Xerava Dailymed
Generic: eravacycline is used for the treatment of Intraabdominal Infections

Go PRO for all pill images
1 Indications And Usage
XERAVA is a tetracycline class antibacterial indicated for the treatment of complicated intra‑abdominal infections in patients 18 years of age and older. (1.1 )
Limitations of Use XERAVA is not indicated for the treatment of complicated urinary tract infections (cUTI). (1.1 )
To reduce the development of drug-resistant bacteria and maintain the effectiveness of XERAVA and other antibacterial drugs, XERAVA should be used only to treat or prevent infections that are proven or strongly suspected to be caused by susceptible bacteria. (1.2 )
1.1 Complicated Intra-abdominal Infections
XERAVA is indicated for the treatment of complicated intra‑abdominal infections (cIAI) caused by susceptible microorganisms: Escherichia coli, Klebsiella pneumoniae, Citrobacter freundii, Enterobacter cloacae, Klebsiella oxytoca, Enterococcus faecalis, Enterococcus faecium, Staphylococcus aureus, Streptococcus anginosus group, Clostridium perfringens, Bacteroides species, and Parabacteroides distasonis in patients 18 years or older [see Microbiology (12.4) and Clinical Studies (14.1)].
Limitations of Use
XERAVA is not indicated for the treatment of complicated urinary tract infections (cUTI) [see Clinical Studies (14.2)].
1.2 Usage
To reduce the development of drug-resistant bacteria and maintain the effectiveness of XERAVA and other antibacterial drugs, XERAVA should be used only to treat or prevent infections that are proven or strongly suspected to be caused by susceptible bacteria. When culture and susceptibility information are available, they should be considered in selecting or modifying antibacterial therapy. In the absence of such data, local epidemiology and susceptibility patterns may contribute to the empiric selection of therapy.
2 Dosage And Administration
- Administer XERAVA for injection 1 mg/kg by intravenous infusion over approximately 60 minutes every 12 hours for a total duration of 4 to 14 days. (
2.1 )- Severe Hepatic Impairment (Child Pugh C): 1 mg/kg XERAVA every 12 hours on Day 1, then 1 mg/kg every 24 hours starting on Day 2 for a total duration of 4 to 14 days. (
2.2 )- Concomitant Use of a Strong Cytochrome P450 Isoenzymes (CYP)3A Inducer: 1.5 mg/kg XERAVA every 12 hours for a total duration of 4 to 14 days. (
2.3 )- See full prescribing information for the preparation of XERAVA (
2.4 )2.1 Recommended Adult Dosage
The recommended dose regimen of XERAVA is 1 mg/kg every 12Â hours. Administer intravenous infusions of XERAVA over approximately 60 minutes every 12Â hours.
The recommended duration of treatment with XERAVA for cIAI is 4 to 14 days. The duration of therapy should be guided by the severity and location of infection and the patient's clinical response.
2.2 Dosage Modifications in Patients with Hepatic Impairment
In patients with severe hepatic impairment (Child Pugh C), administer XERAVA 1 mg/kg every 12Â hours on Day 1 followed by XERAVA 1 mg/kg every 24 hours starting on Day 2 for a total duration of 4 to 14 days. No dosage adjustment is warranted in patients with mild to moderate hepatic impairment (Child Pugh A and Child Pugh B) [see Use in Specific Populations (8.6) and Clinical Pharmacology (12.3)].
2.3 Dosage Modifications in Patients with Concomitant Use of a Strong Cytochrome P450 Isoenzymes (CYP) 3A Inducer
With concomitant use of a strong CYP3A inducer, administer XERAVA 1.5 mg/kg every 12Â hours for a total duration of 4 to 14 days. No dosage adjustment is warranted in patients with concomitant use of a weak or moderate CYP3A inducer [see Drug Interactions (7.1) and Clinical Pharmacology (12.3)].
2.5 Drug Compatibilities
XERAVA is compatible with 0.9% Sodium Chloride Injection, USP. The compatibility of XERAVA with other drugs and infusion solutions has not been established. XERAVA should not be mixed with other drugs or added to solutions containing other drugs.
2.4 Preparation and Administration
XERAVA is for intravenous infusion only. Each vial is for a single dose only.
Preparation
XERAVA is supplied as a sterile yellow to orange dry powder in a single-dose vial that must be reconstituted and further diluted prior to intravenous infusion as outlined below. XERAVA does not contain preservatives. Aseptic technique must be used for reconstitution and dilution as follows:
- Calculate the dose of XERAVA based on the patient weight; 1 mg/kg actual body weight. Prepare the required dose for intravenous infusion, by reconstituting the appropriate number of vials needed. Reconstitute each vial of XERAVA with 5 mL of Sterile Water for Injection, USP or with 5 mL of 0.9% Sodium Chloride Injection, USP, which will deliver the following:
- XERAVA 50 mg vial will deliver 50 mg (10 mg/mL) of eravacycline (free base equivalents).
- XERAVA 100 mg vial will deliver 100 mg (20 mg/mL) of eravacycline (free base equivalents)
- Swirl the vial gently until the powder has dissolved entirely. Avoid shaking or rapid movement as it may cause foaming. The reconstituted XERAVA solution should be a clear, pale yellow to orange solution. Do not use the solution if you notice any particles or the solution is cloudy. Reconstituted solution is not for direct injection. The stability of the solution after reconstitution in the vial has been demonstrated for 1 hour at room temperature (not to exceed 25°C/77°F). If the reconstituted solution in the vial is not diluted in the infusion bag within 1 hour, the reconstituted vial content must be discarded.
- The reconstituted XERAVA solution is further diluted for intravenous infusion to a target concentration of 0.3 mg/mL, in a 0.9% Sodium Chloride Injection, USP infusion bag before intravenous infusion. To dilute the reconstituted solution, withdraw the full or partial reconstituted vial content from each vial and add it into the infusion bag to generate an infusion solution with a target concentration of 0.3 mg/mL (within a range of 0.2 to 0.6 mg/mL). Do not shake the bag.
