution should be used when prescribing voriconazole tablets to patients with hypersensitivity to other azoles.
Coadministration of pimozide, quinidine or ivabradine with voriconazole tablets is contraindicated because increased plasma concentrations of these drugs can lead to QT prolongation and rare occurrences of torsade de pointes [see Drug Interactions (7)]. - Coadministration of voriconazole tablets with sirolimus is contraindicated because voriconazole tablets significantly increases sirolimus concentrations [see Drug Interactions (7) and Clinical Pharmacology (12.3)].
Coadministration of voriconazole tablets with rifampin, carbamazepine, long-acting barbiturates, and St John’s Wort is contraindicated because these drugs are likely to decrease plasma voriconazole concentrations significantly [see Drug Interactions (7) and Clinical Pharmacology (12.3)]. - Coadministration of standard doses of voriconazole with efavirenz doses of 400 mg every 24 hours or higher is contraindicated, because efavirenz significantly decreases plasma voriconazole concentrations in healthy subjects at these doses. Voriconazole also significantly increases efavirenz plasma concentrations [see Drug Interactions (7) and Clinical Pharmacology (12.3)].
- Coadministration of voriconazole tablets with high-dose ritonavir (400 mg every 12 hours) is contraindicated because ritonavir (400 mg every 12 hours) significantly decreases plasma voriconazole concentrations. Coadministration of voriconazole and low-dose ritonavir (100 mg every 12 hours) should be avoided, unless an assessment of the benefit/risk to the patient justifies the use of voriconazole [see Drug Interactions (7) and Clinical Pharmacology (12.3)].
- Coadministration of voriconazole tablets with rifabutin is contraindicated since voriconazole tablets significantly increases rifabutin plasma concentrations and rifabutin also significantly decreases voriconazole plasma concentrations [see Drug Interactions (7) and Clinical Pharmacology (12.3)].
- Coadministration of voriconazole tablets with ergot alkaloids (ergotamine and dihydroergotamine) is contraindicated because voriconazole may increase the plasma concentration of ergot alkaloids, which may lead to ergotism [see Drug Interactions (7)].    
Coadministration of voriconazole tablets with naloxegol is contraindicated because voriconazole may increase plasma concentrations of naloxegol which may precipitate opioid withdrawal symptoms [see Drug Interactions (7)]. Coadministration of voriconazole tablets with tolvaptan is contraindicated because voriconazole may increase tolvaptan plasma concentrations and increase risk of adverse reactions [see Drug Interactions (7)]. Coadministration of voriconazole tablets with venetoclax at initiation and during the ramp-up phase is contraindicated in patients with chronic lymphocytic leukemia (CLL) or small lymphocytic lymphoma (SLL) due to the potential for increased risk of tumor lysis syndrome [see Drug Interactions (7)]. Coadministration of voriconazole tablets with lurasidone is contraindicated since it may result in significant increases in lurasidone exposure and the potential for serious adverse reactions [see Drug Interactions (7)].
- Hypersensitivity to voriconazole or its excipients (
4 )- Coadministration with pimozide or quinidine, sirolimus or ivabradine due to risk of serious adverse reactions (
4 ,7 )- Coadministration with rifampin, carbamazepine, long-acting barbiturates, efavirenz, ritonavir, rifabutin, ergot alkaloids, and St. John’s Wort due to risk of loss of efficacy (
4 ,7 )- Coadministration with naloxegol, tolvaptan, and lurasidone due to risk of adverse reactions (
4 ,7 )- Coadministration of voriconazole tablets with venetoclax at initiation and during the ramp-up phase in patients with chronic lymphocytic leukemia (CLL) or small lymphocytic lymphoma (SLL) due to increased risk of adverse reactions (
4 ,7 )
5 Warnings And Precautions
- Hepatic Toxicity: Serious hepatic reactions reported. Evaluate liver function tests at start of and during voriconazole therapy (
5.1 )- Arrhythmias and QT Prolongation: Correct potassium, magnesium and calcium prior to use; caution patients with proarrhythmic conditions (
5.2 )- Visual Disturbances (including optic neuritis and papilledema): Monitor visual function if treatment continues beyond 28 days (
5.4 )- Severe Cutaneous Adverse Reactions: Discontinue for exfoliative cutaneous reactions (
5.5 )- Photosensitivity: Avoid sunlight due to risk of photosensitivity (
5.6 )- Adrenal Dysfunction: Carefully monitor patients receiving voriconazole tablets and corticosteroids (via all routes of administration) for adrenal dysfunction both during and after voriconazole tablets treatment. Instruct patients to seek immediate medical care if they develop signs and symptoms of Cushing’s syndrome or adrenal insufficiency (
5.8 )- Embryo-Fetal Toxicity: Voriconazole can cause fetal harm when administered to a pregnant woman. Inform pregnant patients of the potential hazard to the fetus. Advise females of reproductive potential to use effective contraception during treatment with voriconazole tablets (
5.9 ,8.1 ,8.3 )- Skeletal Adverse Reactions: Fluorosis and periostitis with long-term voriconazole therapy. Discontinue if these adverse reactions occur (
5.12 )- Clinically Significant Drug Interactions: Review patient’s concomitant medications (
5.13 ,7 )- Patients with Hereditary Galactose Intolerance, Lapp Lactase Deficiency or Glucose-Galactose Malabsorption: Voriconazole tablets should not be given to these patients because it contains lactose (
5.14 )5.1 Hepatic Toxicity
In clinical trials, there have been uncommon cases of serious hepatic reactions during treatment with voriconazole (including clinical hepatitis, cholestasis and fulminant hepatic failure, including fatalities). Instances of hepatic reactions were noted to occur primarily in patients with serious underlying medical conditions (predominantly hematological malignancy). Hepatic reactions, including hepatitis and jaundice, have occurred among patients with no other identifiable risk factors. Liver dysfunction has usually been reversible on discontinuation of therapy [see Adverse Reactions (6.1)] .
A higher frequency of liver enzyme elevations was observed in the pediatric population [see Adverse Reactions (6.1)] . Hepatic function should be monitored in both adult and pediatric patients.
Measure serum transaminase levels and bilirubin at the initiation of voriconazole therapy and monitor at least weekly for the first month of treatment. Monitoring frequency can be reduced to monthly during continued use if no clinically significant changes are noted. If liver function tests become markedly elevated compared to baseline, voriconazole tablets should be discontinued unless the medical judgment of the benefit/risk of the treatment for the patient justifies continued use [see Dosage and Administration (2.5) and Adverse Reactions (6.1)] .
5.2 Arrhythmias and QT Prolongation
Some azoles, including voriconazole, have been associated with prolongation of the QT interval on the electrocardiogram. During clinical development and postmarketing surveillance, there have been rare cases of arrhythmias, (including ventricular arrhythmias such as torsade de pointes), cardiac arrests and sudden deaths in patients taking voriconazole. These cases usually involved seriously ill patients with multiple confounding risk factors, such as history of cardiotoxic chemotherapy, cardiomyopathy, hypokalemia and concomitant medications that may have been contributory.
Voriconazole should be administered with caution to patients with potentially proarrhythmic conditions, such as:
- Congenital or acquired QT prolongation
- Cardiomyopathy, in particular when heart failure is present
- Sinus bradycardia
- Existing symptomatic arrhythmias
- Concomitant medicinal product that is known to prolong QT interval [see contraindications (4), Drug Interactions (7), and Clinical Pharmacology (12.3)]
Rigorous attempts to correct potassium, magnesium and calcium should be made before starting and during voriconazole therapy [see Clinical Pharmacology (12.3)].
5.4 Visual Disturbances
The effect of voriconazole tablets on visual function is not known if treatment continues beyond 28 days. There have been postmarketing reports of prolonged visual adverse reactions, including optic neuritis and papilledema. If treatment continues beyond 28 days, visual function including visual acuity, visual field, and color perception should be monitored [see Adverse Reactions (6.2)].
5.5 Severe Cutaneous Adverse Reactions
Severe cutaneous adverse reactions (SCARs), such as Stevens-Johnson syndrome (SJS), toxic epidermal necrolysis (TEN), and drug reaction with eosinophilia and systemic symptoms (DRESS), which can be life-threatening or fatal, have been reported during treatment with voriconazole tablets. If a patient develops a severe cutaneous adverse reaction, voriconazole tablets should be discontinued [see Adverse Reaction (6.1, 6.2)].
5.6 Photosensitivity
Voriconazole has been associated with photosensitivity skin reaction. Patients, including pediatric patients, should avoid exposure to direct sunlight during voriconazole treatment and should use measures such as protective clothing and sunscreen with high sun protection factor (SPF). If phototoxic reactions occur, the patient should be referred to a dermatologist and voriconazole tablets discontinuation should be considered. If voriconazole tablets are continued despite the occurrence of phototoxicity-related lesions, dermatologic evaluation should be performed on a systematic and regular basis to allow early detection and management of premalignant lesions. Squamous cell carcinoma of the skin (including cutaneous SCC in situ, or Bowen’s disease) and melanoma have been reported during long-term voriconazole tablets therapy in patients with photosensitivity skin reactions. If a patient develops a skin lesion consistent with premalignant skin lesions, squamous cell carcinoma or melanoma, voriconazole tablets should be discontinued. In addition, voriconazole has been associated with photosensitivity related skin reactions such as pseudoporphyria, cheilitis, and cutaneous lupus erythematosus, as well as increased risk of skin toxicity with concomitant use of methotrexate, a drug associated with ultraviolet (UV) reactivation. There is the potential for this risk to be observed with other drugs associated with ultraviolet (UV) reactivation. Patients should avoid strong, direct sunlight during voriconazole therapy.
The frequency of phototoxicity reactions is higher in the pediatric population. Because squamous cell carcinoma has been reported in patients who experience photosensitivity reactions, stringent measures for photoprotection are warranted in children. In children experiencing photoaging injuries such as lentigines or ephelides, sun avoidance and dermatologic follow-up are recommended even after treatment discontinuation.
5.7 Renal Toxicity
Acute renal failure has been observed in patients undergoing treatment with voriconazole tablets. Patients being treated with voriconazole are likely to be treated concomitantly with nephrotoxic medications and may have concurrent conditions that may result in decreased renal function.
Patients should be monitored for the development of abnormal renal function. This should include laboratory evaluation of serum creatinine [see Clinical Pharmacology (12.3) and Dosage and Administration (2.6)].
5.8 Adrenal Dysfunction
Reversible cases of azole-induced adrenal insufficiency have been reported in patients receiving azoles, including voriconazole tablets. Adrenal insufficiency has been reported in patients receiving azoles with or without concomitant corticosteroids. In patients receiving azoles without corticosteroids adrenal insufficiency is related to direct inhibition of steroidogenesis by azoles. In patients taking corticosteroids, voriconazole associated CYP3A4 inhibition of their metabolism may lead to corticosteroid excess and adrenal suppression [see Drug Interactions (7) and Clinical Pharmacology (12.3)]. Cushing’s syndrome with and without subsequent adrenal insufficiency has also been reported in patients receiving voriconazole tablets concomitantly with corticosteroids.
Patients receiving voriconazole tablets and corticosteroids (via all routes of administration) should be carefully monitored for adrenal dysfunction both during and after voriconazole tablets treatment. Patients should be instructed to seek immediate medical care if they develop signs and symptoms of Cushing’s syndrome or adrenal insufficiency.
5.9 Embryo-Fetal Toxicity
Voriconazole can cause fetal harm when administered to a pregnant woman.
In animals, voriconazole administration was associated with fetal malformations, embryotoxicity, increased gestational length, dystocia and embryomortality [see Use in Specific Populations (8.1)].
If voriconazole tablets are used during pregnancy, or if the patient becomes pregnant while taking voriconazole tablets, inform the patient of the potential hazard to the fetus. Advise females of reproductive potential to use effective contraception during treatment with voriconazole tablets[see Use in Specific Populations (8.3)].
5.10 Laboratory Tests
Electrolyte disturbances such as hypokalemia, hypomagnesemia and hypocalcemia should be corrected prior to initiation of and during voriconazole tablets therapy.
Patient management should include laboratory evaluation of renal (particularly serum creatinine) and hepatic function (particularly liver function tests and bilirubin).
5.11 Pancreatitis
Pancreatitis has been observed in patients undergoing treatment with voriconazole tablets  [see Adverse Reactions (6.1, 6.2)]. Patients with risk factors for acute pancreatitis (e.g., recent chemotherapy, hematopoietic stem cell transplantation [HSCT]) should be monitored for the development of pancreatitis during voriconazole tablets treatment.
5.12 Skeletal Adverse Reactions
Fluorosis and periostitis have been reported during long-term voriconazole therapy. If a patient develops skeletal pain and radiologic findings compatible with fluorosis or periostitis, voriconazole should be discontinued [see Adverse Reactions (6.2)].
5.13 Clinically Significant Drug Interactions
See Table 10 for a uling of drugs that may significantly alter voriconazole concentrations. Also, see Table 11 for a uling of drugs that may interact with voriconazole resulting in altered pharmacokinetics or pharmacodynamics of the other drug [see Contraindications (4) and Drug Interactions (7)].
5.14 Galactose Intolerance
Voriconazole tablets contain lactose and should not be given to patients with rare hereditary problems of galactose intolerance, Lapp lactase deficiency or glucose-galactose malabsorption.
6 Adverse Reactions
The following serious adverse reactions are described elsewhere in the labeling:
Hepatic Toxicity [see Warnings and Precautions (5.1)]
Arrhythmias and QT Prolongation [see Warnings and Precautions (5.2)]
Visual Disturbances [see Warnings and Precautions (5.4)]
Severe Cutaneous Adverse Reactions [see Warnings and Precautions (5.5)]
Photosensitivity [see Warnings and Precautions (5.6)]
Renal Toxicity [see Warnings and Precautions (5.7)]
- Adult Patients: The most common adverse reactions (incidence ‚Č•2%) were visual disturbances, fever, nausea, rash, vomiting, chills, headache, liver function test abnormal, tachycardia, hallucinations (
6 )- Pediatric Patients: The most common adverse reactions (incidence ‚Č• 5%) were visual disturbances, pyrexia, vomiting, epistaxis, nausea, rash, abdominal pain, diarrhea, hypertension, hypokalemia, cough, headache, thrombocytopenia, ALT abnormal, hypotension, peripheral edema, hyperglycemia, tachycardia, dyspnea, hypocalcemia, hypophosphatemia, LFT abnormal, mucosal inflammation, photophobia, abdominal distention, constipation, dizziness, hallucinations, hemoptysis, hypoalbuminemia, hypomagnesemia, renal impairment, upper respiratory tract infection (
6 )
To report SUSPECTED ADVERSE REACTIONS, contact Ajanta Pharma USA Inc. at 855-664-7744 or FDA at 1-800-FDA-1088 orwww.fda.gov/medwatch .
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
Clinical Trials Experience in Adults
Overview
The most frequently reported adverse reactions (see Table 4) in the adult therapeutic trials were visual disturbances (18.7%), fever (5.7%), nausea (5.4%), rash (5.3%), vomiting (4.4%), chills (3.7%), headache (3.0%), liver function test increased (2.7%), tachycardia (2.4%), hallucinations (2.4%). The adverse reactions which most often led to discontinuation of voriconazole therapy were elevated liver function tests, rash, and visual disturbances [see Warning and Precautions (5.1, 5.4)  and Adverse Reactions (6.1) ].
The data described in Table¬†4 reflect exposure to voriconazole in 1655 patients in nine therapeutic studies. This represents a heterogeneous population, including immunocompromised patients, e.g., patients with hematological malignancy or HIV and non-neutropenic patients. This subgroup does not include healthy subjects and patients treated in the compassionate use and non-therapeutic studies. This patient population was 62% male, had a mean age of 46 years (range 11-90, including 51 patients aged 12-18 years), and was 78% White and 10% Black. Five hundred sixty one patients had a duration of voriconazole therapy of greater than 12 weeks, with 136 patients receiving voriconazole for over six months. Table¬†4 includes all adverse reactions which were reported at an incidence of ‚Č•2% during voriconazole therapy in the all therapeutic studies population, studies 307/602 and 608 combined, or study 305, as well as events of concern which occurred at an incidence of <2%.
In study 307/602, 381 patients (196 on voriconazole, 185 on amphotericin B) were treated to compare voriconazole to amphotericin B followed by other licensed antifungal therapy (OLAT) in the primary treatment of patients with acute IA. The rate of discontinuation from voriconazole study medication due to adverse reactions was 21.4% (42/196 patients). In study 608, 403 patients with candidemia were treated to compare voriconazole (272 patients) to the regimen of amphotericin B followed by fluconazole (131 patients). The rate of discontinuation from voriconazole study medication due to adverse reactions was 19.5% out of 272 patients. Study 305 evaluated the effects of oral voriconazole (200 patients) and oral fluconazole (191 patients) in the treatment of EC. The rate of discontinuation from voriconazole study medication in Study 305 due to adverse reactions was 7% (14/200 patients). Laboratory test abnormalities for these studies are discussed under Clinical Laboratory Values below.
Table 4: Adverse Reactions Rate ‚Č• 2% on Voriconazole or Adverse Reactions¬†of Concern in Therapeutic Studies Population, Studies 307/602-608 Combined, or Study 305. Possibly Related to Therapy or Causality Unknown‚Ć ‚Ć Study 307/602: IA; Study 608: candidemia; Study 305: EC¬†* Studies 303, 304, 305, 307, 309, 602, 603, 604, 608¬†**Amphotericin B followed by other licensed antifungal therapy ¬† *** See Warnings and Precautions (5.4) ¬† Therapeutic Studies* Studies 307/602 and 608 (IV/ oral therapy) Study 305 (oral therapy) ¬† Voriconazole N=1655 Voriconazole N=468 Ampho B** N=185 Ampho B‚Üí Fluconazole N=131 Voriconazole N=200 Fluconazole N=191 ¬† N (%) N (%) N (%) N (%) N (%) N (%) Special Senses*** ¬† ¬† ¬† ¬† ¬† ¬† Abnormal vision 310 (18.7) 63 (13.5) 1 (0.5) 0 31 (15.5) 8 (4.2) Photophobia 37 (2.2) 8 (1.7) 0 0 5 (2.5) 2 (1.0) Chromatopsia 20 (1.2) 2 (0.4) 0 0 2 (1.0) 0 Body as a Whole ¬† ¬† ¬† ¬† ¬† ¬† Fever 94 (5.7) 8 (1.7) 25 (13.5) 5 (3.8) 0 0 Chills 61 (3.7) 1 (0.2) 36 (19.5) 8 (6.1) 1 (0.5) 0 Headache 49 (3.0) 9 (1.9) 8 (4.3) 1 (0.8) 0 1 (0.5) Cardiovascular System ¬† ¬† ¬† ¬† ¬† ¬† Tachycardia 39 (2.4) 6 (1.3) 5 (2.7) 0 0 0 Digestive System ¬† ¬† ¬† ¬† ¬† ¬† Nausea 89 (5.4) 18 (3.8) 29 (15.7) 2 (1.5) 2 (1.0) 3 (1.6) Vomiting¬† 72 (4.4) 15 (3.2) 18 (9.7) 1 (0.8) 2 (1.0) 1 (0.5) Liver function tests abnormal 45 (2.7) 15 (3.2) 4 (2.2) 1 (0.8) 6 (3.0) 2 (1.0) Cholestatic jaundice 17 (1.0) 8 (1.7) 0 1 (0.8) 3 (1.5) 0 Metabolic and Nutritional Systems ¬† ¬† ¬† ¬† ¬† ¬† Alkaline phosphatase increased 59 (3.6) 19 (4.1) 4 (2.2) 3 (2.3) 10 (5.0) 3 (1.6) Hepatic enzymes increased 30 (1.8) 11 (2.4) 5 (2.7) 1 (0.8) 3 (1.5) 0 SGOT increased 31 (1.9) 9 (1.9) 0 1 (0.8) 8 (4.0) 2 (1.0) SGPT increased 29 (1.8) 9 (1.9) 1 (0.5) 2 (1.5) 6 (3.0) 2 (1.0) Hypokalemia 26 (1.6) 3 (0.6) 36 (19.5) 16 (12.2) 0 0 Bilirubinemia 15 (0.9) 5 (1.1) 3 (1.6) 2 (1.5) 1 (0.5) 0 Creatinine increased 4 (0.2) 0 59 (31.9) 10 (7.6) 1 (0.5) 0 Nervous System ¬† ¬† ¬† ¬† ¬† ¬† Hallucinations 39 (2.4) 13 (2.8) 1 (0.5) 0 0 0 Skin and Appendages ¬† ¬† ¬† ¬† ¬† ¬† Rash 88 (5.3) 20 (4.3) 7 (3.8) 1 (0.8) 3 (1.5) 1 (0.5) Urogenital ¬† ¬† ¬† ¬† ¬† ¬† Kidney function abnormal 10 (0.6) 6 (1.3) 40 (21.6) 9 (6.9) 1 (0.5) 1 (0.5) Acute kidney failure 7 (0.4) 2 (0.4) 11 (5.9) 7 (5.3) 0 0
Visual Disturbances
Voriconazole treatment-related visual disturbances are common. In therapeutic trials, approximately 21% of patients experienced abnormal vision, color vision change and/or photophobia. Visual disturbances may be associated with higher plasma concentrations and/or doses.
The mechanism of action of the visual disturbance is unknown, although the site of action is most likely to be within the retina. In a study in healthy subjects investigating the effect of 28-day treatment with voriconazole on retinal function, voriconazole caused a decrease in the electroretinogram (ERG) waveform amplitude, a decrease in the visual field, and an alteration in color perception. The ERG measures electrical currents in the retina. These effects were noted early in administration of voriconazole and continued through the course of study drug treatment. Fourteen days after the end of dosing, ERG, visual fields and color perception returned to normal [see Warnings and Precautions (5.4) ].
Dermatological Reactions
Dermatological reactions were common in patients treated with voriconazole. The mechanism underlying these dermatologic adverse reactions remains unknown.
Severe cutaneous adverse reactions (SCARs), including Stevens-Johnson syndrome (SJS), toxic epidermal necrolysis (TEN), and drug reaction with eosinophilia and systemic symptoms (DRESS) have been reported during treatment with voriconazole tablets. Erythema multiforme has also been reported during treatment with voriconazole tablets. [see Warnings and Precautions (5.5) and Adverse Reaction (6.2)].
Voriconazole tablets have also been associated with additional photosensitivity related skin reactions such as pseudoporphyria, cheilitis, and cutaneous lupus erythematosus [see Warnings and Precautions (5.6) and Adverse Reactions (6.2)].
Less Common Adverse Reactions
The following adverse reactions occurred in <2% of all voriconazole-treated patients in all therapeutic studies (N=1655). This uling includes events where a causal relationship to voriconazole cannot be ruled out or those which may help the physician in managing the risks to the patients. The ul does not include events included in Table 4 above and does not include every event reported in the voriconazole clinical program.
Body as a Whole: abdominal pain, abdomen enlarged, allergic reaction, anaphylactoid reaction, ascites, asthenia, back pain, chest pain, cellulitis, edema, face edema, flank pain, flu syndrome, graft versus host reaction, granuloma, infection, bacterial infection, fungal infection, injection site pain, injection site infection/inflammation, mucous membrane disorder, multi-organ failure, pain, pelvic pain, peritonitis, sepsis, substernal chest pain.
Cardiovascular: atrial arrhythmia, atrial fibrillation, AV block complete, bigeminy, bradycardia, bundle branch block, cardiomegaly, cardiomyopathy, cerebral hemorrhage, cerebral ischemia, cerebrovascular accident, congestive heart failure, deep thrombophlebitis, endocarditis, extrasystoles, heart arrest, hypertension, hypotension, myocardial infarction, nodal arrhythmia, palpitation, phlebitis, postural hypotension, pulmonary embolus, QT interval prolonged, supraventricular extrasystoles, supraventricular tachycardia, syncope, thrombophlebitis, vasodilatation, ventricular arrhythmia, ventricular fibrillation, ventricular tachycardia (including torsade de pointes) [see Warnings and Precautions (5.2)].
Digestive: anorexia, cheilitis, cholecystitis, cholelithiasis, constipation, diarrhea, duodenal ulcer perforation, duodenitis, dyspepsia, dysphagia, dry mouth, esophageal ulcer, esophagitis, flatulence, gastroenteritis, gastrointestinal hemorrhage, GGT/LDH elevated, gingivitis, glossitis, gum hemorrhage, gum hyperplasia, hematemesis, hepatic coma, hepatic failure, hepatitis, intestinal perforation, intestinal ulcer, jaundice, enlarged liver, melena, mouth ulceration, pancreatitis, parotid gland enlargement, periodontitis, proctitis, pseudomembranous colitis, rectal disorder, rectal hemorrhage, stomach ulcer, stomatitis, tongue edema.
Endocrine: adrenal cortex insufficiency, diabetes insipidus, hyperthyroidism, hypothyroidism.
Hemic and Lymphatic: agranulocytosis, anemia (macrocytic, megaloblastic, microcytic, normocytic), aplastic anemia, hemolytic anemia, bleeding time increased, cyanosis, DIC, ecchymosis, eosinophilia, hypervolemia, leukopenia, lymphadenopathy, lymphangitis, marrow depression, pancytopenia, petechia, purpura, enlarged spleen, thrombocytopenia, thrombotic thrombocytopenic purpura.
Metabolic and Nutritional: albuminuria, BUN increased, creatine phosphokinase increased, edema, glucose tolerance decreased, hypercalcemia, hypercholesteremia, hyperglycemia, hyperkalemia, hypermagnesemia, hypernatremia, hyperuricemia, hypocalcemia, hypoglycemia, hypomagnesemia, hyponatremia, hypophosphatemia, peripheral edema, uremia.
Musculoskeletal: arthralgia, arthritis, bone necrosis, bone pain, leg cramps, myalgia, myasthenia, myopathy, osteomalacia, osteoporosis.
Nervous System: abnormal dreams, acute brain syndrome, agitation, akathisia, amnesia, anxiety, ataxia, brain edema, coma, confusion, convulsion, delirium, dementia, depersonalization, depression, diplopia, dizziness, encephalitis, encephalopathy, euphoria, Extrapyramidal Syndrome, grand mal convulsion, Guillain-Barré syndrome, hypertonia, hypesthesia, insomnia, intracranial hypertension, libido decreased, neuralgia, neuropathy, nystagmus, oculogyric crisis, paresthesia, psychosis, somnolence, suicidal ideation, tremor, vertigo.
Respiratory System: cough increased, dyspnea, epistaxis, hemoptysis, hypoxia, lung edema, pharyngitis, pleural effusion, pneumonia, respiratory disorder, respiratory distress syndrome, respiratory tract infection, rhinitis, sinusitis, voice alteration.
Skin and Appendages: alopecia, angioedema, contact dermatitis, discoid lupus erythematosis, eczema, erythema multiforme, exfoliative dermatitis, fixed drug eruption, furunculosis, herpes simplex, maculopapular rash, melanoma, melanosis, photosensitivity skin reaction, pruritus, pseudoporphyria, psoriasis, skin discoloration, skin disorder, skin dry, Stevens-Johnson syndrome, squamous cell carcinoma (including cutaneous SCC in situ, or Bowen’s disease), sweating, toxic epidermal necrolysis, urticaria.
Special Senses: abnormality of accommodation, blepharitis, color blindness, conjunctivitis, corneal opacity, deafness, ear pain, eye pain, eye hemorrhage, dry eyes, hypoacusis, keratitis, keratoconjunctivitis, mydriasis, night blindness, optic atrophy, optic neuritis, otitis externa, papilledema, retinal hemorrhage, retinitis, scleritis, taste loss, taste perversion, tinnitus, uveitis, visual field defect.
Urogenital: anuria, blighted ovum, creatinine clearance decreased, dysmenorrhea, dysuria, epididymitis, glycosuria, hemorrhagic cystitis, hematuria, hydronephrosis, impotence, kidney pain, kidney tubular necrosis, metrorrhagia, nephritis, nephrosis, oliguria, scrotal edema, urinary incontinence, urinary retention, urinary tract infection, uterine hemorrhage, vaginal hemorrhage.
Clinical Laboratory Values in Adults
The overall incidence of transaminase increases >3x upper limit of normal (not necessarily comprising an adverse reaction) was 17.7% (268/1514) in adult subjects treated with voriconazole for therapeutic use in pooled clinical trials. Increased incidence of liver function test abnormalities may be associated with higher plasma concentrations and/or doses. The majority of abnormal liver function tests either resolved during treatment without dose adjustment or resolved following dose adjustment, including discontinuation of therapy.
Voriconazole has been infrequently associated with cases of serious hepatic toxicity including cases of jaundice and rare cases of hepatitis and hepatic failure leading to death. Most of these patients had other serious underlying conditions.
Liver function tests should be evaluated at the start of and during the course of voriconazole  tablets therapy. Patients who develop abnormal liver function tests during voriconazole tablets therapy should be monitored for the development of more severe hepatic injury. Patient management should include laboratory evaluation of hepatic function (particularly liver function tests and bilirubin). Discontinuation of voriconazole tablets must be considered if clinical signs and symptoms consistent with liver disease develop that may be attributable to voriconazole tablets [see Warnings and Precautions (5.1)].
Acute renal failure has been observed in severely ill patients undergoing treatment with voriconazole  tablets. Patients being treated with voriconazole are likely to be treated concomitantly with nephrotoxic medications and may have concurrent conditions that can result in decreased renal function. It is recommended that patients are monitored for the development of abnormal renal function. This should include laboratory evaluation of serum creatinine.
Tables 5 to 7 show the number of patients with hypokalemia and clinically significant changes in renal and liver function tests in three randomized, comparative multicenter studies. In study 305, patients with EC were randomized to either oral voriconazole or oral fluconazole. In study 307/602, patients with definite or probable IA were randomized to either voriconazole or amphotericin B therapy. In study 608, patients with candidemia were randomized to either voriconazole or the regimen of amphotericin B followed by fluconazole.
Table 5: Protocol 305 ‚Äď Patients with Esophageal CandidiasisClinically Significant Laboratory Test Abnormalities *¬†¬†¬†¬† Without regard to baseline valuen = number of patients with a clinically significant abnormality while on study therapyN = total number of patients with at least one observation of the given lab test while on study therapyAST = Aspartate aminotransferase; ALT= alanine aminotransferaseULN = upper limit of normal ¬† Criteria* Voriconazole Fluconazole ¬† ¬† n/N (%) n /N (%) T. Bilirubin >1.5x ULN 8/185 (4.3) 7/186 (3.8) AST >3.0x ULN 38/187 (20.3) 15/186 (8.1) ALT >3.0x ULN 20/187 (10.7) 12/186 (6.5) Alkaline Phosphatase >3.0x ULN 19/187 (10.2) 14/186 (7.5)
Table 6: Protocol 307/602 ‚Äď Primary Treatment of Invasive Aspergillosis Clinically Significant Laboratory Test Abnormalities *¬†¬†¬†¬†¬†¬†¬† Without regard to baseline value **¬†¬†¬†¬†¬† Amphotericin B followed by other licensed antifungal therapyn = number of patients with a clinically significant abnormality while on study therapyN = total number of patients with at least one observation of the given lab test while on study therapyAST = Aspartate aminotransferase; ALT= alanine aminotransferaseULN = upper limit of normalLLN = lower limit of normal ¬† Criteria* Voriconazole Amphotericin B** ¬† ¬† n/N (%) n/N (%) T. Bilirubin >1.5x ULN 35/180 (19.4) 46/173 (26.6) AST >3.0x ULN 21/180 (11.7) 18/174 (10.3) ALT >3.0x ULN 34/180 (18.9) 40/173 (23.1) Alkaline Phosphatase >3.0x ULN 29/181 (16.0) 38/173 (22.0) Creatinine >1.3x ULN 39/182 (21.4) 102/177 (57.6) Potassium <0.9x LLN 30/181 (16.6) 70/178 (39.3)
Table 7: Protocol 608 ‚Äď Treatment of Candidemia Clinically Significant Laboratory Test Abnormalities * ¬†¬†¬†¬†¬†¬†Without regard to baseline valuen = number of patients with a clinically significant abnormality while on study therapyN = total number of patients with at least one observation of the given lab test while on study therapyAST = Aspartate aminotransferase; ALT= alanine aminotransferaseULN = upper limit of normalLLN = lower limit of normal ¬† Criteria* ¬† Voriconazole ¬† Amphotericin B followed by Fluconazole ¬† ¬† n/N (%) n/N (%) T. Bilirubin >1.5x ULN 50/261 (19.2) 31/115 (27.0) AST >3.0x ULN 40/261 (15.3) 16/116 (13.8) ALT >3.0x ULN 22/261 (8.4) 15/116 (12.9) Alkaline Phosphatase >3.0x ULN 59/261 (22.6) 26/115 (22.6) Creatinine >1.3x ULN 39/260 (15.0) 32/118 (27.1) Potassium <0.9x LLN 43/258 (16.7) 35/118 (29.7)
Clinical Trials Experience in Pediatric Patients
The safety of voriconazole was investigated in 105 pediatric patients aged 2 to less than 18 years, including 52 pediatric patients less than 18 years of age who were enrolled in the adult therapeutic studies.
Serious Adverse Reactions and Adverse Reactions Leading to Discontinuation
In clinical studies, serious adverse reactions occurred in 46% (48/105) of voriconazole treated pediatric patients. Treatment discontinuations due to adverse reactions occurred in 12 /105 (11%) of all patients. Hepatic adverse reactions (i.e. ALT increased; liver function test abnormal; jaundice) 6% (6/105) accounted for the majority of voriconazole treatment discontinuations.
Most Common Adverse Reactions
The most common adverse reactions occurring in ‚Č•5% of pediatric patients receiving voriconazole tablets in the pooled pediatric clinical trials are displayed by body system, in Table 8.