- The diluted solutions must be infused within 24 hours if stored at room temperature (not to exceed 25°C/77°F) or within 10 days if stored refrigerated at 2°C to 8°C (36°F to 46°F). Reconstituted XERAVA solutions and diluted XERAVA infusion solutions should not be frozen.
- Visually inspect the diluted XERAVA solution for particulate matter and discoloration prior to administration (the XERAVA infusion solution for administration is clear and ranges from light yellow to orange). Discard unused portions of the reconstituted and diluted solution.
Administration of the Intravenous Infusion
The diluted XERAVA solution is administered as an intravenous infusion over approximately 60 minutes.
XERAVA may be administered intravenously through a dedicated line or through a Y-site. If the same intravenous line is used for sequential infusion of several drugs, the line should be flushed before and after infusion of XERAVA with 0.9% Sodium Chloride Injection, USP.
3 Dosage Forms And Strengths
XERAVA for injection is a yellow to orange, sterile, preservative-free, lyophilized powder in single-dose vials for reconstitution and further dilution. XERAVA is available in two strengths:
- 50 mg of eravacycline (equivalent to 63.5 mg eravacycline dihydrochloride)
- 100 mg of eravacycline (equivalent to 127 mg eravacycline dihydrochloride)
For injection: 50 mg and 100 mg of eravacycline (equivalent to 63.5 mg and 127 mg eravacycline dihydrochloride, respectively) as a lyophilized powder in a single-dose vial for reconstitution and further dilution. (3 )
4 Contraindications
XERAVA is contraindicated for use in patients with known hypersensitivity to eravacycline, tetracycline-class antibacterial drugs, or to any of the excipients [see Warnings and Precautions (5.1) and Adverse Reactions (6)].
Known hypersensitivity to eravacycline, tetracycline-class antibacterial drugs, or any of the excipients in XERAVA (4 ,5 ,6 )
5 Warnings And Precautions
- Hypersensitivity Reactions: Life-threatening hypersensitivity (anaphylactic) reactions have been reported with tetracycline antibacterial drugs, including XERAVA. Avoid use in patients with known hypersensitivity to tetracyclines. (
5.1 )- Tooth Discoloration and Enamel Hypoplasia: The use of XERAVA during tooth development (last half of pregnancy, infancy and childhood to the age of 8 years) may cause permanent discoloration of the teeth (yellow-gray-brown) and enamel hypoplasia. (
5.2 ,8.1 ,8.4 )- Inhibition of Bone Growth: The use of XERAVA during the second and third trimester of pregnancy, infancy and childhood up to the age of 8 years may cause reversible inhibition of bone growth (
5.3 ,8.1 ,8.4 ).- Clostridium difficile-associated diarrhea: Evaluate if diarrhea occurs. (
5.4 )5.1 Hypersensitivity Reactions
Life-threatening hypersensitivity (anaphylactic) reactions have been reported with XERAVA [see Adverse Reactions (6.1)]. XERAVA is structurally similar to other tetracycline-class antibacterial drugs and should be avoided in patients with known hypersensitivity to tetracycline-class antibacterial drugs. Discontinue XERAVA if an allergic reaction occurs.
5.2 Tooth Discoloration and Enamel Hypoplasia
The use of XERAVA during tooth development (last half of pregnancy, infancy and childhood to the age of 8 years) may cause permanent discoloration of the teeth (yellow-grey-brown). This adverse reaction is more common during long-term use of the tetracycline class drugs, but it has been observed following repeated short-term courses. Enamel hypoplasia has also been reported with tetracycline class drugs. Advise the patient of the potential risk to the fetus if XERAVA is used during the second or third trimester of pregnancy [see Use in Specific Populations (8.1, 8.4)].
5.3 Inhibition of Bone Growth
The use of XERAVA during the second and third trimester of pregnancy, infancy and childhood up to the age of 8 years may cause reversible inhibition of bone growth. All tetracyclines form a stable calcium complex in any bone-forming tissue. A decrease in fibula growth rate has been observed in premature infants given oral tetracycline in doses of 25 mg/kg every 6 hours. This reaction was shown to be reversible when the drug was discontinued. Advise the patient of the potential risk to the fetus if XERAVA is used during the second or third trimester of pregnancy [see Use in Specific Populations (8.1, 8.4)].
5.4 Associated Diarrhea
Clostridium difficile associated diarrhea (CDAD) has been reported with use of nearly all antibacterial agents, and may range in severity from mild diarrhea to fatal colitis. Treatment with antibacterial agents alters the normal flora of the colon leading to overgrowth of C. difficile.
C. difficile produces toxins A and B which contribute to the development of CDAD. Hypertoxin producing strains of C. difficile cause increased morbidity and mortality, as these infections can be refractory to antimicrobial therapy and may require colectomy. CDAD must be considered in all patients who present with diarrhea following antibacterial drug use. Careful medical history is necessary since CDAD has been reported to occur over two months after the administration of antibacterial agents.
If CDAD is suspected or confirmed, ongoing antibacterial drug use not directed against C. difficile may need to be discontinued. Appropriate fluid and electrolyte management, protein supplementation, antibacterial drug treatment of C. difficile, and surgical evaluation should be instituted as clinically indicated.
5.6 Potential for Microbial Overgrowth
XERAVA use may result in overgrowth of non-susceptible organisms, including fungi. If such infections occur, discontinue XERAVA and institute appropriate therapy.
5.5 Tetracycline Class Adverse Reactions
XERAVA is structurally similar to tetracycline-class antibacterial drugs and may have similar adverse reactions. Adverse reactions including photosensitivity, pseudotumor cerebri, and anti‑anabolic action which has led to increased BUN, azotemia, acidosis, hyperphosphatemia, pancreatitis, and abnormal liver function tests, have been reported for other tetracycline-class antibacterial drugs, and may occur with XERAVA. Discontinue XERAVA if any of these adverse reactions is suspected.