                                                                                                                                                         
Table 8: Adverse Reactions Occurring in ‚Č•5% of Pediatric Patients ReceivingVoriconazole tablets in the Pooled Pediatric Clinical Trials a Reflects all adverse reactions and not treatment-related only. b Pooled reports include such terms as: amaurosis (partial or total blindness without visible change in the eye); asthenopia (eye strain); chromatopsia (abnormally colored vision); color blindness; diplopia; photopsia; retinal disorder; vision blurred, visual acuity decreased, visual brightness; visual impairment. Several patients had more than one visual disturbance. c Pooled reports include such terms as: abdominal pain and abdominal pain, upper.d Pooled reports include such terms as: ALT abnormal and ALT increased. e Pooled reports include such terms as: hallucination; hallucination, auditory; hallucination, visual. Several patients had both visual and auditory hallucinations. f Pooled reports include such terms as: renal failure and a single patient with renal impairment. g Pooled reports include such terms as: rash; rash generalized; rash macular; rash maculopapular; rash pruritic.Abbreviations: ALT = alanine aminotransferase; LFT = liver function test ¬†Body System ¬† ¬†Adverse Reaction ¬† ¬†Pooled Pediatric Dataa ¬†N=105 ¬†n (%) ¬† ¬†Blood and Lymphatic Systems Disorders ¬† ¬†Thrombocytopenia ¬† ¬†10 (10)¬† ¬†Cardiac Disorders ¬† ¬†Tachycardia ¬† ¬†7 (7)¬† ¬†Eye Disorders ¬† ¬†Visual Disturbancesb ¬† ¬†27 (26)¬† ¬†Photophobia¬† ¬†6 (6)¬† ¬†Gastrointestinal Disorders ¬† ¬†Vomiting ¬† ¬†21 (20)¬† ¬†Nausea¬† ¬†14 (13)¬† ¬†Abdominal painc ¬† ¬†13 (12)¬† ¬†Diarrhea¬† ¬†12 (11)¬† ¬†Abdominal distention ¬† ¬†5 (5)¬† ¬†Constipation ¬† ¬†5 (5)¬† General Disorders and Administration Site Conditions¬† ¬†Pyrexia ¬† ¬†25 (25)¬† ¬†Peripheral edema ¬† ¬†9 (9)¬† ¬†Mucosal inflammation ¬† ¬†6 (6)¬† ¬†Infections and Infestations ¬† ¬†Upper respiratory tract infection ¬† ¬†5 (5)¬† ¬†Investigations ¬† ¬†ALT abnormald ¬† ¬†9 (9)¬† ¬†LFT abnormal ¬† ¬†6 (6)¬† ¬†Metabolism and Nutrition Disorders ¬† ¬†Hypokalemia¬† ¬†11 (11)¬† ¬†Hyperglycemia¬† ¬†7 (7)¬† ¬†Hypocalcemia ¬† ¬†6 (6)¬† ¬†Hypophosphotemia ¬† ¬†6 (6)¬† ¬†Hypoalbuminemia ¬† ¬†5 (5)¬† ¬†Hypomagnesemia ¬† ¬†5 (5)¬† ¬†Nervous System Disorders ¬† ¬†Headache ¬† ¬†10 (10)¬† ¬†Dizziness ¬† ¬†5 (5)¬† ¬†Psychiatric Disorders ¬† ¬†Hallucinationse ¬† ¬†5 (5)¬† ¬†Renal and Urinary Disorders ¬† ¬†Renal impairmentf ¬† ¬†5 (5)¬† ¬†Respiratory Disorders ¬† ¬†Epistaxis ¬† ¬†17 (16)¬† ¬†Cough¬† ¬†10 (10)¬† ¬†Dyspnea ¬† ¬†6 (6)¬† ¬†Hemoptysis ¬† ¬†5 (5)¬† ¬†Skin and Subcutaneous Tissue Disorders ¬† ¬†Rashg ¬† ¬†14 (13)¬† ¬†Vascular Disorders ¬† ¬†Hypertension ¬† ¬†12 (11)¬† ¬†Hypotension ¬† ¬†9 (9)¬†
The following adverse reactions with incidence less than 5% were reported in 105 pediatric patients treated with voriconazole:
Blood and Lymphatic System Disorders: anemia, leukopenia, pancytopenia
Cardiac Disorders: bradycardia, palpitations, supraventricular tachycardia
Eye Disorders: dry eye, keratitis
Ear and Labyrinth Disorders: tinnitus, vertigo
Gastrointestinal Disorders: abdominal tenderness, dyspepsia
General Disorders and Administration Site Conditions: asthenia, catheter site pain, chills, hypothermia, lethargy
Hepatobiliary Disorders: cholestasis, hyperbilirubinemia, jaundice
Immune System Disorders: hypersensitivity, urticaria
Infections and Infestations: conjunctivitis
Laboratory Investigations: AST increased, blood creatinine increased, gamma-glutamyl transferase increased
Metabolism and Nutrition Disorders: hypercalcemia, hypermagnesemia, hyperphosphatemia, hypoglycemia
Musculoskeletal and Connective Tissue Disorders: arthralgia, myalgia
Nervous System Disorders: ataxia, convulsion, dizziness, nystagmus, paresthesia, syncope
Psychiatric Disorders: affect lability, agitation, anxiety, depression, insomnia
Respiratory Disorders: bronchospasm, nasal congestion, respiratory failure, tachypnea
Skin and Subcutaneous Tissue Disorders: alopecia, dermatitis (allergic, contact, and exfoliative), pruritus
Vascular Disorders: flushing, phlebitis
Hepatic-Related Adverse Reactions in Pediatric Patients
The frequency of hepatic-related adverse reactions in pediatric patients exposed to voriconazole in therapeutic studies was numerically higher than that of adults (28.6% compared to 24.1%, respectively). The higher frequency of hepatic adverse reactions in the pediatric population was mainly due to an increased frequency of liver enzyme elevations (21.9% in pediatric patients compared to 16.1% in adults), including transaminase elevations (ALT and AST combined) 7.6% in the pediatric patients compared to 5.1% in adults.
Clinical Laboratory Values in Pediatric Patients
The overall incidence of transaminase increases >3x upper limit of normal was 27.2% (28/103) in pediatric and 17.7% (268/1514) in adult patients treated with voriconazole in pooled clinical trials. The majority of abnormal liver function tests either resolved on treatment with or without dose adjustment or after voriconazole discontinuation.
A higher frequency of clinically significant liver laboratory abnormalities, irrespective of baseline laboratory values (>3x ULN ALT or AST), was consistently observed in the combined therapeutic pediatric population (15.5% AST and 22.5% ALT) when compared to adults (12.9% AST and 11.6% ALT). The incidence of bilirubin elevation was comparable between adult and pediatric patients. The incidence of hepatic abnormalities in pediatric patients is shown in Table 9.
Table 9: Incidence of Hepatic Abnormalities among Pediatric Subjects n = number of patients with a clinically significant abnormality while on study therapyN = total number of patients with at least one observation of the given lab test while on study therapyAST = Aspartate aminotransferase; ALT = alanine aminotransferaseULN = upper limit of normal      Criteria    n/N (%)    Total bilirubin    >1.5x ULN   19/102 (19)   AST    >3.0x ULN   16/103 (16)   ALT    >3.0x ULN   23/102 (23)   Alkaline Phosphatase    >3.0x ULN   8/97 (8)  6.2 Postmarketing Experience in Adult and Pediatric Patients
The following adverse reactions have been identified during post-approval use of voriconazole. Because these reactions are reported voluntarily from a population of uncertain size, it is not always possible to reliably estimate their frequency or establish a causal relationship to drug exposure.
Dermatological Reactions
Increased risk of skin toxicity with concomitant use of methotrexate, a drug associated with UV reactivation, was observed in postmarketing reports [see Warnings and Precautions (5.6) and Adverse Reactions (6.1)].
Adults
Skeletal: fluorosis and periostitis have been reported during long-term voriconazole therapy [see Warnings and Precautions (5.12)].
Eye disorders: prolonged visual adverse reactions, including optic neuritis and papilledema [see Warnings and Precautions (5.4)].
Skin and Appendages: drug reaction with eosinophilia and systemic symptoms (DRESS) has been reported [see Warnings and Precautions (5.5) and Adverse Reactions (6.1)].
  Endocrine disorders: adrenal insufficiency, Cushing’s syndrome (when voriconazole has been used concomitantly with corticosteroids) [see Warnings and Precautions (5.8)].
Pediatric Patients
There have been postmarketing reports of pancreatitis in pediatric patients.
7 Drug Interactions
Voriconazole is metabolized by cytochrome P450 isoenzymes, CYP2C19, CYP2C9, and CYP3A4. Therefore, inhibitors or inducers of these isoenzymes may increase or decrease voriconazole plasma concentrations, respectively. Voriconazole is a strong inhibitor of CYP3A4, and also inhibits CYP2C19 and CYP2C9. Therefore, voriconazole may increase the plasma concentrations of substances metabolized by these CYP450 isoenzymes.  
Tables 10 and 11 provide the clinically significant interactions between voriconazole and other medical products.
Table 10:Effect of Other Drugs on Voriconazole Pharmacokinetics [see Clinical Pharmacology (12.3)] ¬†Drug/Drug Class ¬†(Mechanism of Interaction by the Drug) ¬† ¬†Voriconazole Plasma Exposure (Cmax and AUC ŌĄ after 200 mg every 12 hours) ¬† ¬†Recommendations for Voriconazole Dosage Adjustment/Comments ¬† *¬†¬†¬† Results based on in vivo clinical studies generally following repeat oral dosing with 200 mg every 12 hours voriconazole to healthy subjects **¬†¬† Results based on in vivo clinical study following repeat oral dosing with 400 mg every 12 hours for 1 day, then 200 mg every 12 hours for at least 2 days voriconazole to healthy subjects¬†***¬†¬† Non-Nucleoside Reverse Transcriptase Inhibitors ¬†Rifampin* and Rifabutin* (CYP450 Induction)¬† ¬†Significantly Reduced¬† ¬†Contraindicated ¬† ¬†Efavirenz (400 mg every 24 hours)** ¬†(CYP450 Induction) ¬†¬†¬†Efavirenz (300 mg every 24 hours)** ¬†(CYP450 Induction) ¬†¬†¬† ¬†Significantly Reduced¬†¬†¬†¬†¬†Slight Decrease in AUCŌĄ ¬† ¬†Contraindicated ¬†¬†¬†¬†¬†When voriconazole is coadministered with efavirenz, voriconazole oral maintenance dose should be increased to 400 mg every 12 hours and efavirenz should be decreased to 300 mg every 24 hours.¬† ¬†High-dose Ritonavir (400 mg every 12 hours)** ¬†(CYP450 Induction) ¬†¬†¬†Low-dose Ritonavir (100 mg every 12 hours)** (CYP450 Induction)¬† ¬†Significantly Reduced ¬†¬†¬†¬†¬†¬†¬†Reduced¬† ¬†Contraindicated ¬†¬†¬†¬†¬†¬†¬†Coadministration of voriconazole and low-dose ritonavir (100 mg every 12 hours) should be avoided, unless an assessment of the benefit/risk to the patient justifies the use of voriconazole.¬† ¬†Carbamazepine¬†(CYP450 Induction)¬† ¬†Not Studied In Vivo or In Vitro, but Likely to Result in Significant Reduction¬† ¬†Contraindicated ¬† ¬†Long Acting Barbiturates (e.g., phenobarbital, mephobarbital) (CYP450 Induction)¬† ¬†Not Studied In Vivo or In Vitro, but Likely to Result in Significant Reduction¬† ¬†Contraindicated ¬† ¬†Phenytoin* ¬†(CYP450 Induction)¬† ¬†Significantly Reduced¬† ¬†Increase voriconazole maintenance dose from 4 mg/kg to 5 mg/kg IV every 12 hours or from 200 mg to 400 mg orally every 12 hours (100 mg to 200 mg orally every 12 hours in patients weighing less than 40 kg).¬† ¬†Letermovir ¬†(CYP2C9/2C19 Induction)¬† ¬†Reduced¬† ¬†If concomitant administration of voriconazole with letermovir cannot be avoided, monitor for reduced effectiveness of voriconazole.¬† ¬†St. John‚Äôs Wort ¬†(CYP450 inducer; P-gp inducer)¬† ¬†Significantly Reduced¬† ¬†Contraindicated ¬† ¬†Oral Contraceptives** ¬†containing ethinyl estradiol and norethindrone (CYP2C19 Inhibition)¬† ¬†Increased¬† ¬†Monitoring for adverse reactions and toxicity related to voriconazole is recommended when coadministered with oral contraceptives.¬† ¬†Fluconazole** (CYP2C9, CYP2C19 and CYP3A4 Inhibition) ¬† ¬†Significantly Increased¬† ¬†Avoid concomitant administration of voriconazole and fluconazole. Monitoring for adverse reactions ¬†and toxicity related to voriconazole is started within 24 hours after the last dose of fluconazole.¬† ¬†Other HIV Protease Inhibitors (CYP3A4 Inhibition) ¬† ¬†In Vivo Studies Showed No Significant Effects of Indinavir on Voriconazole Exposure¬†¬†¬†In Vitro Studies Demonstrated Potential for Inhibition of Voriconazole Metabolism ¬†(Increased Plasma Exposure)¬† ¬†No dosage adjustment in the voriconazole dosage needed when coadministered with indinavir.¬†¬†¬†Frequent monitoring for adverse¬†reactions¬†and toxicity related to voriconazole when coadministered with other HIV protease inhibitors.¬† ¬†Other NNRTIs*** ¬†(CYP3A4 Inhibition or CYP450 Induction) ¬† ¬†In Vitro Studies Demonstrated Potential for Inhibition of Voriconazole Metabolism by Delavirdine and Other NNRTIs (Increased Plasma Exposure)¬†¬†¬†A Voriconazole-Efavirenz Drug Interaction Study Demonstrated the Potential for the Metabolism of Voriconazole to be Induced by Efavirenz and Other NNRTIs ¬†(Decreased Plasma Exposure)¬† ¬†Frequent monitoring for adverse reactions and toxicity related to voriconazole.¬†¬†¬†¬†¬†¬†¬†¬†¬†Careful assessment of voriconazole effectiveness.¬†
 
                                                                                                                                                          
Table 11:Effect of Voriconazole on Pharmacokinetics of Other Drugs [see Clinical Pharmacology (12.3)] ¬†Drug/Drug Class ¬†(Mechanism of Interaction by Voriconazole) ¬† ¬†Drug Plasma Exposure ¬†(Cmax and AUC ŌĄ ) ¬† ¬†Recommendations for Drug Dosage Adjustment/Comments ¬† *¬†¬†¬†¬†¬† Results based on in vivo clinical studies generally following repeat oral dosing with 200 mg BID voriconazole to healthy subjects **¬†¬†¬†¬† Results based on in vivo clinical study following repeat oral dosing with 400 mg every 12 hours for1 day, then 200 mg every 12 hours¬† for at least 2 days voriconazole to healthy subjects ***¬†¬†¬† Results based on in vivo clinical study following repeat oral dosing with 400 mg every 12 hours for 1 day, then 200 mg every 12 hours¬† for 4 days voriconazole to subjects receiving a methadone maintenance dose (30-100 mg every 24 hours) ****¬†¬†¬† Non-Steroidal Anti-Inflammatory Drug¬†*****¬† Non-Nucleoside Reverse Transcriptase Inhibitors ¬†Sirolimus* ¬†(CYP3A4 Inhibition) ¬† ¬†Significantly Increased¬† ¬†Contraindicated ¬† ¬†Rifabutin* ¬†(CYP3A4 Inhibition) ¬† ¬†Significantly Increased¬† ¬†Contraindicated ¬† ¬†Efavirenz (400 mg every 24 hours)** (CYP3A4 Inhibition) ¬†¬†¬†Efavirenz (300 mg every 24 hours)** (CYP3A4 Inhibition) ¬†¬†¬† ¬†Significantly Increased¬†¬†¬†¬†¬†Slight Increase in AUC ŌĄ ¬† ¬†Contraindicated ¬†¬†¬†¬†¬†When voriconazole is coadministered with efavirenz, voriconazole oral maintenance dose should be increased to 400 mg every 12 hours and efavirenz should be decreased to 300 mg every 24 hours.¬† ¬†High-dose Ritonavir (400 mg every 12 hours )**(CYP3A4 Inhibition) ¬†¬†¬†¬†¬†Low-dose Ritonavir (100 mg every 12 hours)**¬† ¬†No Significant Effect of Voriconazole on Ritonavir Cmax or AUCŌĄ ¬†¬†¬†¬†¬†Slight Decrease in Ritonavir Cmax and AUCŌĄ ¬† ¬†Contraindicated ¬†because of significant reduction of voriconazole Cmax and AUCŌĄ. ¬†¬†¬†Coadministration of voriconazole and low-dose ritonavir (100 mg every 12 hours) should be avoided (due to the reduction in voriconazole Cmax and AUCŌĄ) unless an assessment of the benefit/risk to the patient justifies the use of voriconazole.