5.7 Development of Drug-Resistant Bacteria
Prescribing XERAVA in the absence of a proven or strongly suspected bacterial infection is unlikely to provide benefit to the patient and increases the risk of the development of drug-resistant bacteria [see Indications and Usage (1.2)].
6 Adverse Reactions
The following clinically significant adverse reactions are described in greater detail in the Warnings and Precautions section:
- Hypersensitivity Reactions [Warning and Precautions (5.1)]
- Tooth Discoloration [Warning and Precautions (5.2)]
- Inhibition of Bone Growth [Warning and Precautions (5.3)]
- Clostridium difficile-Associated Diarrhea [Warning and Precautions (5.4)]
- Tetracycline Class Adverse Reactions [Warning and Precautions (5.5)]
Most common adverse reactions (incidence ≥ 3%) are infusion site reactions, nausea, and vomiting. (6.1 )
To report SUSPECTED ADVERSE REACTIONS, contact Tetraphase Pharmaceuticals Inc., at 1-833-7-XERAVA (1-833-793-7282) or FDA at 1-800-FDA-1088 orwww.fda.gov/medwatch.
6.1 Clinical Trials Experience
Because clinical trials are conducted under widely varying conditions, adverse reaction rates observed in the clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not reflect the rates observed in practice.
XERAVA was evaluated in 3 active-controlled clinical trials (Trial 1, Trial 2, and Trial 3) in adults with cIAI. These trials included two Phase 3 trials (Trial 1 and Trial 2) and one Phase 2 trial (Trial 3, NCT01265784). The Phase 3 trials included 520 patients treated with XERAVA and 517 patients treated with comparator antibacterial drugs (ertapenem or meropenem). The median age of patients treated with XERAVA was 56 years, ranging between 18 and 93 years old; 30% were age 65 years and older. Patients treated with XERAVA were predominantly male (57%) and Caucasian (98%). The XERAVA-treated population included 31% obese patients (BMI ≥ 30 kg/m 2) and 8% with baseline moderate to severe renal impairment (calculated creatinine clearance 15 to less than 60 mL/min). Among the trials, 66 (13%) of patients had baseline moderate hepatic impairment (Child Pugh B); patients with severe hepatic impairment (Child Pugh C) were excluded from the trials.
Adverse Reactions Leading to Discontinuation
Treatment discontinuation due to an adverse reaction occurred in 2% (11/520) of patients receiving XERAVA and 2% (11/517) of patients receiving the comparator. The most commonly reported adverse reactions leading to discontinuation of XERAVA were related to gastrointestinal disorders.
Most Common Adverse Reactions
Adverse reactions occurring at 3% or greater in patients receiving XERAVA were infusion site reactions, nausea, and vomiting.
Table 1 uls adverse reactions occurring in ≥ 1% of patients receiving XERAVA and with incidences greater than the comparator in the Phase 3 cIAI clinical trials. A similar adverse reaction profile was observed in the Phase 2 cIAI clinical trial (Trial 3).
Table 1. Selected Adverse Reactions Reported in ≥ 1% of Patients Receiving XERAVA in the Phase 3 cIAI Trials (Trial 1 and Trial 2) Adverse Reactions XERAVA a N=520 n (%) Comparators b N=517 n (%) Abbreviations: IV=intravenous a XERAVA dose equals 1 mg/kg every 12 hours IV. b Comparators include ertapenem 1 g every 24 hours IV and meropenem 1 g every 8 hours IV. c Infusion site reactions include: catheter/vessel puncture site pain, infusion site extravasation, infusion site hypoaesthesia, infusion/injection site phlebitis, infusion site thrombosis, injection site/vessel puncture site erythema, phlebitis, phlebitis superficial, thrombophlebitis, and vessel puncture site swelling.
Infusion site reactions c
40 (7.7)
10 (1.9)
Nausea
34 (6.5)
3 (0.6)
Vomiting
19 (3.7)
13 (2.5)
Diarrhea
12 (2.3)
8 (1.5)
Hypotension
7 (1.3)
2 (0.4)
Wound dehiscence
7 (1.3)
1 (0.2)
Other Adverse Reactions of XERAVA
The following selected adverse reactions were reported in XERAVA-treated patients at a rate of less than 1% in the Phase 3 trials:
Cardiac disorders: palpitations
Gastrointestinal System: acute pancreatitis, pancreatic necrosis
General Disorders and Administrative Site Conditions: chest pain
Immune system disorders: hypersensitivity
Laboratory Investigations: increased amylase, increased lipase, increased alanine aminotransferase, prolonged activated partial thromboplastin time, decreased renal clearance of creatinine, increased gamma-glutamyltransferase, decreased white blood cell count, neutropenia
Metabolism and nutrition disorders: hypocalcemia
Nervous System: dizziness, dysgeusia
Psychiatric disorders: anxiety, insomnia, depression
Respiratory, Thoracic, and Mediastinal System: pleural effusion, dyspnea
Skin and subcutaneous tissue disorders: rash, hyperhidrosis
7 Drug Interactions
- Concomitant use of strong CYP3A inducers decreases the exposure of eravacycline. Increase XERAVA dose with concomitant use of a strong CYP3A inducer (
2.3 ,7.1 ,12.3 )- Patients who are on anticoagulant therapy may require downward adjustment of their anticoagulant dosage (
7.2 )7.1 Effect of Strong CYP3A Inducers on XERAVA
Concomitant use of strong CYP3A inducers decreases the exposure of eravacycline, which may reduce the efficacy of XERAVA [see Clinical Pharmacology (12.3)]. Increase XERAVA dose in patients with concomitant use of a strong CYP3A inducer [see Dosage and Administration (2.3)].
7.2 Anticoagulant Drugs
Because tetracyclines have been shown to depress plasma prothrombin activity, patients who are on anticoagulant therapy may require downward adjustment of their anticoagulant dosage.