¬† ¬†Pimozide, Quinidine, Ivabradine (CYP3A4 Inhibition) ¬† ¬†Not Studied In Vivo or In Vitro, but Drug Plasma Exposure Likely to be Increased¬† ¬†Contraindicated because of potential for QT prolongation and rare occurrence of torsade de pointes. ¬† ¬†Ergot Alkaloids¬†(CYP450 Inhibition) ¬† ¬†Not Studied In Vivo or In Vitro, but Drug Plasma Exposure Likely to be Increased¬† ¬†Contraindicated ¬† ¬†Naloxegol ¬†(CYP3A4 Inhibition) ¬† ¬†Not Studied In Vivo or In Vitro, but Drug Plasma Exposure Likely to be Increased which may Increase the Risk of Adverse Reactions¬† ¬†Contraindicated ¬† ¬†Tolvaptan ¬†(CYP3A4 Inhibition) ¬† ¬†Although Not Studied Clinically, Voriconazole is Likely to Significantly Increase the Plasma Concentrations of Tolvaptan¬† ¬†Contraindicated ¬† ¬†Venetoclax (CYP3A4 Inhibition) ¬†Not studied In Vivo or In Vitro, but Venetoclax Plasma Exposure Likely to be Significantly Increased Coadministration of voriconazole is contraindicated at initiation and during the ramp-up phase in patients with chronic lymphocytic leukemia (CLL) or small lymphocytic lymphoma (SLL). Refer to the venetoclax labeling for safety monitoring and dose reduction in the steady daily dosing phase in CLL/SLL patients. For patients with acute myeloid leukemia (AML), dose reduction and safety monitoring are recommended across all dosing phases when coadministering voriconazole tablets with venetoclax. Refer to the venetoclax prescribing information for dosing instructions. Lemborexant (CYP3A4 Inhibition) Not Studied In Vivo or In Vitro, but Drug Plasma Exposure Likely to be Increased Avoid concomitant use of voriconazole tablets with lemborexant. Glasdegib (CYP3A4 Inhibition) Not Studied In Vivo or In Vitro, but Drug Plasma Exposure Likely to be Increased Consider alternative therapies. If concomitant use cannot be avoided, monitor patients for increased risk of adverse reactions including QTc interval prolongation. Tyrosine kinase inhibitors (including but not limited to axitinib, bosutinib, cabozantinib, ceritinib, cobimetinib, dabrafenib, dasatinib, nilotinib, sunitinib, ibrutinib, ribociclib) (CYP3A4 Inhibition) ¬†Not Studied In Vivo or In Vitro, but Drug Plasma Exposure Likely to be Increased Avoid concomitant use of voriconazole tablets. If concomitant use cannot be avoided, dose reduction of the tyrosine kinase inhibitor is recommended. Refer to the prescribing information for the relevant product. Lurasidone (CYP3A4 Inhibition) Not Studied In Vivo or In Vitro, but Voriconazole is Likely to Significantly Increase the Plasma Concentrations of Lurasidone Contraindicated ¬†Cyclosporine* ¬†(CYP3A4 Inhibition) ¬† ¬†AUCŌĄ Significantly Increased; No Significant Effect on Cmax ¬† ¬†When initiating therapy with voriconazole tablets in patients already receiving cyclosporine, reduce the cyclosporine dose to one-half of the starting dose and follow with frequent monitoring of cyclosporine blood levels. Increased cyclosporine levels have been associated with nephrotoxicity. When voriconazole tablets are discontinued, cyclosporine concentrations must be frequently monitored and the dose increased as necessary.¬† ¬†Methadone*** (CYP3A4 Inhibition) ¬† ¬†Increased¬† ¬†Increased plasma concentrations of methadone have been associated with toxicity including QT prolongation. Frequent monitoring for adverse reactions and toxicity related to methadone is recommended during coadministration. Dose reduction of methadone may be needed.¬† ¬†Fentanyl (CYP3A4 Inhibition) ¬† ¬†Increased¬† ¬†Reduction in the dose of fentanyl and other long-acting opiates metabolized by CYP3A4 should be considered when coadministered with voriconazole tablets. Extended and frequent monitoring for opiate-associated adverse reactions may be necessary. ¬†Alfentanil ¬†(CYP3A4 Inhibition) ¬† ¬†Significantly Increased¬† ¬†An increase in the incidence of delayed and persistent alfentanil-associated nausea and vomiting were observed when coadministered with voriconazole tablets. Reduction in the dose of alfentanil and other opiates metabolized by CYP3A4 (e.g., sufentanil) should be considered when coadministered with voriconazole tablets. A longer period for monitoring respiratory and other opiate-associated adverse reactions may be necessary.¬† ¬†Oxycodone (CYP3A4 Inhibition) ¬† ¬†Significantly Increased¬† ¬†Increased visual effects (heterophoria and miosis) of oxycodone were observed when coadministered with voriconazole tablets. Reduction in the dose of oxycodone and other long-acting opiates metabolized by CYP3A4 should be considered when coadministered with voriconazole tablets. Extended and frequent monitoring for opiate-associated adverse reactions¬†may be necessary.¬† ¬†NSAIDs**** including ibuprofen and diclofenac ¬†(CYP2C9 Inhibition) ¬†¬†¬† ¬†Increased¬† ¬†Frequent monitoring for adverse reactions¬†and toxicity related to NSAIDs. Dose reduction of NSAIDs may be needed. ¬† ¬†Tacrolimus* ¬†(CYP3A4 Inhibition) ¬† ¬†Significantly Increased¬† ¬†When initiating therapy with voriconazole tablets in patients already receiving tacrolimus, reduce the tacrolimus dose to one-third of the starting dose and follow with frequent monitoring of tacrolimus blood levels. Increased tacrolimus levels have been associated with nephrotoxicity. When voriconazole tablets are discontinued, tacrolimus concentrations must be frequently monitored and the dose increased as necessary.¬† ¬†Phenytoin* ¬†(CYP2C9 Inhibition) ¬† ¬†Significantly Increased¬† ¬†Frequent monitoring of phenytoin plasma concentrations and frequent monitoring of adverse effects related to phenytoin.¬† ¬†Oral Contraceptives containing ethinyl estradiol and norethindrone ¬†(CYP3A4 Inhibition)** ¬† ¬†Increased¬† ¬†Monitoring for adverse reactions¬†related to oral contraceptives is recommended during coadministration.¬† ¬†Prednisolone and other corticosteroids ¬†(CYP3A4 Inhibition) ¬† ¬†In Vivo Studies Showed No Significant Effects of voriconazole tablets on Prednisolone Exposure¬†¬†¬†¬†¬†Not Studied In vitro or In vivo for Other Corticosteroids, but Drug Exposure Likely to be Increased ¬† ¬†No dosage adjustment for prednisolone when coadministered with voriconazole tablets [see Clinical Pharmacology (12.3)]. ¬†¬†¬†Monitor for potential adrenal dysfunction when voriconazole tablets is administered with other corticosteroids [See Warnings and Precautions (5.8)]. ¬† ¬†Warfarin* ¬†(CYP2C9 Inhibition)Other Oral Coumarin Anticoagulants (CYP2C9/3A4 Inhibition) ¬† ¬†Prothrombin Time Significantly IncreasedNot Studied In Vivo or In Vitro for other Oral Coumarin Anticoagulants, but Drug Plasma Exposure Likely to be Increased¬† ¬† ¬†If patients receiving coumarin preparations are treated simultaneously with voriconazole, the prothrombin time or other suitable anticoagulation tests should be monitored at close intervals and the dosage of anticoagulants adjusted accordingly.¬† ¬†Ivacaftor ¬†(CYP3A4 Inhibition) ¬† ¬†Not Studied In Vivo or In Vitro, but Drug Plasma Exposure Likely to be Increased which may Increase the Risk of Adverse Reactions ¬† ¬†Dose reduction of ivacaftor is recommended. Refer to the prescribing information for ivacaftor ¬† Eszopiclone (CYP3A4 Inhibition) ¬†Not Studied In Vivo or In Vitro, but Drug Plasma Exposure Likely to be Increased which may Increase the Sedative Effect of Eszopiclone ¬†Dose reduction of eszopiclone is recommended. Refer to the prescribing information for eszopiclone. ¬†Omeprazole* ¬†(CYP2C19/3A4 Inhibition) ¬† ¬†Significantly Increased¬† ¬†When initiating therapy with voriconazole tablets in patients already receiving omeprazole doses of 40 mg or greater, reduce the omeprazole dose by one-half. The metabolism of other proton pump inhibitors that are CYP2C19 substrates may also be inhibited by voriconazole and may result in increased plasma concentrations of other proton pump inhibitors.¬† ¬†Other HIV Protease Inhibitors ¬†(CYP3A4 Inhibition) ¬† ¬†In Vivo Studies Showed No Significant Effects on Indinavir Exposure¬†¬†¬†¬†¬†In Vitro Studies Demonstrated Potential for Voriconazole to Inhibit Metabolism¬†(Increased Plasma Exposure)¬† ¬†No dosage adjustment for indinavir when coadministered with voriconazole tablets.¬†¬†¬†¬†¬†Frequent monitoring for adverse¬†reactions and toxicity related to other HIV protease inhibitors.¬† ¬†Other NNRTIs***** ¬†(CYP3A4 Inhibition) ¬† ¬†A Voriconazole-Efavirenz Drug Interaction Study Demonstrated the Potential for Voriconazole to Inhibit Metabolism of Other NNRTIs (Increased Plasma Exposure)¬† ¬†Frequent monitoring for adverse reactions¬†and toxicity related to NNRTI.¬† Tretinoin (CYP3A4 Inhibition) Although Not Studied, Voriconazole may Increase Tretinoin Concentrations and Increase the Risk of Adverse Reactions Frequent monitoring for signs and symptoms of pseudotumor cerebri or hypercalcemia.¬† ¬†Midazolam (CYP3A4 Inhibition) Other benzodiazepines including triazolam and alprazolam (CYP3A4 Inhibition)¬† ¬†Significantly Increased In Vitro Studies Demonstrated Potential for Voriconazole to Inhibit Metabolism (Increased Plasma Exposure)¬† ¬† Increased plasma exposures may increase the risk of adverse reactions and toxicities related to benzodiazepines. Refer to drug-specific labeling for details. ¬† ¬† ¬†HMG-CoA Reductase Inhibitors (Statins) (CYP3A4 Inhibition) ¬† ¬†In Vitro Studies Demonstrated Potential for Voriconazole to Inhibit Metabolism¬†(Increased Plasma Exposure)¬† ¬†Frequent monitoring for adverse reactions¬†and toxicity related to statins. Increased statin concentrations in plasma have been associated with rhabdomyolysis. Adjustment of the statin dosage may be needed.¬† ¬†Dihydropyridine Calcium Channel Blockers ¬†(CYP3A4 Inhibition) ¬† ¬†In Vitro Studies Demonstrated Potential for Voriconazole to Inhibit Metabolism¬†(Increased Plasma Exposure)¬† ¬†Frequent monitoring for adverse reactions and toxicity related to calcium channel blockers. Adjustment of calcium channel blocker dosage may be needed.¬† ¬†Sulfonylurea Oral Hypoglycemics (CYP2C9 Inhibition) ¬† ¬†Not Studied In Vivo or In Vitro, but Drug Plasma Exposure Likely to be Increased¬† ¬†Frequent monitoring of blood glucose and for signs and symptoms of hypoglycemia. Adjustment of oral hypoglycemic drug dosage may be needed.¬† ¬†Vinca Alkaloids (CYP3A4 Inhibition) ¬† ¬†Not Studied In Vivo or In Vitro, but Drug Plasma Exposure Likely to be Increased¬† ¬†Frequent monitoring for adverse reactions and toxicity (i.e., neurotoxicity) related to vinca alkaloids. Reserve azole antifungals, including voriconazole, for patients receiving a vinca alkaloid who have no alternative antifungal treatment options.¬† ¬†Everolimus ¬†(CYP3A4 Inhibition)¬† ¬†Not Studied In Vivo or In Vitro, but Drug Plasma Exposure Likely to be Increased¬† ¬†Concomitant administration of voriconazole and everolimus is not recommended.¬†
- CYP3A4, CYP2C9, and CYP2C19 inhibitors and inducers: Adjust voriconazole tablets dosage and monitor for adverse reactions or lack of efficacy (
4 ,7 )- Voriconazole tablets may increase the concentrations and activity of drugs that are CYP3A4, CYP2C9 and CYP2C19 substrates. Reduce dosage of these other drugs and monitor for adverse reactions (
4 ,7 )- Phenytoin or Efavirenz: With co-administration, increase maintenance oral and intravenous dosage of voriconazole (
2.3 ,2.7 ,7 )
8 Use In Specific Populations
- Pediatrics: Safety and effectiveness in patients younger than 2 years has not been established (
8.4 )
 
8.1 Pregnancy
Risk Summary
Voriconazole can cause fetal harm when administered to a pregnant woman. There are no available data on the use of voriconazole in pregnant women. In animal reproduction studies, oral voriconazole was associated with fetal malformations in rats and fetal toxicity in rabbits. Cleft palates and hydronephrosis/hydroureter were observed in rat pups exposed to voriconazole during organogenesis at and above 10 mg/kg (0.3 times the RMD of 200 mg every 12 hours based on body surface area comparisons). In rabbits, embryomortality, reduced fetal weight and increased incidence of skeletal variations, cervical ribs and extrasternal ossification sites were observed in pups when pregnant rabbits were orally dosed at 100 mg/kg (6 times the RMD based on body surface area comparisons) during organogenesis. Rats exposed to voriconazole from implantation to weaning experienced increased gestational length and dystocia, which were associated with increased perinatal pup mortality at the 10 mg/kg dose [see Data]. If this drug is used during pregnancy, or if the patient becomes pregnant while taking this drug, inform the patient of the potential hazard to the fetus [see Warnings and Precautions (5.9)].
The background risk of major birth defects and miscarriage for the indicated populations is unknown. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2-4% and 15-20% respectively.
Data
Animal Data
Voriconazole was administered orally to pregnant rats during organogenesis (gestation days 6-17) at 10, 30, and 60 mg/kg/day. Voriconazole was associated with increased incidences of the malformations hydroureter and hydronephrosis at 10 mg/kg/day or greater, approximately 0.3 times the RMD based on body surface area comparisons, and cleft palate at 60 mg/kg, approximately 2 times the  RMD based on body surface area comparisons. Reduced ossification of sacral and caudal vertebrae, skull, pubic, and hyoid bone, supernumerary ribs, anomalies of the sternebrae, and dilatation of the ureter/renal pelvis were also observed at doses of 10 mg/kg or greater. There was no evidence of maternal toxicity at any dose.
Voriconazole was administered orally to pregnant rabbits during the period of organogenesis (gestation days 7-19) at 10, 40, and 100 mg/kg/day. Voriconazole was associated with increased post-implantation loss and decreased fetal body weight, in association with maternal toxicity (decreased body weight gain and food consumption) at 100 mg/kg/day (6 times the RMD based on body surface area comparisons). Fetal skeletal variations (increases in the incidence of cervical rib and extra sternebral ossification sites) were observed at 100 mg/kg/day.
In a peri-and postnatal toxicity study in rats, voriconazole was administered orally to female rats from implantation through the end of lactation at 1, 3, and 10 mg/kg/day. Voriconazole prolonged the duration of gestation and labor and produced dystocia with related increases in maternal mortality and decreases in perinatal survival of F1 pups at 10 mg/kg/day, approximately 0.3 times the RMD.
8.2 Lactation
Risk Summary
No data are available regarding the presence of voriconazole in human milk, the effects of voriconazole on the breastfed infant, or the effects on milk production. The developmental and health benefits of breastfeeding should be considered along with the mother’s clinical need for voriconazole and any potential adverse effects on the breastfed child from voriconazole or from the underlying maternal condition.
8.3 Females and Males of Reproductive Potential
Contraception
Advise females of reproductive potential to use effective contraception during treatment with voriconazole tablets. The coadministration of voriconazole with the oral contraceptive, Ortho-Novum¬ģ (35 mcg ethinyl estradiol and 1 mg norethindrone), results in an interaction between these two drugs, but is unlikely to reduce the contraceptive effect. Monitoring for adverse reactions associated with oral contraceptives and voriconazole is recommended [see Drug Interactions ( 7 ) and Clinical Pharmacology ( 12.3 )].
8.4 Pediatric Use
The safety and effectiveness of voriconazole have been established in pediatric patients 2 years of age and older based on evidence from adequate and well-controlled studies in adult and pediatric patients and additional pediatric pharmacokinetic and safety data. A total of 105 pediatric patients aged 2 to less than 12 [N=26] and aged 12 to less than 18 [N=79] from two, non-comparative Phase 3 pediatric studies and eight adult therapeutic trials provided safety information for voriconazole use in the pediatric population [see Adverse Reactions (6.1), Clinical Pharmacology (12.3), and Clinical Studies (14)].
Safety and effectiveness in pediatric patients below the age of 2 years has not been established. Therefore, voriconazole is not recommended for pediatric patients less than 2 years of age.
A higher frequency of liver enzyme elevations was observed in the pediatric patients [see Dosage and Administration (2.5), Warnings and Precautions (5.1), and Adverse Reactions (6.1)].