8 Use In Specific Populations
Lactation: Breastfeeding is not recommended during treatment with XERAVA. (8.2 )
8.1 Pregnancy
XERAVA, like other tetracycline-class antibacterial drugs, may cause discoloration of deciduous teeth and reversible inhibition of bone growth when administered during the second and third trimester of pregnancy [see Warnings and Precautions (5.1, 5.2), Data, Use in Specific Populations (8.4)]. The limited available data with XERAVA use in pregnant women are insufficient to inform drug‑associated risk of major birth defects and miscarriages. Animal studies indicate that eravacycline crosses the placenta and is found in fetal plasma; doses greater than approximately 3- and 2.8- times the clinical exposure, based on AUC in rats and rabbits, respectively, administered during the period of organogenesis, were associated with decreased ossification, decreased fetal body weight, and/or increased post-implantation loss [see Data].
The estimated background risk of major birth defects and miscarriage for the indicated population is unknown. All pregnancies have a background risk of birth defect, loss, or other adverse outcomes. In the U.S. general population, the estimated background risk of major birth defects and miscarriage in clinically recognized pregnancies is 2 to 4% and 15 to 20%, respectively.
Animal Data
Embryo-fetal development studies in rats and rabbits reported no treatment-related effects at approximately 3 and 2.8 times the clinical exposure (based on AUC). Dosing was during the period of organogenesis, i.e., gestation days 7-17 in rats and gestation days 7-19 in rabbits. Higher doses, approximately 8.6 and 6.3 times the clinical exposure (based on AUC) in rats and rabbits, respectively, were associated with fetal effects including increased post-implantation loss, reduced fetal body weights, and delays in skeletal ossification in both species, and abortion in the rabbit.
A peri-natal and post-natal rat toxicity study demonstrated that eravacycline crosses the placenta and is found in fetal plasma following intravenous administration to the dams. This study did not demonstrate anatomical malformations, but there were early decreases in pup weight that were later comparable to controls and a non-significant trend toward increased stillbirths or dead pups during lactation. F1 males from dams treated with 10 mg/kg/day eravacycline that continued to fertility testing had decreased testis and epididymis weights at approximately post-natal day 111 that may have been at least partially related to lower body weights in this group.
Tetracyclines cross the placenta, are found in fetal tissues, and can have toxic effects on the developing fetus (often related to retardation of skeletal development). Evidence of embryotoxicity also has been noted in animals treated early in pregnancy.
8.2 Lactation
It is not known whether XERAVA is excreted in human breast milk. Eravacycline (and its metabolites) is excreted in the milk of lactating rats (see Data). Tetracyclines are excreted in human milk; however, the extent of absorption of tetracyclines, including eravacycline, by the breastfed infant is not known. There are no data on the effects of XERAVA on the breastfed infant, or the effects on milk production. Because there are other antibacterial drug options available to treat cIAI in lactating women and because of the potential for serious adverse reactions, including tooth discoloration and inhibition of bone growth, advise patients that breastfeeding is not recommended during treatment with XERAVA and for 4 days (based on half-life) after the last dose.
Animal Data
Eravacycline (and its metabolites) was excreted in the milk of lactating rats on post-natal day 15 following intravenous administration of 3, 5, and 10 mg/kg/day eravacycline.
8.3 Females and Males of Reproductive Potential
Based on animal studies, XERAVA can lead to impaired spermiation and sperm maturation, resulting in abnormal sperm morphology and poor motility. The effect is reversible in rats. The long-term effects of XERAVA on male fertility have not been studied [see Nonclinical Toxicology (13.1)].
8.4 Pediatric Use
The safety and effectiveness of XERAVA in pediatric patients have not been established.
Due to the adverse effects of the tetracycline-class of drugs, including XERAVA on tooth development and bone growth, use of XERAVA in pediatric patients less than 8 years of age is not recommended [see Warnings and Precautions (5.1, 5.2)]
8.5 Geriatric Use
Of the total number of patients with cIAI who received XERAVA in Phase 3 clinical trials (n = 520), 158 subjects were ≥ 65 years of age, while 59 subjects were ≥ 75 years of age. No overall differences in safety or efficacy were observed between these subjects and younger subjects.
No clinically relevant differences in the pharmacokinetics of eravacycline were observed with respect to age in a population pharmacokinetic analysis of eravacycline [see Clinical Pharmacology (12.3)].
8.6 Hepatic Impairment
No dosage adjustment is warranted for XERAVA in patients with mild to moderate hepatic impairment (Child Pugh A and Child Pugh B). Adjust XERAVA dosage in patients with severe hepatic impairment (Child Pugh C) [see Dosage and Administration (2.2) and Clinical Pharmacology (12.3)].
8.7 Renal Impairment
No dosage adjustment is necessary for XERAVA in patients with renal impairment [see Clinical Pharmacology (12.3)].
10 Overdosage
No reports of overdose were reported in clinical trials. In the case of suspected overdose, XERAVA should be discontinued and the patient monitored for adverse reactions. Hemodialysis is not expected to remove significant quantities of XERAVA [see Clinical Pharmacology (12.3)].
11 Description
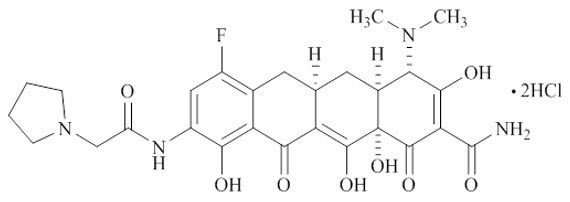
XERAVA contains eravacycline, a synthetic tetracycline-class antibacterial agent for intravenous administration. Chemically, eravacycline is a C7-, C9-substituted sancycline derivative. The chemical name of eravacycline dihydrochloride is [(4S,4aS,5aR,12aS)-4-(dimethylamino)-7-fluoro-3,10,12,12a-tetrahydroxy-1,11-dioxo-9-[2‑(pyrrolidin-1-yl) acetamido]-1,4,4a,5,5a,6,11,12a-octahydrotetracene-2-carboxamide] dihydrochloride. The molecular formula for eravacycline dihydrochloride is C27H31FN4O8•2HCl, and its molecular weight is 631.5.