 
The frequency of phototoxicity reactions is higher in the pediatric population. Squamous cell carcinoma has been reported in patients who experience photosensitivity reactions. Stringent measures for photoprotection are warranted. Sun avoidance and dermatologic follow-up are recommended in pediatric patients experiencing photoaging injuries, such as lentigines or ephelides, even after treatment discontinuation [see Warnings and Precautions (5.6)].
 Voriconazole has not been studied in pediatric patients with hepatic or renal impairment [see Dosage and Administration (2.5, 2.6)]. Hepatic function and serum creatinine levels should be closely monitored in pediatric patients [see Dosage and Administration (2.6) and Warnings and Precautions (5.1, 5.10)].
8.5 Geriatric Use
In multiple dose therapeutic trials of voriconazole, 9.2% of patients were ‚Č•65 years of age and 1.8% of patients were ‚Č•75 years of age. In a study in healthy subjects, the systemic exposure (AUC) and peak plasma concentrations (Cmax) were increased in elderly males compared to young males. Pharmacokinetic data obtained from 552 patients from 10 voriconazole therapeutic trials showed that voriconazole plasma concentrations in the elderly patients were approximately 80% to 90% higher than those in younger patients after either IV or oral administration. However, the overall safety profile of the elderly patients was similar to that of the young so no dosage adjustment is recommended [see Clinical Pharmacology ( 12.3 )].
10 Overdosage
In clinical trials, there were three cases of accidental overdose. All occurred in pediatric patients who received up to five times the recommended intravenous dose of voriconazole. A single adverse reaction of photophobia of 10 minutes duration was reported.
There is no known antidote to voriconazole.
Voriconazole is hemodialyzed with clearance of 121 mL/min. The intravenous vehicle, SBECD, is hemodialyzed with clearance of 55 mL/min. In an overdose, hemodialysis may assist in the removal of voriconazole and SBECD from the body.
11 Description
Voriconazole USP, an azole antifungal agent is available as film-coated tablets for oral administration. The structural formula is:
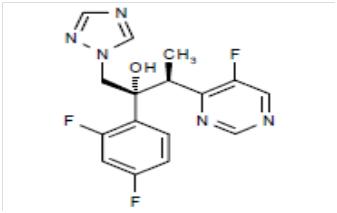
Voriconazole is designated chemically as (őĪR,ő≤S)-őĪ-(2,4-difluorophenyl)-5- fluoro- ő≤ -methyl- őĪ -(1H-1,2,4-traizol-l-ylmethyl)-4-pyrimidineethanol with an empirical formula of C16H14F3N5O and a molecular weight of 349.31 g/mole.
Voriconazole drug substance is a white or almost white powder.
Voriconazole tablets contain 50 mg or 200 mg of voriconazole. The inactive ingredients include croscarmellose sodium, lactose monohydrate, magnesium stearate, povidone, pregelatinized starch, and a coating containing hypromellose, lactose monohydrate, titanium dioxide, and triacetin.
12 Clinical Pharmacology
12.1 Mechanism of Action
Voriconazole USP is an antifungal drug [see Microbiology (12.4) ].
12.2 Pharmacodynamics
Exposure-Response Relationship For Efficacy and Safety
In 10 clinical trials (N=1121), the median values for the average and maximum voriconazole plasma concentrations in individual patients across these studies was 2.51 őľg/mL (inter-quartile range 1.21 to 4.44 őľg/mL) and 3.79 őľg/mL (inter-quartile range 2.06 to 6.31 őľg/mL), respectively. A pharmacokinetic-pharmacodynamic analysis of patient data from 6 of these 10 clinical trials (N=280) could not detect a positive association between mean, maximum or minimum plasma voriconazole concentration and efficacy. However, pharmacokinetic/pharmacodynamic analyses of the data from all 10 clinical trials identified positive associations between plasma voriconazole concentrations and rate of both liver function test abnormalities and visual disturbances [see Adverse Reactions (6)].
Cardiac Electrophysiology
A placebo-controlled, randomized, crossover study to evaluate the effect on the QT interval of healthy male and female subjects was conducted with three single oral doses of voriconazole and ketoconazole. Serial ECGs and plasma samples were obtained at specified intervals over a 24-hour post dose observation period. The placebo-adjusted mean maximum increases in QTc from baseline after 800, 1200, and 1600 mg of voriconazole and after ketoconazole 800 mg were all <10 msec. Females exhibited a greater increase in QTc than males, although all mean changes were <10 msec. Age was not found to affect the magnitude of increase in QTc. No subject in any group had an increase in QTc of ‚Č•60 msec from baseline. No subject experienced an interval exceeding the potentially clinically relevant threshold of 500 msec. However, the QT effect of voriconazole combined with drugs known to prolong the QT interval is unknown [see Contraindications (4) and Drug Interactions (7)].
12.3 Pharmacokinetics
The pharmacokinetics of voriconazole has been characterized in healthy subjects, special populations and patients.
The pharmacokinetics of voriconazole are non-linear due to saturation of its metabolism. The interindividual variability of voriconazole pharmacokinetics is high. Greater than proportional increase in exposure is observed with increasing dose. It is estimated that, on average, increasing the oral dose from 200 mg every 12 hours to 300 mg every 12 hours leads to an approximately 2.5-fold increase in exposure (AUCŌĄ); similarly, increasing the intravenous dose from 3 mg/kg every 12 hours to 4 mg/kg every 12 hours produces an approximately 2.5-fold increase in exposure (Table 12).
Table 12: Geometric Mean (%CV) Plasma Voriconazole Pharmacokinetic Parameters in Adults Receiving Different Dosing Regimens ¬† 6 mg/kg IV (loading dose) 3 mg/kg IV¬† every 12 hours 4 mg/kg IV every 12 hours 400 mg Oral (loading dose) 200 mg Oral¬† every 12 hours¬† ¬† 300 mg Oral¬† every 12 hours¬† ¬† N 35 23 40 17 48 16 AUC12 (őľg¬∑h/mL) 13.9 (32) 13.7 (53) 33.9 (54) 9.31 (38) 12.4 (78) 34.0 (53) Cmax (őľg/mL) 3.13 (20) 3.03 (25) 4.77 (36) 2.30 (19) 2.31 (48) 4.74 (35) Cmin (őľg/mL) -¬≠- 0.46 (97) 1.73 (74) -¬≠- 0.46 (120) 1.63 (79)
Note: Parameters were estimated based on non-compartmental analysis from 5 pharmacokinetic studies.
AUC12 = area under the curve over 12 hour dosing interval, Cmax = maximum plasma concentration, Cmin = minimum plasma concentration. CV = coefficient of variation.
When the recommended intravenous loading dose regimen is administered to healthy subjects, plasma concentrations close to steady state are achieved within the first 24 hours of dosing (e.g., 6 mg/kg IV every 12 hours on day 1 followed by 3 mg/kg IV every 12 hours). Without the loading dose, accumulation occurs during twice daily multiple dosing with steady state plasma voriconazole concentrations being achieved by day 6 in the majority of subjects.
Absorption
The pharmacokinetic properties of voriconazole are similar following administration by the intravenous and oral routes. Based on a population pharmacokinetic analysis of pooled data in healthy subjects (N=207), the oral bioavailability of voriconazole is estimated to be 96% (CV 13%). Bioequivalence was established between the 200 mg tablet and the 40 mg/mL oral suspension when administered as a 400 mg every 12 hours loading dose followed by a 200 mg every 12 hours maintenance dose.
Maximum plasma concentrations (Cmax) are achieved 1-2 hours after dosing. When multiple doses of voriconazole are administered with high-fat meals, the mean Cmax and AUCŌĄ are reduced by 34% and 24%, respectively when administered as a tablet and by 58% and 37% respectively when administered as the oral suspension [see Dosage and Administration (2)].
In healthy subjects, the absorption of voriconazole is not affected by coadministration of oral ranitidine, cimetidine, or omeprazole, drugs that are known to increase gastric pH.
Distribution
The volume of distribution at steady state for voriconazole is estimated to be 4.6 L/kg, suggesting extensive distribution into tissues. Plasma protein binding is estimated to be 58% and was shown to be independent of plasma concentrations achieved following single and multiple oral doses of 200 mg or 300 mg (approximate range: 0.9-15 őľg/mL). Varying degrees of hepatic and renal impairment do not affect the protein binding of voriconazole.
Elimination
Metabolism
In vitro studies showed that voriconazole is metabolized by the human hepatic cytochrome P450 enzymes, CYP2C19, CYP2C9 and CYP3A4 [see Drug Interactions (7)].
In vivo studies indicated that CYP2C19 is significantly involved in the metabolism of voriconazole. This enzyme exhibits genetic polymorphism [see Clinical Pharmacology (12.5)].
The major metabolite of voriconazole is the N-oxide, which accounts for 72% of the circulating radiolabelled metabolites in plasma. Since this metabolite has minimal antifungal activity, it does not contribute to the overall efficacy of voriconazole.
Excretion
Voriconazole is eliminated via hepatic metabolism with less than 2% of the dose excreted unchanged in the urine. After administration of a single radiolabelled dose of either oral or IV voriconazole, preceded by multiple oral or IV dosing, approximately 80% to 83% of the radioactivity is recovered in the urine. The majority (>94%) of the total radioactivity is excreted in the first 96 hours after both oral and intravenous dosing.
As a result of non-linear pharmacokinetics, the terminal half-life of voriconazole is dose dependent and therefore not useful in predicting the accumulation or elimination of voriconazole.
Specific Populations
Male and Female Patients
In a multiple oral dose study, the mean Cmax and AUCŌĄ for healthy young females were 83% and 113% higher, respectively, than in healthy young males (18-45 years), after tablet dosing. In the same study, no significant differences in the mean Cmax and AUCŌĄ were observed between healthy elderly males and healthy elderly females (>65 years). In a similar study, after dosing with the oral suspension, the mean AUC for healthy young females was 45% higher than in healthy young males whereas the mean Cmax was comparable between genders. The steady state trough voriconazole concentrations (Cmin) seen in females were 100% and 91% higher than in males receiving the tablet and the oral suspension, respectively.
In the clinical program, no dosage adjustment was made on the basis of gender. The safety profile and plasma concentrations observed in male and female subjects were similar. Therefore, no dosage adjustment based on gender is necessary.
Geriatric Patients
In an oral multiple dose study the mean Cmax and AUCŌĄ in healthy elderly males (‚Č•65 years) were 61% and 86% higher, respectively, than in young males (18-45 years). No significant differences in the mean Cmax and AUCŌĄ were observed between healthy elderly females (‚Č•65 years) and healthy young females (18-45 years).
In the clinical program, no dosage adjustment was made on the basis of age. An analysis of pharmacokinetic data obtained from 552 patients from 10 voriconazole clinical trials showed that the median voriconazole plasma concentrations in the elderly patients (>65 years) were approximately 80% to 90% higher than those in the younger patients (‚ȧ65 years) after either IV or oral administration. However, the safety profile of voriconazole in young and elderly subjects was similar and, therefore, no dosage adjustment is necessary for the elderly [see Use in Special Populations ( 8.5 )].
 
Pediatric Patients
The recommended doses in pediatric patients were based on a population pharmacokinetic analysis of data obtained from 112 immunocompromised pediatric patients aged 2 to less than 12 years and 26 immunocompromised pediatric patients aged 12 to less than 17 years.
A comparison of the pediatric and adult population pharmacokinetic data indicated that the predicted total exposure (AUC12) in pediatric patients aged 2 to less than 12 years following administration of a 9 mg/kg intravenous loading dose was comparable to that in adults following a 6 mg/kg intravenous loading dose. The predicted total exposures in pediatric patients aged 2 to less than 12 years following intravenous maintenance doses of 4 and 8 mg/kg twice daily were comparable to those in adults following 3 and 4 mg/kg IV twice daily, respectively.
The predicted total exposure in pediatric patients aged 2 to less than 12 years following an oral maintenance dose of 9 mg/kg (maximum of 350 mg) twice daily was comparable to that in adults following 200 mg oral twice daily. An 8 mg/kg intravenous dose will provide voriconazole exposure approximately 2-fold higher than a 9 mg/kg oral dose in pediatric patients aged 2 to less than 12 years.
Voriconazole exposures in the majority of pediatric patients aged 12 to less than 17 years were comparable to those in adults receiving the same dosing regimens. However, lower voriconazole exposure was observed in some pediatric patients aged 12 to less than 17 years with low body weight compared to adults [see Dosage and Administration ( 2.4 )].
Limited voriconazole trough plasma samples were collected in pediatric patients aged 2 to less than 18 years with IA or invasive candidiasis including candidemia, and EC in two prospective, open-label, non-comparative, multicenter clinical studies. In eleven pediatric patients aged 2 to less than 12 years and aged 12 to 14 years, with body weight less than 50 kg, who received 9 mg/kg intravenously every 12 hours as a loading dose on the first day of treatment, followed by 8 mg/kg every 12 hours as an intravenous maintenance dose, or 9 mg/kg every 12 hours as an oral maintenance dose, the mean trough concentration of voriconazole was 3.6 mcg/mL (range 0.3 to 10.7 mcg/mL). In four pediatric patients aged 2 to less than 12 years and aged 12 to 14 years, with body weight less than 50 kg, who received 4 mg/kg intravenously every 12 hours, the mean trough concentration of voriconazole was 0.9 mcg/mL (range 0.3 to 1.6 mcg/mL) [see Clinical Studies ( 14.5 )].
Patients with Hepatic Impairment
After a single oral dose (200 mg) of voriconazole in 8 patients with mild (Child-Pugh Class A) and 4 patients with moderate (Child-Pugh Class B) hepatic impairment, the mean systemic exposure (AUC) was 3.2-fold higher than in age and weight matched controls with normal hepatic function. There was no difference in mean peak plasma concentrations (Cmax) between the groups. When only the patients with mild (Child-Pugh Class A) hepatic impairment were compared to controls, there was still a 2.3-fold increase in the mean AUC in the group with hepatic impairment compared to controls.
In an oral multiple dose study, AUCŌĄ was similar in 6 subjects with moderate hepatic impairment (Child-Pugh Class B) given a lower maintenance dose of 100 mg twice daily compared to 6 subjects with normal hepatic function given the standard 200 mg twice daily maintenance dose. The mean peak plasma concentrations (Cmax) were 20% lower in the hepatically impaired group. No pharmacokinetic data are available for patients with severe hepatic cirrhosis (Child-Pugh Class C) [see Dosage and Administration ( 2.5 )].
Patients with Renal Impairment
In a single oral dose (200 mg) study in 24 subjects with normal renal function and mild to severe renal impairment, systemic exposure (AUC) and peak plasma concentration (Cmax) of voriconazole were not significantly affected by renal impairment. Therefore, no adjustment is necessary for oral dosing in patients with mild to severe renal impairment.
In a multiple dose study of IV voriconazole (6 mg/kg IV loading dose x 2, then 3 mg/kg IV x 5.5 days) in 7 patients with moderate renal dysfunction (creatinine clearance 30-50 mL/min), the systemic exposure (AUC) and peak plasma concentrations (Cmax) were not significantly different from those in 6 subjects with normal renal function.
However, in patients with moderate renal dysfunction (creatinine clearance 30-50 mL/min), accumulation of the intravenous vehicle, SBECD, occurs. The mean systemic exposure (AUC) and peak plasma concentrations (Cmax) of SBECD were increased 4-fold and almost 50%, respectively, in the moderately impaired group compared to the normal control group.
A pharmacokinetic study in subjects with renal failure undergoing hemodialysis showed that voriconazole is dialyzed with clearance of 121 mL/min. The intravenous vehicle, SBECD, is hemodialyzed with clearance of 55 mL/min. A 4-hour hemodialysis session does not remove a sufficient amount of voriconazole to warrant dose adjustment [see Dosage and Administration ( 2.6 )].
Patients at Risk of Aspergillosis
The observed voriconazole pharmacokinetics in patients at risk of aspergillosis (mainly patients with malignant neoplasms of lymphatic or hematopoietic tissue) were similar to healthy subjects.
Drug Interaction Studies  
Effects of Other Drugs on Voriconazole
Voriconazole is metabolized by the human hepatic cytochrome P450 enzymes CYP2C19, CYP2C9, and CYP3A4. Results of in vitro metabolism studies indicate that the affinity of voriconazole is highest for CYP2C19, followed by CYP2C9, and is appreciably lower for CYP3A4. Inhibitors or inducers of these three enzymes may increase or decrease voriconazole systemic exposure (plasma concentrations), respectively.
The systemic exposure to voriconazole is significantly reduced by the concomitant administration of the following agents and their use is contraindicated:
Rifampin (potent CYP450 inducer)‚ÄďRifampin (600 mg once daily) decreased the steady state Cmax and AUCŌĄ of voriconazole (200 mg every 12 hours x 7 days) by an average of 93% and 96%, respectively, in healthy subjects. Doubling the dose of voriconazole to 400 mg every 12 hours does not restore adequate exposure to voriconazole during coadministration with rifampin [see Contraindications ( 4 ) ].
Ritonavir (potent CYP450 inducer; CYP3A4 inhibitor and substrate)‚ÄďThe effect of the coadministration of voriconazole and ritonavir (400 mg and 100 mg) was investigated in two separate studies. High-dose ritonavir (400 mg every 12 hours for 9 days) decreased the steady state Cmax and AUCŌĄ of oral voriconazole (400¬†¬† mg every 12 hours for 1 day, then 200 mg every 12 hours for 8 days) by an average of 66% and 82%, respectively, in healthy subjects. Low-dose ritonavir (100 mg every 12 hours for 9 days) decreased the steady state Cmax and AUCŌĄ of oral voriconazole (400 mg every 12 hours for 1 day, then 200 mg every 12 hours for 8 days) by an average of 24% and 39%, respectively, in healthy subjects. Although repeat oral administration of voriconazole did not have a significant effect on steady state Cmax and AUCŌĄ of high-dose ritonavir in healthy subjects, steady state Cmax and AUCŌĄ of low-dose ritonavir decreased slightly by 24% and 14% respectively, when administered concomitantly with oral voriconazole in healthy subjects [see Contraindications ( 4 ) ].
St. John‚Äôs Wort (CYP450 inducer; P-gp inducer)‚ÄďIn an independent published study in healthy volunteers who were given multiple oral doses of St. John‚Äôs Wort (300 mg LI 160 extract three times daily for 15 days) followed by a single 400 mg oral dose of voriconazole, a 59% decrease in mean voriconazole AUC0-‚ąě was observed. In contrast, coadministration of single oral doses of St. John‚Äôs Wort and voriconazole had no appreciable effect on voriconazole AUC0-‚ąě. Long-term use of St. John‚Äôs Wort could lead to reduced voriconazole exposure¬†[see Contraindications ( 4 )].
Significant drug interactions that may require voriconazole dosage adjustment, or frequent monitoring of voriconazole-related adverse reactions/toxicity:
Fluconazole (CYP2C9, CYP2C19 and CYP3A4 inhibitor): Concurrent administration of oral voriconazole (400 mg every 12 hours¬† for 1 day, then 200 mg every 12 hours for 2.5 days) and oral fluconazole (400 mg on day 1, then 200 mg every 24 hours¬† for 4 days) to 6 healthy male subjects resulted in an increase in Cmax and AUCŌĄ of voriconazole by an average of 57% (90% CI: 20%, 107%) and 79% (90% CI: 40%, 128%), respectively. In a follow-on clinical study involving 8 healthy male subjects, reduced dosing and/or frequency of voriconazole and fluconazole did not eliminate or diminish this effect [see Drug Interactions ( 7 )].
Letermovir (CYP2C9/2C19 inducer)‚ÄďCoadministration of oral letermovir with oral voriconazole decreased the steady state Cmax and AUC0-12 of voriconazole by an average of 39% and 44%, respectively [see Drug Interactions ( 7 )].
Minor or no significant pharmacokinetic interactions that do not require dosage adjustment:
Cimetidine (non-specific CYP450 inhibitor and increases gastric pH)‚ÄďCimetidine (400 mg every 12 hours x 8 days) increased voriconazole steady state Cmax and AUCŌĄ by an average of 18% (90% CI: 6%, 32%) and 23% (90% CI: 13%, 33%), respectively, following oral doses of 200 mg every 12 hours x 7 days to healthy subjects.
Ranitidine (increases gastric pH)‚ÄďRanitidine (150 mg every 12 hours) had no significant effect on voriconazole Cmax and AUCŌĄ following oral doses of 200 mg every 12 hours x 7 days to healthy subjects.
Macrolide antibiotics ‚ÄďCoadministration of erythromycin (CYP3A4 inhibitor; 1 gram every 12 hours for 7 days) or azithromycin (500 mg every 24 hours for 3 days) with voriconazole 200 mg every 12 hours for 14 days had no significant effect on voriconazole steady state Cmax and AUCŌĄ in healthy subjects. The effects of voriconazole on the pharmacokinetics of either erythromycin or azithromycin are not known.
Effects of Voriconazole on Other Drugs
In vitro studies with human hepatic microsomes show that voriconazole inhibits the metabolic activity of the cytochrome P450 enzymes CYP2C19, CYP2C9, and CYP3A4. In these studies, the inhibition potency of voriconazole for CYP3A4 metabolic activity was significantly less than that of two other azoles, ketoconazole and itraconazole. In vitro studies also show that the major metabolite of voriconazole, voriconazole N-oxide, inhibits the metabolic activity of CYP2C9 and CYP3A4 to a greater extent than that of CYP2C19. Therefore, there is potential for voriconazole and its major metabolite to increase the systemic exposure (plasma concentrations) of other drugs metabolized by these CYP450 enzymes.
The systemic exposure of the following drugs is significantly increased by coadministration of voriconazole and their use is contraindicated:
Sirolimus (CYP3A4 substrate)‚ÄďRepeat dose administration of oral voriconazole (400 mg every 12 hours¬† for 1 day, then 200 mg every 12 hours¬† for 8 days) increased the Cmax and AUC of sirolimus (2 mg single dose) an average of 7-fold (90% CI: 5.7, 7.5) and 11-fold (90% CI: 9.9, 12.6), respectively, in healthy male subjects [see Contraindications ( 4 ) ].
Coadministration of voriconazole with the following agents results in increased exposure to these drugs. Therefore, careful monitoring and/or dosage adjustment of these drugs is needed:
Alfentanil (CYP3A4 substrate)‚ÄďCoadministration of multiple doses of oral voriconazole (400 mg every 12 hours on day 1, 200 mg every 12 hours on day 2) with a single 20 mcg/kg intravenous dose of alfentanil with concomitant naloxone resulted in a 6-fold increase in mean alfentanil ¬†¬†AUC0-‚ąě and a 4-fold prolongation of mean alfentanil elimination half-life, compared to when alfentanil was given alone [see Drug Interactions ( 7 )].
 
Fentanyl (CYP3A4 substrate): In an independent published study, concomitant use of voriconazole (400 mg every 12 hours on Day 1, then 200 mg every 12 hours on Day 2) with a single intravenous dose of fentanyl (5 őľg/kg) resulted in an increase in the mean AUC0-‚ąě of fentanyl by 1.4-fold (range 0.81- to 2.04-fold) [see Drug Interactions ( 7 )].
Oxycodone (CYP3A4 substrate): In an independent published study, coadministration of multiple doses of oral voriconazole (400 mg every 12 hours, on Day 1 followed by five doses of 200 mg every 12 hours¬† on Days 2 to 4) with a single 10 mg oral dose of oxycodone on Day 3 resulted in an increase in the mean Cmax and AUC0‚Äď‚ąě of oxycodone by 1.7-fold (range 1.4- to 2.2-fold) and 3.6-fold (range 2.7- to 5.6-fold), respectively. The mean elimination half-life of oxycodone was also increased by 2.0-fold (range 1.4- to 2.5-fold) ¬†[see Drug Interactions ( 7 )].
Cyclosporine (CYP3A4 substrate)‚ÄďIn stable renal transplant recipients receiving chronic cyclosporine therapy, concomitant administration of oral voriconazole (200 mg every 12 hours¬† for 8 days) increased cyclosporine Cmax and AUCŌĄ an average of 1.1 times (90% CI: 0.9, 1.41) and 1.7 times (90% CI: 1.5, 2.0), respectively, as compared to when cyclosporine was administered without voriconazole ¬†[see Drug Interactions ( 7 )].
Methadone (CYP3A4, CYP2C19, CYP2C9 substrate)‚ÄďRepeat dose administration of oral voriconazole (400 mg every 12 hours for 1 day, then 200 mg every 12 hours for 4 days) increased the Cmax and AUCŌĄ of pharmacologically active Rmethadone by 31% (90% CI: 22%, 40%) and 47% (90% CI: 38%, 57%), respectively, in subjects receiving a methadone maintenance dose (30-100 mg every 24 hours). The Cmax and AUC of (S)-methadone increased by 65% (90% CI: 53%, 79%) and 103% (90% CI: 85%, 124%), respectively¬†[see Drug Interactions ( 7 )].
Tacrolimus (CYP3A4 substrate)‚ÄďRepeat oral dose administration of voriconazole (400 mg every 12 hours¬† x 1 day, then 200 mg every 12 hours¬† x 6 days) increased tacrolimus (0.1 mg/kg single dose) Cmax and AUCŌĄ in healthy subjects by an average of 2-fold (90% CI: 1.9, 2.5) and 3-fold (90% CI: 2.7, 3.8), respectively [see Drug Interactions ( 7 )].
Warfarin (CYP2C9 substrate)‚ÄďCoadministration of voriconazole (300 mg every 12 hours x 12 days) with warfarin (30 mg single dose) significantly increased maximum prothrombin time by approximately 2 times that of placebo in healthy subjects [see Drug Interactions ( 7 )].
Non-Steroidal Anti-Inflammatory Drugs (NSAIDs; CYP2C9 substrates): In two independent published studies, single doses of ibuprofen (400 mg) and diclofenac (50 mg) were coadministered with the last dose of voriconazole (400 mg every 12 hours  on Day 1, followed by 200 mg every 12 hours on Day 2). Voriconazole increased the mean Cmax and AUC of the pharmacologically active isomer, S (+)-ibuprofen by 20% and 100%, respectively. Voriconazole increased the mean Cmax and AUC of diclofenac by 114% and 78%, respectively [see Drug Interactions ( 7 )].
No significant pharmacokinetic interactions were observed when voriconazole was coadministered with the following agents. Therefore, no dosage adjustment for these agents is recommended:
Prednisolone (CYP3A4 substrate)‚ÄďVoriconazole (200 mg every 12 hours¬† x 30 days) increased Cmax and AUC of prednisolone (60 mg single dose) by an average of 11% and 34%, respectively, in healthy subjects [see Warnings and Precautions ( 5.8 )].
Digoxin (P-glycoprotein mediated transport)‚ÄďVoriconazole (200 mg every 12 hours x 12 days) had no significant effect on steady state Cmax and AUCŌĄ of digoxin (0.25 mg once daily for 10 days) in healthy subjects.
Mycophenolic acid (UDP-glucuronyl transferase substrate)‚ÄďVoriconazole (200 mg every 12 hours x 5 days) had no significant effect on the Cmax and AUCŌĄ of mycophenolic acid and its major metabolite, mycophenolic acid glucuronide after administration of a 1 gram single oral dose of mycophenolate mofetil.
Two-Way Interactions
Concomitant use of the following agents with voriconazole is contraindicated:
Rifabutin (potent CYP450 inducer)‚ÄďRifabutin (300 mg once daily) decreased the Cmax and AUCŌĄ of voriconazole at 200 mg twice daily by an average of 67% (90% CI: 58%, 73%) and 79% (90% CI: 71%, 84%), respectively, in healthy subjects. During coadministration with rifabutin (300 mg once daily), the steady state Cmax and AUCŌĄ of voriconazole following an increased dose of 400 mg twice daily were on average approximately 2 times higher, compared with voriconazole alone at 200 mg twice daily. Coadministration of voriconazole at 400 mg twice daily with rifabutin 300 mg twice daily increased the Cmax and AUCŌĄ of rifabutin by an average of 3-times (90% CI: 2.2, 4.0) and 4 times (90% CI: 3.5, 5.4), respectively, compared to rifabutin given alone [see Contraindications ( 4 )] .
Significant drug interactions that may require dosage adjustment, frequent monitoring of drug levels and/or frequent monitoring of drug-related adverse  reactions/toxicity:
Efavirenz, a non-nucleoside reverse transcriptase inhibitor(CYP450 inducer; CYP3A4 inhibitor and substrate) ‚Äď Standard doses of Voriconazole and efavirenz (400 mg every 24 hours or higher) must not be coadministered [see Drug Interactions (7)]. Steady state efavirenz (400 mg PO every 24 hours) decreased the steady state Cmax and AUCŌĄ of voriconazole (400 mg PO every 12 hours for 1 day, then 200 mg PO every 12 hours for 8 days) by an average of 61% and 77%, respectively, in healthy male subjects. Voriconazole at steady state (400 mg PO every 12 hours for 1 day, then 200 mg every 12 hours for 8 days) increased the steady state Cmax and AUCŌĄ of efavirenz (400 mg PO every 24 hours for 9 days) by an average of 38% and 44%, respectively, in healthy subjects.
The pharmacokinetics of adjusted doses of voriconazole and efavirenz were studied in healthy male subjects following administration of voriconazole (400 mg PO every 12 hours on Days 2 to 7) with efavirenz (300 mg PO every 24 hours on Days 1-7), relative to steady-state administration of voriconazole (400 mg for 1 day, then 200 mg PO every 12 hours for 2 days) or efavirenz (600 mg every 24 hours for 9 days). Coadministration of voriconazole 400 mg every 12 hours¬† with efavirenz 300 mg every 24 hours, decreased voriconazole AUCŌĄ by 7% (90% CI: -23%, 13%) and increased Cmax by 23% (90% CI: -1%, 53%); efavirenz AUCŌĄ was increased by 17% (90% CI: 6%, 29%) and Cmax was equivalent¬†[ see Dosage and Administration ( 2.7 ), Contraindications ( 4 ), and Drug Interactions ( 7 )].
Phenytoin (CYP2C9 substrate and potent CYP450 inducer)‚ÄďRepeat dose administration of phenytoin (300 mg once daily) decreased the steady state Cmax and AUCŌĄ of orally administered voriconazole (200 mg every 12 hours x 14 days) by an average of 50% and 70%, respectively, in healthy subjects. Administration of a higher voriconazole dose (400 mg every 12 hours x 7 days) with phenytoin (300 mg once daily) resulted in comparable steady state voriconazole Cmax and AUCŌĄ estimates as compared to when voriconazole was given at 200 mg every 12 hours without phenytoin[see Dosage and Administration ( 2.7 ), and Drug Interactions ( 7 )].
Repeat dose administration of voriconazole (400 mg every 12 hours x 10 days) increased the steady state Cmax and AUCŌĄ of phenytoin (300 mg once daily) by an average of 70% and 80%, respectively, in healthy subjects. The increase in phenytoin Cmax and AUC when coadministered with voriconazole may be expected to be as high as 2 times the Cmax and AUC estimates when phenytoin is given without¬†voriconazole [see Drug Interactions ( 7 )]. ¬†
Omeprazole (CYP2C19 inhibitor; CYP2C19 and CYP3A4 substrate)‚ÄďCoadministration of omeprazole (40 mg once daily x 10 days) with oral voriconazole (400 mg every 12 hours x 1 day, then 200 mg every 12 hours x 9 days) increased the steady state Cmax and AUCŌĄ of voriconazole by an average of 15% (90% CI: 5%, 25%) and 40% (90% CI: 29%, 55%), respectively, in healthy subjects. No dosage adjustment of voriconazole is recommended.
Coadministration of voriconazole (400 mg every 12 hours x 1 day, then 200 mg x 6 days) with omeprazole (40 mg once daily x 7 days) to healthy subjects significantly increased the steady state Cmax and AUCŌĄ of omeprazole an average of 2 times (90% CI: 1.8, 2.6) and 4 times (90% CI: 3.3, 4.4), respectively [see Drug Interactions ( 7 )]. ¬†
Oral Contraceptives (CYP3A4 substrate; CYP2C19 inhibitor)-Coadministration of oral voriconazole (400 mg every 12 hours¬† for 1 day, then 200 mg every 12 hours¬† for 3 days) and oral contraceptive (Ortho-Novum1/35¬ģ consisting of 35 mcg ethinyl estradiol and 1 mg norethindrone, every 24 hours) to healthy female subjects at steady state increased the Cmax and AUCŌĄ of ethinyl estradiol by an average of 36% (90% CI: 28%, 45%) and 61% (90% CI: 50%, 72%), respectively, and that of norethindrone by 15% (90% CI: 3%, 28%) and 53% (90% CI: 44%, 63%), respectively in healthy subjects. Voriconazole Cmax and AUCŌĄ increased by an average of 14% (90% CI: 3%, 27%) and 46% (90% CI: 32%, 61%), respectively [see Drug Interactions ( 7 )]. ¬†
No significant pharmacokinetic interaction was seen and no dosage adjustment of these drugs is recommended:
Indinavir (CYP3A4 inhibitor and substrate)‚ÄďRepeat dose administration of indinavir (800 mg TID for 10 days) had no significant effect on voriconazole Cmax and AUC following repeat dose administration (200 mg every 12 hours¬† for 17 days) in healthy subjects.
Repeat dose administration of voriconazole (200 mg every 12 hours for 7 days) did not have a significant effect on steady state Cmax and AUCŌĄ of indinavir following repeat dose administration (800 mg TID for 7 days) in healthy subjects.
12.4 Microbiology
Mechanism of Action
Voriconazole is an azole antifungal drug. The primary mode of action of voriconazole is the inhibition of fungal cytochrome P-450-mediated 14 alpha-lanosterol demethylation, an essential step in fungal ergosterol biosynthesis. The accumulation of 14 alpha-methyl sterols correlates with the subsequent loss of ergosterol in the fungal cell wall and may be responsible for the antifungal activity of voriconazole.