The following represents the chemical structure of eravacycline dihydrochloride:
XERAVA is a sterile, preservative-free, yellow to orange, lyophilized powder in a glass single-dose vial for intravenous infusion after reconstitution and dilution. XERAVA is supplied in two (2) strengths as follows:
- Each 50 mg single-dose vial contains 50 mg of eravacycline (equivalent to 63.5 mg of eravacycline dihydrochloride) and the excipient, mannitol (150 mg). Sodium hydroxide and hydrochloric acid are used as needed for pH adjustment to 5.5 to 7.0.
- Each 100 mg single-dose vial contains 100 mg of eravacycline (equivalent to 127 mg of eravacycline dihydrochloride) and the excipient, mannitol (150 mg). Sodium hydroxide and hydrochloric acid are used as needed for pH adjustment to 5.5 to 7.0.
12 Clinical Pharmacology
12.1 Mechanism of Action
Eravacycline is an antibacterial drug [see Microbiology (12.4)].
12.2 Pharmacodynamics
The AUC divided by the MIC of eravacycline has been shown to be the best predictor of activity. Based on the flat exposure-response relationship observed in clinical studies, eravacycline exposure achieved with the recommended dosage regimen appears to be on the plateau of the exposure-response curve.
Cardiac Electrophysiology
The effect of XERAVA on the QTc interval was evaluated in a Phase 1 randomized, placebo and positive controlled, double-blind, single-dose, crossover thorough QTc study in 60 healthy adult subjects. At the 1.5 mg/kg single dose (1.5 times the maximum approved recommended dose), XERAVA did not prolong the QTc interval to any clinically relevant extent.
12.3 Pharmacokinetics
Following single-dose intravenous administration, eravacycline AUC and Cmax increase approximately dose-proportionally over doses from 1 mg/kg to 3 mg/kg (3 times the approved dose).
The mean exposure of eravacycline after single and multiple intravenous infusions (approximately 60Â minutes) of 1Â mg/kg administered to healthy adults every 12Â hours is presented in Table 2.
There is approximately 45% accumulation following intravenous dosing of 1Â mg/kg every 12Â hours.
Table 2 Mean (%CV) Plasma Exposure of Eravacycline After Single and Multiple Intravenous Dose in Healthy Adults Abbreviations: Cmax = maximum observed plasma concentration, CV = coefficient of variation; AUC0-12 = area under the plasma concentration-time curve from time 0 to 12 hours. a AUC of day 1 equals AUC0-12 after the first dose of eravacycline. b AUC of day 10 equals steady state AUC0-12.
Exposure [Arithmetic Mean (%CV)]
Cmax (ng/mL)
AUC0-12 (ng∙h/mL)
Day 1
2125 (15)
4305 (14)a
Day 10
1825 (16)
6309 (15)b
Distribution
Protein binding of eravacycline to human plasma proteins increases with increasing plasma concentrations, with 79% to 90% (bound) at plasma concentrations ranging from 100 to 10,000Â ng/mL. The volume of distribution at steady-state is approximately 321 L.
Elimination
The mean elimination half-life is 20 hours.
Metabolism
Eravacycline is metabolized primarily by CYP3A4- and FMO-mediated oxidation.
Excretion
Following a single intravenous dose of radiolabeled eravacycline 60Â mg, approximately 34% of the dose is excreted in urine and 47% in feces as unchanged eravacycline (20% in urine and 17% in feces) and metabolites.
Specific Populations
No clinically significant differences in the pharmacokinetics of eravacycline were observed based on age (18-86 years), sex, and race.
Patients with Renal Impairment
The geometric least square mean Cmax for eravacycline was increased by 8.8% for subjects with end stage renal disease (ESRD) versus healthy subjects with 90% CI -19.4, 45.2. The geometric least square mean AUC0-inf for eravacycline was decreased by 4.0% for subjects with ESRD versus healthy subjects with 90% CI -14.0, 12.3 [see Use in Specific Populations (8.7)].
Patients with Hepatic Impairment
Eravacycline Cmax was 13.9%, 16.3%, and 19.7% higher in subjects with mild (Child-Pugh Class A), moderate (Child-Pugh Class B), and severe (Child‑Pugh Class C) hepatic impairment versus healthy subjects, respectively. Eravacycline AUC0-inf was 22.9%, 37.9%, and 110.3% higher in subjects with mild, moderate, and severe hepatic impairment versus healthy subjects, respectively [see Dosage and Administration (2.2) and Use in Specific Populations (8.6)].
Drug Interaction Studies
Clinical Studies
Concomitant use of rifampin (strong CYP3A4/3A5 inducer) decreased eravacycline AUC by 35% and increased eravacycline clearance by 54% [see Dosage and Administration (2.3) and Drug Interactions (7.1)].
Concomitant use of itraconazole (strong CYP3A inhibitor) increased eravacycline Cmax by 5% and AUC by 32%, and decreased eravacycline clearance by 32%.
In Vitro Studies
Eravacycline is not an inhibitor of CYP1A2, 2B6, 2C8, 2C9, 2C19, 2D6, or 3A4/5. Eravacycline is not an inducer of CYP1A2, 2B6, or 3A4.
Eravacycline is not a substrate for P-glycoprotein (P-gp), breast cancer resistance protein (BCRP), bile salt export pump (BSEP), organic anion transporter peptide (OATP)1B1, OATP1B3, organic ion transporter (OAT)1, OAT3, OCT1, OCT2, multidrug and toxin extrusion (protein) (MATE)1, or MATE2-K transporters.
Eravacycline is not an inhibitor of BCRP, BSEP, OATP1B1, OATP1B3, OAT1, OAT3, OCT1, OCT2, MATE1, or MATE2-K transporters.
12.4 Microbiology
Mechanism of Action
Eravacycline is a fluorocycline antibacterial within the tetracycline class of antibacterial drugs. Eravacycline disrupts bacterial protein synthesis by binding to the 30SÂ ribosomal subunit thus preventing the incorporation of amino acid residues into elongating peptide chains.
In general, eravacycline is bacteriostatic against gram-positive bacteria (e.g., Staphylococcus aureus and Enterococcus faecalis); however, in vitro bactericidal activity has been demonstrated against certain strains of Escherichia coli and Klebsiella pneumoniae.
Resistance
Eravacycline resistance in some bacteria is associated with upregulated, non-specific intrinsic multidrug-resistant (MDR) efflux, and target-site modifications such as to the 16s rRNA or certain 30S ribosomal proteins (e.g., S10).
The C7 and C9Â substitutions in eravacycline are not present in any naturally occurring or semisynthetic tetracyclines and the substitution pattern imparts microbiological activities including in vitro activity against gram-positive and gram-negative strains expressing certain tetracycline-specific resistance mechanism(s) [i.e., efflux mediated by tet(A), tet(B), and tet(K); ribosomal protection as encoded by tet(M) and tet(Q)].
Activity of eravacycline was demonstrated in vitro against Enterobacteriaceae in the presence of certain beta-lactamases, including extended spectrum β-lactamases, and AmpC. However, some beta-lactamase-producing isolates may confer resistance to eravacycline via other resistance mechanisms.
The overall frequency of spontaneous mutants in the gram-positive organisms tested was in the range of 10-9 to 10-10 at 4 times the eravacycline Minimum Inhibitory Concentration (MIC). Multistep selection of gram-negative strains resulted in a 16- to 32-times increase in eravacycline MIC for one isolate of Escherichia coli and Klebsiella pneumoniae, respectively. The frequency of spontaneous mutations in K. pneumoniae was 10-7 to 10‑8 at 4 times the eravacycline MIC.
Interaction with Other Antimicrobials
In vitro studies have not demonstrated antagonism between XERAVA and other commonly used antibacterial drugs for the indicated pathogens.
Antimicrobial Activity
XERAVA has been shown to be active against most isolates of the following microorganisms, both in vitro and in clinical infections [see Indications and Usage (1)]:
Aerobic bacteria
   Gram-positive bacteria
   Enterococcus faecalis    Enterococcus faecium    Staphylococcus aureus    Streptococcus anginosus group
   Gram-negative bacteria   Citrobacter freundii    Enterobacter cloacae    Escherichia coli    Klebsiella oxytoca    Klebsiella pneumoniae
Anaerobic bacteria   Gram-positive bacteria   Clostridium perfringens
   Gram-negative bacteria   Bacteroides caccae    Bacteroides fragilis    Bacteroides ovatus    Bacteroides thetaiotaomicron    Bacteroides uniformis    Bacteroides vulgatus    Parabacteroides distasonis
The following in vitro data are available, but their clinical significance is unknown. At least 90 percent of the following bacteria exhibit an in vitro minimum inhibitory concentration (MIC) less than or equal to the susceptible breakpoint for eravacycline against isolates of similar genus or organism group. However, the efficacy of eravacycline in treating clinical infections caused by these bacteria has not been established in adequate and well-controlled clinical trials.
Aerobic bacteria
   Gram-positive bacteria   Streptococcus salivarius group
   Gram-negative bacteria   Citrobacter koseri    Enterobacter aerogenes
Susceptibility Test Methods
For specific information regarding susceptibility test interpretive criteria, and associated test methods and quality control standards recognized by FDA for this drug, please see https://www.fda.gov/STIC.
13 Nonclinical Toxicology
13.1 Carcinogenesis, Mutagenesis, Impairment of Fertility
Carcinogenicity studies with eravacycline have not been conducted. However, there has been evidence of oncogenic activity in rats in studies with the related antibacterial drugs, oxytetracycline (adrenal and pituitary tumors) and minocycline (thyroid tumors).
Eravacycline was not genotoxic in a standard battery of assays, including an in vitro mammalian cell mutation assay, an in vitro clastogenicity assay, and an in vivo rat bone marrow micronucleus assay.
There are no human data on the effect of eravacycline on fertility. Eravacycline did not affect mating or fertility in male rats following intravenous administration at a dose approximating a clinical dose of 0.65 mg/kg/day (approximately 1.5 times the clinical exposure, based on AUC determined in a separate study), however, eravacycline administration at higher doses was associated with adverse reactions on male fertility and spermatogenesis that were at least partially reversible after a 70-day recovery period (1Â spermatogenic cycle). Decreased sperm counts, abnormal sperm morphology, and reduced sperm motility were observed with testicular effects (impaired spermiation and sperm maturation). There were no adverse reactions on mating or fertility in female rats administered intravenous eravacycline at a dose approximating a clinical dose of 3.2 mg/kg/day (approximately 18 times the clinical exposure based on AUC determined in a separate study in unmated females).
Decreased sperm counts and eravacycline‑related lesions noted in the testes and epididymides were seen in general toxicology studies in rats and were reversible. These findings were anticipated effects for a tetracycline-class compound.
13.2 Animal Toxicology and/or Pharmacology
In repeated dose toxicity studies in rats, dogs and monkeys, lymphoid depletion/atrophy of lymph nodes, spleen and thymus, decreased erythrocytes, reticulocytes, leukocytes, and platelets (dog and monkey), in association with bone marrow hypocellularity, and adverse gastrointestinal effects (dog and monkey) were observed with eravacycline. These findings were reversible or partially reversible during recovery periods of 3 to 7 weeks.
Bone discoloration, which was not fully reversible over recovery periods of up to 7 weeks, was observed in rats and monkeys after 13 weeks of dosing and in the juvenile rat study after dosing over post-natal days 21-70.
Intravenous administration of eravacycline has been associated with a histamine response in rat and dog studies.