Resistance
A potential for development of resistance to voriconazole is well known. The mechanisms of resistance may include mutations in the gene ERG11 (encodes for the target enzyme, lanosterol 14-őĪ-demethylase), upregulation of genes encoding the ATP-binding cassette efflux transporters i.e., Candida drug resistance (CDR) pumps and reduced access of the drug to the target, or some combination of those mechanisms. The frequency of drug resistance development for the various fungi for which this drug is indicated is not known.
Fungal isolates exhibiting reduced susceptibility to fluconazole or itraconazole may also show reduced susceptibility to voriconazole, suggesting cross-resistance can occur among these azoles. The relevance of cross-resistance and clinical outcome has not been fully characterized. Clinical cases where azole cross-resistance is demonstrated may require alternative antifungal therapy.
Antimicrobial Activity
Voriconazole has been shown to be active against most isolates of the following microorganisms, both in vitro and in clinical infections.
Aspergillus fumigatus
Aspergillus flavus
Aspergillus niger
Aspergillus terreus
Candida albicans
Candida glabrata (In clinical studies, the voriconazole MIC90 was 4 őľg/mL) *
Candida krusei
Candida parapsilosis
Candida tropicalis
Fusarium spp. including Fusarium solani
Scedosporium apiospermum
* In clinical studies, voriconazole MIC90 for C. glabrata baseline isolates was 4 őľg/mL; 13/50 (26%) C. glabrata baseline isolates were resistant (MIC ‚Č•4 őľg/mL) to voriconazole. However, based on 1054 isolates tested in surveillance studies the MIC90 was 1 őľg/mL.
The following data are available, but their clinical significance is unknown. At least 90 percent of the following fungi exhibit an in vitro minimum inhibitory concentration (MIC) less than or equal to the susceptible breakpoint for voriconazole against isolates of similar genus or organism group. However, the effectiveness of voriconazole in treating clinical infections due to these fungi has not been established in adequate and well-controlled clinical trials:
Candida lusitaniae
Candida guilliermondii
Susceptibility Testing
For specific information regarding susceptibility test interpretive criteria and associated test methods and quality control standards recognized by FDA for this drug, please see: https://www.fda.gov/STIC.
12.5 Pharmacogenomics
CYP2C19, significantly involved in the metabolism of voriconazole, exhibits genetic polymorphism. Approximately 15-20% of Asian populations may be expected to be poor metabolizers. For Caucasians and Blacks, the prevalence of poor metabolizers is 3-5%. Studies conducted in Caucasian and Japanese healthy subjects have shown that poor metabolizers have, on average, 4-fold higher voriconazole exposure (AUCŌĄ) than their homozygous extensive metabolizer counterparts. Subjects who are heterozygous extensive metabolizers have, on average, 2-fold higher voriconazole exposure than their homozygous extensive metabolizer counterparts [see Clinical Pharmacology ( 12.3 )].
13 Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Two-year carcinogenicity studies were conducted in rats and mice. Rats were given oral doses of 6, 18 or 50 mg/kg voriconazole, or 0.2, 0.6, or 1.6 times the recommended maintenance dose on a body surface area basis. Hepatocellular adenomas were detected in females at 50 mg/kg and hepatocellular carcinomas were found in males at 6 and 50 mg/kg. Mice were given oral doses of 10, 30 or 100 mg/kg voriconazole, or 0.1, 0.4, or 1.4 times the RMD on a body surface area basis. In mice, hepatocellular adenomas were detected in males and females and hepatocellular carcinomas were detected in males at 1.4 times the RMD of voriconazole.
Voriconazole demonstrated clastogenic activity (mostly chromosome breaks) in human lymphocyte cultures in vitro. Voriconazole was not genotoxic in the Ames assay, CHO HGPRT assay, the mouse micronucleus assay or the in vivo DNA repair test (Unscheduled DNA Synthesis assay).
Voriconazole administration induced no impairment of male or female fertility in rats dosed at 50 mg/kg, or 1.6 times the RMD.
14 Clinical Studies
Voriconazole administered orally or parenterally, has been evaluated as primary or salvage therapy in 520 patients aged 12 years and older with infections caused by Aspergillus spp., Fusarium spp., and Scedosporium spp.
14.1 Invasive Aspergillosis (IA)
Voriconazole was studied in patients for primary therapy of IA (randomized, controlled study 307/602), for primary and salvage therapy of aspergillosis (non-comparative study 304) and for treatment of patients with IA who were refractory to, or intolerant of, other antifungal therapy (non-comparative study 309/604).
Study 307/602 ‚Äď Primary Therapy of Invasive Aspergillosis
The efficacy of voriconazole compared to amphotericin B in the primary treatment of acute IA was demonstrated in 277 patients treated for 12 weeks in a randomized, controlled study (Study 307/602). The majority of study patients had underlying hematologic malignancies, including bone marrow transplantation. The study also included patients with solid organ transplantation, solid tumors, and AIDS. The patients were mainly treated for definite or probable IA of the lungs. Other aspergillosis infections included disseminated disease, CNS infections and sinus infections. Diagnosis of definite or probable IA was made according to criteria modified from those established by the National Institute of Allergy and Infectious Diseases Mycoses Study Group/European Organisation for Research and Treatment of Cancer (NIAID MSG/EORTC).
Voriconazole was administered intravenously with a loading dose of 6 mg/kg every 12 hours for the first 24 hours followed by a maintenance dose of 4 mg/kg every 12 hours for a minimum of 7 days. Therapy could then be switched to the oral formulation at a dose of 200 mg every 12 hours. Median duration of IV voriconazole therapy was 10 days (range 2-85 days). After IV voriconazole therapy, the median duration of PO voriconazole therapy was 76 days (range 2-232 days).
Patients in the comparator group received conventional amphotericin B as a slow infusion at a daily dose of 1.0-1.5 mg/kg/day. Median duration of IV amphotericin therapy was 12 days (range 1-85 days). Treatment was then continued with OLAT, including itraconazole and lipid amphotericin B formulations. Although initial therapy with conventional amphotericin B was to be continued for at least two weeks, actual duration of therapy was at the discretion of the investigator. Patients who discontinued initial randomized therapy due to toxicity or lack of efficacy were eligible to continue in the study with OLAT treatment.
A satisfactory global response at 12 weeks (complete or partial resolution of all attributable symptoms, signs, radiographic/bronchoscopic abnormalities present at baseline) was seen in 53% of voriconazole treated patients compared to 32% of amphotericin B treated patients (Table 15). A benefit of voriconazole compared to amphotericin B on patient survival at Day 84 was seen with a 71% survival rate on voriconazole compared to 58% on amphotericin B (Table 13).
Table 13 also summarizes the response (success) based on mycological confirmation and species.
Table 13: Overall Efficacy and Success by Species in the Primary Treatment of Acute Invasive Aspergillosis Study 307/602 a Assessed by independent Data Review Committee (DRC) b Proportion of subjects alive c Amphotericin B followed by other licensed antifungal therapy d Difference and corresponding 95% confidence interval are stratified by protocol e Not all mycologically confirmed specimens were speciated f Some patients had more than one species isolated at baseline   Voriconazole Ampho B c Stratified Difference (95% CI)d   n/N (%) n/N (%)   Efficacy as Primary Therapy       Satisfactory Global Response a 76/144 (53) 42/133 (32) 21.8%(10.5%, 33.0%)p<0.0001 Survival at Day 84 b 102/144 (71) 77/133 (58) 13.1%(2.1%, 24.2%) Success by Species       Success n/N (%)   Overall success 76/144 (53) 42/133 (32)   Mycologically confirmed e 37/84 (44) 16/67 (24)   Aspergillus spp. f       A. fumigatus 28/63 (44) 12/47 (26)   A. flavus 3/6 4/9   A. terreus 2/3 0/3   A. niger 1/4 0/9   A. nidulans 1/1 0/0  
Study 304 ‚Äď Primary and Salvage Therapy of Aspergillosis
In this non-comparative study, an overall success rate of 52% (26/50) was seen in patients treated with voriconazole for primary therapy. Success was seen in 17/29 (59%) with Aspergillus fumigatus infections and 3/6 (50%) patients with infections due to non-fumigatus species [A. flavus (1/1); A. nidulans (0/2); A. niger (2/2); A. terreus (0/1)]. Success in patients who received voriconazole as salvage therapy is presented in Table 14.
Study 309/604 ‚Äď Treatment of Patients with Invasive Aspergillosis who were Refractory to, or Intolerant of, other Antifungal Therapy
Additional data regarding response rates in patients who were refractory to, or intolerant of, other antifungal agents are also provided in Table 16. In this non-comparative study, overall mycological eradication for culture-documented infections due to fumigatus and non-fumigatus species of Aspergillus was 36/82 (44%) and 12/30 (40%), respectively, in voriconazole treated patients. Patients had various underlying diseases and species other than A. fumigatus contributed to mixed infections in some cases.
For patients who were infected with a single pathogen and were refractory to, or intolerant of, other antifungal agents, the satisfactory response rates for voriconazole in studies 304 and 309/604 are presented in Table 14.
Table 14: Combined Response Data in Salvage Patients with Single Aspergillus Species (Studies 304 and 309/604)   Success n/N A. fumigatus 43/97 (44%) A. flavus 5/12 A. nidulans 1/3 A. niger 4/5 A. terreus 3/8 A. versicolor 0/1
Nineteen patients had more than one species of Aspergillus isolated. Success was seen in 4/17 (24%) of these patients.
14.2 Candidemia in Non-neutropenic Patients and Other Deep Tissue Infections
Voriconazole was compared to the regimen of amphotericin B followed by fluconazole in Study 608, an open label, comparative study in nonneutropenic patients with candidemia associated with clinical signs of infection. Patients were randomized in 2:1 ratio to receive either voriconazole (n=283) or the regimen of amphotericin B followed by fluconazole (n=139). Patients were treated with randomized study drug for a median of 15 days. Most of the candidemia in patients evaluated for efficacy was caused by C. albicans (46%), followed by C. tropicalis (19%), C. parapsilosis (17%), C. glabrata (15%), and C. krusei (1%).
An independent Data Review Committee (DRC), blinded to study treatment, reviewed the clinical and mycological data from this study, and generated one assessment of response for each patient. A successful response required all of the following: resolution or improvement in all clinical signs and symptoms of infection, blood cultures negative for Candida, infected deep tissue sites negative for Candida or resolution of all local signs of infection, and no systemic antifungal therapy other than study drug. The primary analysis, which counted DRC-assessed successes at the fixed time point (12 weeks after End of Therapy [EOT]), demonstrated that voriconazole was comparable to the regimen of amphotericin B followed by fluconazole (response rates of 41% and 41%, respectively) in the treatment of candidemia. Patients who did not have a 12-week assessment for any reason were considered a treatment failure.
The overall clinical and mycological success rates by Candida species in Study 150-608 are presented in Table 15.
Table 15: Overall Success Rates Sustained From EOT To The Fixed 12-Week Follow-Up Time Point By Baseline Pathogena,b a A few patients had more than one pathogen at baseline.  b Patients who did not have a 12-week assessment for any reason were considered a treatment failure. Baseline Pathogen   Clinical and Mycological Success (%)  Voriconazole  Amphotericin B --> Fluconazole  C. albicans   46/107 (43%) 30/63 (48%) C. tropicalis 17/53 (32%) 1/16 (6%) C. parapsilosis 24/45 (53%) 10/19 (53%) C. glabrata 12/36 (33%) 7/21 (33%) C. krusei 1/4 0/1
In a secondary analysis, which counted DRC-assessed successes at any time point (EOT, or 2, 6, or 12 weeks after EOT), the response rates were 65% for voriconazole and 71% for the regimen of amphotericin B followed by fluconazole.
In Studies 608 and 309/604 (non-comparative study in patients with invasive fungal infections who were refractory to, or intolerant of, other antifungal agents), voriconazole was evaluated in 35 patients with deep tissue Candida infections. A favorable response was seen in 4 of 7 patients with intra-abdominal infections, 5 of 6 patients with kidney and bladder wall infections, 3 of 3 patients with deep tissue abscess or wound infection, 1 of 2 patients with pneumonia/pleural space infections, 2 of 4 patients with skin lesions, 1 of 1 patients with mixed intraabdominal and pulmonary infection, 1 of 2 patients with suppurative phlebitis, 1 of 3 patients with hepatosplenic infection, 1 of 5 patients with osteomyelitis, 0 of 1 with liver infection, and 0 of 1 with cervical lymph node infection.
14.3 Esophageal Candidiasis (EC)
The efficacy of oral voriconazole 200 mg twice daily compared to oral fluconazole 200 mg once daily in the primary treatment of EC was demonstrated in Study 150-305, a double-blind, double-dummy study in immunocompromised patients with endoscopically-proven EC. Patients were treated for a median of 15 days (range 1 to 49 days). Outcome was assessed by repeat endoscopy at end of treatment (EOT). A successful response was defined as a normal endoscopy at EOT or at least a 1 grade improvement over baseline endoscopic score. For patients in the Intent-to-Treat (ITT) population with only a baseline endoscopy, a successful response was defined as symptomatic cure or improvement at EOT compared to baseline. Voriconazole and fluconazole (200 mg once daily) showed comparable efficacy rates against EC, as presented in Table 16.
Table 16: Success Rates in Patients Treated for Esophageal Candidiasis a Confidence Interval for the difference (Voriconazole ‚Äď Fluconazole) in success rates. b PP (Per Protocol) patients had confirmation of Candida esophagitis by endoscopy, received at least 12 days of treatment, and had a repeat endoscopy at EOT (end of treatment). c ¬†ITT (Intent to Treat) patients without endoscopy or clinical assessment at EOT were treated as failures. Population Voriconazole Fluconazole Difference % (95% CI)a PPb 113/115 (98.2%) 134/141 (95.0%) 3.2 (-1.1, 7.5) ITTc 175/200 (87.5%) 171/191 (89.5%) -2.0 (-8.3, 4.3)
Microbiologic success rates by Candida species are presented in Table 17.
Table 17: Clinical And Mycological Outcome By Baseline Pathogen in Patients with Esophageal Candidiasis (Study-150-305) a  Some patients had more than one species isolated at baseline. b  Patients with endoscopic and/or mycological assessment at end of therapy. Pathogena Voriconazole Fluconazole Favorable endoscopic responseb Mycological eradicationb Favorable endoscopic responseb Mycological eradicationb Success/Total (%) Eradication/Total (%) Success/Total (%)   Eradication/ Total (%) C. albicans 134/140 (96%) 90/107 (84%) 147/156 (94%) 91/115 (79%) C. glabrata 8/8 (100%) 4/7 (57%) 4/4 (100%) 1/4 (25%) C. krusei 1/1 1/1 2/2 (100%) 0/0 14.4 Other Serious Fungal Pathogens
In pooled analyses of patients, voriconazole was shown to be effective against the following additional fungal pathogens:
Scedosporium apiospermum - Successful response to voriconazole therapy was seen in 15 of 24 patients (63%). Three of these patients relapsed within 4 weeks, including 1 patient with pulmonary, skin and eye infections, 1 patient with cerebral disease, and 1 patient with skin infection. Ten patients had evidence of cerebral disease and 6 of these had a successful outcome (1 relapse). In addition, a successful response was seen in 1 of 3 patients with mixed organism infections.
Fusarium spp. - Nine of 21 (43%) patients were successfully treated with voriconazole. Of these 9 patients, 3 had eye infections, 1 had an eye and blood infection, 1 had a skin infection, 1 had a blood infection alone, 2 had sinus infections, and 1 had disseminated infection (pulmonary, skin, hepatosplenic). Three of these patients (1 with disseminated disease, 1 with an eye infection and 1 with a blood infection) had Fusarium solani and were complete successes. Two of these patients relapsed, 1 with a sinus infection and profound neutropenia and 1 post surgical patient with blood and eye infections.
14.5 Pediatric Studies
A total of 22 patients aged 12 to 18 years with IA were included in the adult therapeutic studies. Twelve out of 22 (55%) patients had successful response after treatment with a maintenance dose of voriconazole 4 mg/kg every 12 hours.
Fifty-three pediatric patients aged 2 to less than 18 years old were treated with voriconazole in two prospective, open-label, noncomparative, multicenter clinical studies.
One study was designed to enroll pediatric patients with IA or infections with rare molds (such as Scedosporium or Fusarium). Patients aged 2 to less than 12 years and 12 to 14 years with body weight less than 50 kg received an intravenous voriconazole loading dose of 9 mg/kg every 12 hours for the first 24-hours followed by an 8 mg/kg intravenous maintenance dose every 12 hours. After completing 7 days of intravenous therapy patients had an option to switch to oral voriconazole. The oral maintenance dose was 9 mg/kg every 12 hours (maximum dose of 350 mg). All other pediatric patients aged 12 to less than 18 years received the adult voriconazole dosage regimen. Patients received voriconazole for at least 6 weeks and up to a maximum of 12 weeks.
The study enrolled 31 patients with possible, proven, or probable IA. Fourteen of 31 patients, 5 of whom were 2 to less than 12 years old and 9 of whom were 12 to less than 18 years old, had proven or probable IA and were included in the modified intent-to-treat (MITT) efficacy analyses. No patients with rare mold were enrolled. A successful global response was defined as resolution or improvement in clinical signs and symptoms and at least 50% resolution of radiological lesions attributed to IA. The overall rate of successful global response at 6 weeks in the MITT population is presented in Table 18 below.