14 Clinical Studies
14.2 Complicated Urinary Tract Infections (cUTI) in Adults
Two randomized, double-blind, active-controlled, clinical trials (Trial 4, NCT01978938, and Trial 5, NCT03032510) evaluated the efficacy and safety of once-daily intravenous eravacycline for the treatment of patients with complicated urinary tract infections (cUTI). Trial 4 included an optional switch from IV to oral therapy with eravacycline. The trials did not demonstrate the efficacy of XERAVA for the combined endpoints of clinical cure and microbiological success in the microbiological intent-to-treat (micro-ITT) population at the test-of-cure visit [see Indications and Usage (1)].
14.1 Complicated Intra-abdominal Infections in Adults
A total of 1,041 adults hospitalized with cIAI were enrolled in two Phase 3, randomized, double-blind, active-controlled, multinational, multicenter trials (Trial 1, NCT01844856, and Trial 2, NCT02784704). These studies compared XERAVA (1 mg/kg intravenous every 12 hours) with either ertapenem (1 g every 24 hours) or meropenem (1 g every 8 hours) as the active comparator for 4 to 14 days of therapy. Complicated intra-abdominal infections included appendicitis, cholecystitis, diverticulitis, gastric/duodenal perforation, intra-abdominal abscess, perforation of intestine, and peritonitis.
The microbiologic intent-to-treat (micro-ITT) population, which included all patients who had at least one baseline intra-abdominal pathogen, consisted of 846 patients in the two trials. Populations in Trial 1 and Trial 2 were similar. The median age was 56 years and 56% were male. The majority of patients (95%) were from Europe; 5% were from the United States. The most common primary cIAI diagnosis was intra-abdominal abscess(es), occurring in 60% of patients. Bacteremia at baseline was present in 8% of patients.
Clinical cure was defined as complete resolution or significant improvement of signs or symptoms of the index infection at the Test of Cure (TOC) visit which occurred 25 to 31 days after randomization. Selected clinical responses were reviewed by a Surgical Adjudication Committee. Table 3 presents the clinical cure rates in the micro-ITT population. Clinical cure rates at the TOC visit for selected pathogens are presented in Table 4.
Table 3 Clinical Cure Rates at TOC in the Phase 3 cIAI Trials, Micro-ITT Population Trial 1 Trial 2 XERAVA a N=220 n (%) Ertapenem b N=226 n (%) XERAVA a N=195 n (%) Meropenem c N=205 n (%) Abbreviations: CI = confidence interval; IV = intravenous; micro-ITT = all randomized subjects who had baseline bacterial pathogens that caused cIAI and against at least one of which the investigational drug has in vitro antibacterial activity; N = number of subjects in the micro-ITT population; n = number within a specific category with a clinical cure based on the Surgical Adjudication Committee assessment (if available); TOC = Test of Cure. a XERAVA dose equals 1 mg/kg every 12 hours IV. b Ertapenem dose equals1 g every 24 hours IV c Meropenem dose equals 1 g every 8 hours IV. d Treatment Difference = Difference in clinical cure rates (eravacycline minus ertapenem or meropenem). Confidence intervals are calculated using the unadjusted Miettinen-Nurminen method
Clinical cure
191 (86.8)
198 (87.6)
177 (90.8)
187 (91.2)
Difference (95% CI) d
-0.80 (-7.1, 5.5)
-0.5 (-6.3, 5.3)
Table 4 Clinical Cure Rates at TOC by Selected Baseline Pathogens in Pooled Phase 3 cIAI Trials, Micro-ITT Population Pathogen XERAVA a N=415 n/N1 (%) Comparators b N=431 n/N1 (%) Abbreviations: IV = intravenous; N = Number of subjects in the micro-ITT Population; N1 = Number of subjects with a specific pathogen; n = Number of subjects with a clinical cure at the TOC visit. Percentages are calculated as 100 × (n/N1); TOC = Test of Cure a XERAVA dose equals 1 mg/kg every 12 hours IV. b Comparators include Ertapenem 1 g every 24 hours IV and Meropenem 1 g every 8 hours IV. c Includes Streptococcus anginosus, Streptococcus constellatus, and Streptococcus intermedius d Includes Bacteroides caccae, Bacteroides fragilis, Bacteroides ovatus, Bacteroides thetaiotaomicron, Bacteroides uniformis, Bacteroides vulgatus, Clostridium perfringens, and Parabacteroides distasonis.
Enterobacteriaceae
271/314 (86.3)
289/325 (88.9)
Citrobacter freundii
19/22 (86.4)
8/10 (80.0)
Enterobacter cloacae complex
17/21 (81.0)
23/24 (95.8)
Escherichia coli
220/253 (87.0)
237/266 (89.1)
Klebsiella oxytoca
14/15 (93.3)
16/19 (84.2)
Klebsiella pneumoniae
37/39 (94.9)
42/50 (84.0)
Enterococcus faecalis
45/54 (83.3)
47/54 (87.0)
Enterococcus faecium
38/45 (84.4)
48/53 (90.6)
Staphylococcus aureus
24/24 (100.0)
12/14 (85.7)
Streptococcus anginosus group c
79/92 (85.9)
50/59 (84.7)
Anaerobes d
186/215 (86.5)
194/214 (90.7)
16 How Supplied/storage And Handling
16.1 How Supplied
XERAVA (eravacycline) for injection is a yellow to orange, sterile, preservative-free powder for reconstitution in single-dose 10-mL clear glass vials with a rubber stopper and an aluminum overseal. XERAVA is supplied in the following configurations:
- XERAVA 50 mg vial contains 50 mg of eravacycline (equivalent to 63.5 mg of eravacycline dihydrochloride). Each vial is packaged in a single-dose vial carton (NDC 71773-050-05) and supplied in a 12-vial carton containing 12 single-dose vial cartons -- NDC 71773-050-12.
- XERAVA 100 mg vial contains 100 mg of eravacycline (equivalent to 127 mg of eravacycline dihydrochloride). Each vial is packaged in a single-dose vial carton (NDC 71773-100-05) and supplied in shrink wrap packaging, containing 12 single-dose vial cartons -- NDC 71773-100-12.
16.2 Storage and Handling
Prior to reconstitution, XERAVA should be stored at 2°C to 8°C (36°F to 46°F) [see Dosage and Administration 2.4]. Keep vial in carton until use.
17 Patient Counseling Information
Serious Allergic Reactions
Advise patients that allergic reactions, including serious allergic reactions, could occur and that serious reactions require immediate treatment. Ask patient about any previous hypersensitivity reactions to antibacterial drugs including tetracycline or other allergens [see Warnings and Precautions ( 5.1)] .
Tooth Discoloration and Inhibition of Bone Growth
Advise patients that XERAVA, like other tetracycline-class drugs, may cause permanent tooth discoloration of deciduous teeth and reversible inhibition of bone growth when administered during the second and third trimesters of pregnancy. Advise patients that they should tell their healthcare provider right away if they become pregnant [see Warnings and Precautions ( 5.2, 5.3) and Use in Specific Populations ( 8.1, 8.4)] .
Lactation
Advise women not to breastfeed during treatment with XERAVA and for 4 days after the last dose [see Use in Specific Populations ( 8.2)] .
Diarrhea
Diarrhea is a common problem caused by antibacterial drugs, including XERAVA, which usually ends when the antibacterial drug is discontinued. Sometimes after starting treatment with antibacterial drug, patients can develop watery and bloody stools (with or without stomach cramps and fever) which may be a sign of a more serious intestinal infection, even as late as 2 or more months after having taken the last dose of the antibacterial drug. If this occurs, instruct patients to contact their healthcare provider as soon as possible [see Warnings and Precautions ( 5.4)] .
Tetracycline Class Adverse Reactions
Inform patients that XERAVA is similar to tetracycline-class antibacterial drugs and may have similar adverse reactions [see Warnings and Precautions ( 5.5)] .
Overgrowth of Non-susceptible Microorganisms
Inform patients that antibacterial drugs including XERAVA may promote the overgrowth of non-susceptible microorganisms, including fungi [see Warnings and Precautions ( 5.6)] .
Antibacterial Resistance
Inform patients that antibacterial drugs including XERAVA should only be used to treat bacterial infections. They do not treat viral infections (for example, the common cold). When XERAVA is prescribed to treat a bacterial infection, patients should be told that although it is common to feel better early in the course of therapy, the medication should be taken exactly as directed. Skipping doses or not completing the full course of therapy may (1) decrease the effectiveness of the immediate treatment and (2) increase the likelihood that bacteria will develop resistance and will not be treatable by XERAVA or other antibacterial drugs in the future [see Warnings and Precautions ( 5.7)] .
Distributed by: Tetraphase Pharmaceuticals, Inc.Waltham, MA 02451
XERAVA® is a registered trademark of Tetraphase Pharmaceuticals, Inc. ©2021, Tetraphase Pharmaceuticals, Inc. All rights reserved.
Principal Display Panel - 50 Mg Carton Label

NDC 71773-050-05 Rx only
XERAVA® (eravacycline) for injection
50 mg per vial
MUST BE RECONSTITUTED THEN DILUTED. For Intravenous Infusion Only. See Prescribing Information for complete directions for use.
1 Single-Dose Vial Not labeled for individual sale
TETRAPHASE PHARMACEUTICALS
Before reconstitution, refrigerate at 2°C to 8°C (36°F-46°F). Protect from light; keep vial in carton until use.
Transfer the reconstituted solution from the vial into the IV bag within 1 hour. Use the diluted solution in the IV bag within 24 hours when stored at room temperature or within 10 days when stored refrigerated. Discard unused portion.
NOTE: Parenteral drug products should be inspected visually for particulate matter after reconstitution.
Each vial provides 50 mg eravacycline equivalent to 63.5 mg eravacycline dihydrochloride. Inactive ingredients: mannitol, sodium hydroxide, and hydrochloric acid. Sterile powder, preservative-free.
Distributed by: Tetraphase Pharmaceuticals, Inc. Waltham, MA 02451 USA
Lot:
Exp:
Principal Display Panel - 100 Mg Carton Label
NDC 71773-100-05 Rx only
XERAVA® (eravacycline) for injection
100 mg per vial
MUST BE RECONSTITUTED THEN DILUTED. For Intravenous Infusion Only. See Prescribing Information for complete directions for use.
1 Single-Dose Vial Not labeled for individual sale
TETRAPHASE PHARMACEUTICALS
Each vial provides 100 mg eravacycline equivalent to 127 mg eravacycline dihydrochloride. Inactive ingredients: mannitol, sodium hydroxide, and hydrochloric acid. Sterile powder, preservative-free.
Before reconstitution, refrigerate at 2°C to 8°C (36°F-46°F). Protect from light; keep vial in carton until use.
Transfer the reconstituted solution into the IV bag within 1 hour. Use the diluted solution in the IV bag within 24 hours when stored at room temperature or within 10 days when stored refrigerated. Discard unused portion.
NOTE: Parenteral drug products should be inspected visually for particulate matter after reconstitution.
Distributed by:
Tetraphase Pharmaceuticals, Inc.
Waltham, MA 02451 USA
LOT
EXP
DISCLAIMER:
"This tool does not provide medical advice, and is for informational and educational purposes only, and is not a substitute for professional medical advice, treatment or diagnosis. Call your doctor to receive medical advice. If you think you may have a medical emergency, please dial 911."
"Do not rely on openFDA to make decisions regarding medical care. While we make every effort to ensure that data is accurate, you should assume all results are unvalidated. We may limit or otherwise restrict your access to the API in line with our Terms of Service."
"This product uses publicly available data from the U.S. National Library of Medicine (NLM), National Institutes of Health, Department of Health and Human Services; NLM is not responsible for the product and does not endorse or recommend this or any other product."
PillSync may earn a commission via links on our site