                 
Table 18: Global Responsea in Patients with Invasive Aspergillosis, Modified Intent-to-Treat (MITT)b Population a Global response rate was defined as the number of subjects with a successful response (complete or partial) as a percentage of all subjects (including subjects with an indeterminate or missing response) at 6 weeks in the MITT population. b The Modified Intent-to-Treat (MITT) population was defined as all subjects who received at least 1 dose of study drug and who were diagnosed with proven or probable IA as defined by the modified EORTC/MSG criteria.   Parameter      Global Response at Week 6    Ages 2-<12 years  N=5    Ages 12-<18 years  N=9    Overall  N=14    Number of successes,  n (%)   2 (40%)   7 (78%)   9 (64%)  
The second study enrolled 22 patients with invasive candidiasis including candidemia (ICC) and EC requiring either primary or salvage therapy. Patients with ICC aged 2 to less than 12 years and 12 to 14 years with body weight less than 50 kg received an intravenous voriconazole loading dose of 9 mg/kg every 12 hours for the first 24 hours followed by an 8 mg/kg intravenous maintenance dose every 12-hours. After completing 5 days of intravenous therapy patients had an option to switch to oral voriconazole. The oral maintenance dose was 9 mg/kg every 12 hours (maximum dose of 350 mg). All other pediatric patients aged 12 to less than 18 years received the adult voriconazole dosage regimen. Voriconazole was administered for at least 14 days after the last positive culture. A maximum of 42 days of treatment was permitted.
Patients with primary or salvage EC aged 2 to less than 12 years and 12 to 14 years with body weight less than 50 kg received an intravenous voriconazole dose of 4 mg/kg every 12 hours followed by an oral voriconazole dose of 9 mg/kg every 12 hours (maximum dose of 350 mg) when criteria for oral switch were met. All other pediatric patients aged 12 to less than 18 years received the adult voriconazole dosage regimen. Voriconazole was administered for at least 7 days after the resolution of clinical signs and symptoms. A maximum of 42 days of treatment was permitted.
For EC, study treatment was initiated without a loading dose of intravenous voriconazole. Seventeen of these patients had confirmed Candida infection and were included in the MITT efficacy analyses. Of the 17 patients included in the MITT analyses, 9 were 2 to less than 12 years old (7 with ICC and 2 with EC) and 8 were 12 to less than18 years old (all with EC). For ICC and EC, a successful global response was defined as clinical cure or improvement with microbiological eradication or presumed eradication. The overall rate of successful global response at EOT in the MITT population is presented in Table 19 below.                       
Table 19: Global Responsea at the End of Treatment in the Treatment of Invasive Candidiasis with Candidemia and Esophageal Candidiasis Modified Intent-to-Treat (MITT) Population b a Global response was determined based on the investigator’s assessment of clinical and microbiological response in the Modified Intent-to-Treat (MITT) analysis population at end of treatment. Subjects with missing data or whose response was deemed indeterminate were considered failures. b The MITT population was defined as all subjects who received at least 1 dose of study drug and who had microbiologically confirmed invasive candidiasis with candidemia (ICC) and EC, or subjects with EC who had at least confirmation of oropharyngeal candidiasis without confirmation on esophagoscopy.   c All subjects with ICC were aged 2 to less than 12.   Parameter         Global Response at End of Treatment     EC   N=10     ICCC   N=7     Ages 2-<12   N=2     Ages 12-<18   N=8     Overall   N=10     Overall   N=7     Number of successes, n (%)     2 (100%)     5 (63%)     7 (70%)     6 (86%)  
16 How Supplied/storage And Handling
16.1 How Supplied
Tablets
Voriconazole¬†50 mg tablets: white, round-shaped, biconvex, film-coated tablets debossed with ‚ÄėV50‚Äô on one side and plain on other side.¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†
  Bottles of 30 with child-resistant closure (NDC 27241-062-03)
Voriconazole 200 mg tablets: white, oval-shaped, biconvex, film-coated tablets debossed with ‚ÄėV200‚Äô on one side and plain on other side.¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†¬†
  Bottles of 30 with child-resistant closure (NDC 27241-063-03)
16.2 Storage
Voriconazole tablets should be stored at 15¬įC to 30¬įC (59¬į F to 86¬įF) [see USP Controlled Room Temperature].
17 Patient Counseling Information
Advise the patient to read the FDA-approved patient labeling (Patient Information).
Visual Disturbances
Patients should be instructed that visual disturbances such as blurring and sensitivity to light may occur with the use of voriconazole tablets.
Photosensitivity
- Advise patients of the risk of photosensitivity (with or without concomitant methotrexate), accelerated photoaging, and skin cancer.
- Advise patients that voriconazole tablets can cause serious photosensitivity and to immediately contact their healthcare provider for new or worsening skin rash.
- Advise patients to avoid exposure to direct sun light and to use measures such as protective clothing and sunscreen with high sun protection factor (SPF).
Embryo-Fetal Toxicity
- Advise female patients of the potential risks to a fetus.
- Advise females of reproductive potential to use effective contraception during treatment with voriconazole tablets.
Marketed by:
Ajanta Pharma USA Inc.
Bridgewater, NJ 08807.
Made in India.
Revised: 11/2022
PATIENT INFORMATION
Voriconazole
(vor‚ÄĚ i kon‚Äô a zole)
Tablets, for oral use
Read the Patient Information that comes with voriconazole tablets  before you start taking it and each time you get a refill. There may be new information. This information does not take the place of talking with your healthcare provider about your condition or treatment.
What are voriconazole tablets?
Voriconazole tablets are a prescription medicine used to treat certain serious fungal infections in your blood and body. These infections are called ‚Äúaspergillosis,‚ÄĚ ‚Äúesophageal candidiasis,‚ÄĚ ‚ÄúScedosporium,‚ÄĚ ‚ÄúFusarium,‚ÄĚ and ‚Äúcandidemia‚ÄĚ.
It is not known if voriconazole  tablets are safe and effective in children younger than 2 years old.
Do not take voriconazole tablets if you:
- are allergic to voriconazole or any of the ingredients in voriconazole tablets. See the end of this leaflet for a complete ul of ingredients in voriconazole tablets.
- are taking any of the following medicines:
o pimozide    o rifampin  o efavirenz  o ergotamine, dihydroergotamine (ergot alkaloids)   o tolvaptan  o venetoclax o quinidineo carbamazepineo ritonaviro St.John’s Wort (herbal supplement)o lurasidone  o sirolimuso long-acting barbiturates like phenobarbitalo rifabutino naloxegolo ivabradine 
Ask your healthcare provider or pharmacist if you are not sure if you are taking any of the medicines uled above.
Do not start taking a new medicine without talking to your healthcare provider or pharmacist.
Before you take voriconazole tablets, tell your healthcare provider about all of your medical conditions, including if you:
- have or ever had heart disease, or an abnormal heart rate or rhythm. Your healthcare provider may order a test to check your heart (EKG) before starting voriconazole tablets.
- have low potassium levels, low magnesium levels, and low calcium levels. Your healthcare provider may do blood tests before starting and during treatment with voriconazole tablets.
- have liver or kidney problems. Your healthcare provider may do blood tests to make sure you can take voriconazole tablets.
- have trouble digesting dairy products, lactose (milk sugar), or regular table sugar.  Voriconazole tablets contain lactose.
- are pregnant or plan to become pregnant. Voriconazole tablets can harm your unborn baby. Talk to your healthcare provider if you are pregnant or plan to become pregnant. Women who can become pregnant should use effective birth control while taking voriconazole tablets. Talk to your healthcare provider about birth control methods that may be right for you. 
- are breastfeeding or plan to breastfeed. It is not known if voriconazole  passes into breast milk. Talk to your healthcare provider about the best way to feed your baby if you take voriconazole tablets.
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins and herbal supplements.
Voriconazole tablets may affect the way other medicines work, and other medicines may affect how voriconazole tablet works.
Know what medicines you take. Keep a ul of them to show your healthcare provider or pharmacist when you get a new medicine.
How should I take voriconazole tablets?
- Voriconazole may be prescribed to you as:
- Voriconazole tablets
- Take voriconazole tablets exactly as your healthcare provider tells you to.
- Take voriconazole tablets at least 1 hour before or at least 1 hour after meals.
- If you take too much voriconazole tablets, call your healthcare provider or go to the nearest hospital emergency room. 
What should I avoid while taking voriconazole tablets?
- You should not drive at night while taking voriconazole tablets. Voriconazole tablets can cause changes in your vision such as blurring or sensitivity to light.
- Do not drive or operate machinery, or do other dangerous activities until you know how voriconazole tablet affects you.
- Avoid direct sunlight. Voriconazole tablets can make your skin sensitive to the sun and the light from sunlamps and tanning beds. You could get a severe sunburn. Use sunscreen and wear a hat and clothes that cover your skin if you have to be in sunlight. Talk to your healthcare provider if you get sunburn.
What are possible side effects of voriconazole tablets?
Voriconazole tablets may cause serious side effects including:
- liver problems. Symptoms of liver problems may include:
o itchy skin   o flu-like symptoms  o yellowing of your eyeso nausea or vomiting o feeling very tired             
- vision changes. Symptoms of vision changes may include:          
- blurred vision
- changes in the way you see colors
- sensitivity to light or sun (photosensitivity). Voriconazole tablets can cause serious photosensitivity. There is an increased chance of skin toxicity while taking voriconazole tablets. This can happen with or without taking other medicines like methotrexate. Photosensitivity reactions may also increase your risk of:
- faster skin aging from the sun
- skin cancer
Call your healthcare provider right away if you get a new skin rash or your skin rash gets worse.
- serious heart problems. Voriconazole tablets may cause changes in your heart rate or rhythm, including your heart stopping (cardiac arrest).
- allergic reactions. Symptoms of an allergic reaction may include:
o fever   o chest tightness   o nausea  o sweatingo trouble breathingo itching   o feels like your heart is beating fast (tachycardia)o feel fainto skin rash
- kidney problems.  Voriconazole tablets may cause new or worse problems with kidney function, including kidney failure. Your healthcare provider should check your kidney function while you are taking voriconazole tablets. Your healthcare provider will decide if you can keep taking voriconazole tablets
- serious skin reactions. Symptoms of serious skin reactions may include:
- rash or hives
- mouth sores
- bulering or peeling of your skin
- trouble swallowing or breathing
- adrenal gland problems:
- Voriconazole tablets may cause reduced adrenal function (adrenal insufficiency).
- Voriconazole tablets may cause overactive adrenal function (Cushing’s syndrome) when voriconazole is used at the same time with corticosteroids.
Symptoms of adrenal insufficiency include: 
o feeling tired o nausea and vomiting o abdominal pain  o lack of energy            o feeling dizzy or lightheaded  o weaknesso weight loss 
Symptoms of Cushing’s syndrome include: 
o weight gain o thinning skin   o excessive hair growth  o fatty hump between the shoulders (buffalo hump) and a rounded face (moon face) o bruising easilyo excessive sweating   o darkening of the skin on the stomach, thighs, breasts, and armso high blood sugar 
- bone problems. Voriconazole tablets may cause weakening of bones and bone pain. Tell your healthcare provider if you have bone pain.
Call your healthcare provider or go to the nearest hospital emergency room right away if you have any of the symptoms uled above.
The most common side effects of voriconazole tablets in adults include: 
o vision changes o nausea o hallucinations (seeing or hearing things that are not there)  o rasho headacheo abnormal liver function testso chills o vomitingo fast heart beat (tachycardia)o fever 
The most common side effects of voriconazole tablets in children include: 
o fever  o diarrhea  o low platelet counts o abnormal liver function tests  o low blood calcium levels  o low blood phosphate levels  o vision changes  o rash  o stomach pain  o high blood pressureo cougho low blood pressureo swelling in the arms and legso high blood sugar levelso headacheo fast heart beat(tachycardia)o nose bleedso low blood potassium levels  o inflammation of mucous membraneso hallucinations (seeing or hearing things that are not there)o coughing up bloodo constipationo low blood magnesium levelso fullness of the stomach areao vomitingo nauseao upper respiratory tract infection 
Tell your healthcare provider if you have any side effect that bothers you or that does not go away.
These are not all the possible side effects of voriconazole tablets.
Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088.
How should I store voriconazole tablets?
- Store voriconazole tablets at room temperature, 59 ¬įF¬†to 86 ¬įF (15¬įC to 30¬įC). Do not refrigerate or freeze.
- Keep voriconazole tablets in a tightly closed container.
- Safely throw away medicine that is out of date or no longer needed.
- Keep voriconazole tablets, as well as all other medicines, out of the reach of children.
General information about the safe and effective use of voriconazole tablets.
Medicines are sometimes prescribed for purposes other than those uled in a Patient Information leaflet. Do not use voriconazole tablets for a condition for which it was not prescribed. Do not give voriconazole tablets to other people, even if they have the same symptoms that you have. It may harm them.
You can ask your healthcare provider or pharmacist for information about voriconazole tablets that is written for health professionals.
What are the ingredients of voriconazole tablets?
Active ingredient: voriconazole USP.
Inactive ingredients:
Voriconazole tablets:croscarmellose sodium, lactose monohydrate, magnesium stearate, povidone, pregelatinized starch, and a coating containing hypromellose, lactose monohydrate, titanium dioxide, and triacetin.
Marketed by: Ajanta Pharma USA Inc. Bridgewater, NJ 08807. Made in India
This Patient Information has been approved by the U.S. Food and Drug Administration.
Revised: 11/2022
Spl Patient Package Insert Section
.
Package Label.principal Display Panel- 50 Mg Bottle Label
NDC 27241-062-03
30 Tablets
Voriconazole Tablets
50 mg
Rx Only ajanta
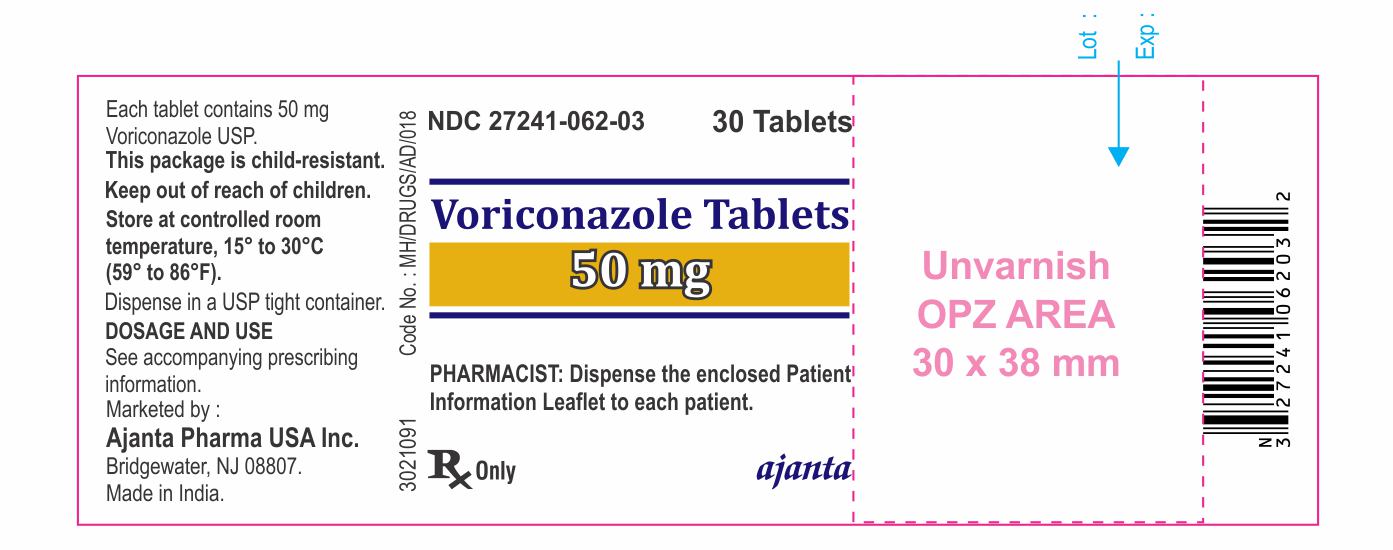
Package Label.principal Display Panel - 200 Mg Bottle Label
NDC 27241-063-03
30 Tablets
Voriconazole Tablets200 mgRx Only ajanta

DISCLAIMER:
"This tool does not provide medical advice, and is for informational and educational purposes only, and is not a substitute for professional medical advice, treatment or diagnosis. Call your doctor to receive medical advice. If you think you may have a medical emergency, please dial 911."
"Do not rely on openFDA to make decisions regarding medical care. While we make every effort to ensure that data is accurate, you should assume all results are unvalidated. We may limit or otherwise restrict your access to the API in line with our Terms of Service."
"This product uses publicly available data from the U.S. National Library of Medicine (NLM), National Institutes of Health, Department of Health and Human Services; NLM is not responsible for the product and does not endorse or recommend this or any other product."
PillSync may earn a commission via links on our site
